Poly(3-hydroxybutyrate) Modified by Plasma and TEMPO-Oxidized Celluloses

 , , , ,
, , , ,
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
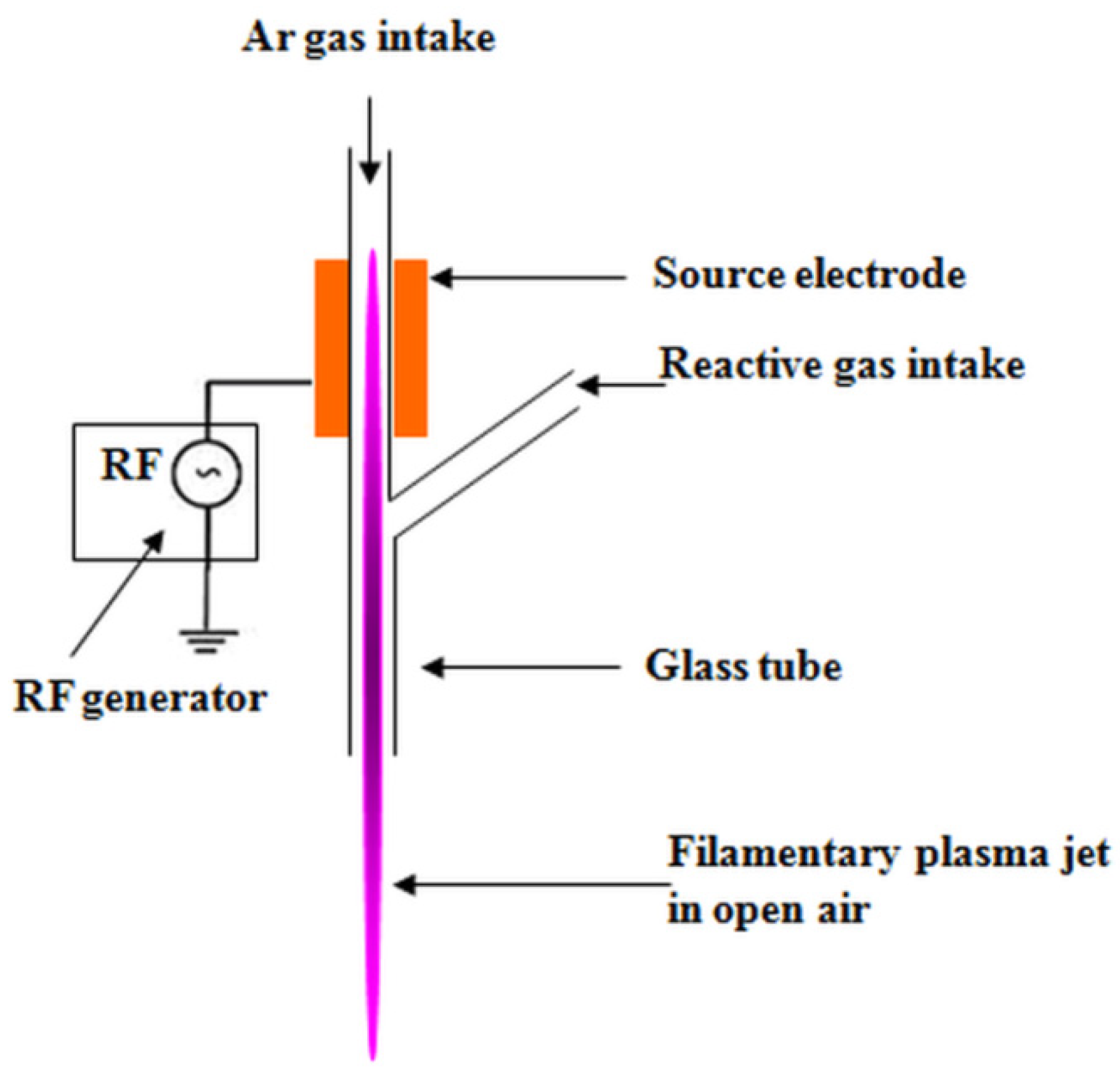
2.2. Plasma Treatment of Cellulose Suspensions
2.3. TEMPO Mediated Oxidation of MCC
2.4. Preparation of PHB Composites with Untreated, Plasma and TEMPO Oxidized MCC
2.5. Characterization
2.5.1. SEM Investigation
2.5.2. Chemical Characterization by FTIR and XPS
2.5.3. Thermal Characterization
2.5.4. Differential Scanning Calorimetry (DSC)
2.5.5. DMA Characterization
3. Results and Discussion
3.1. Characterization of MCC, MCC-P and MCC-T
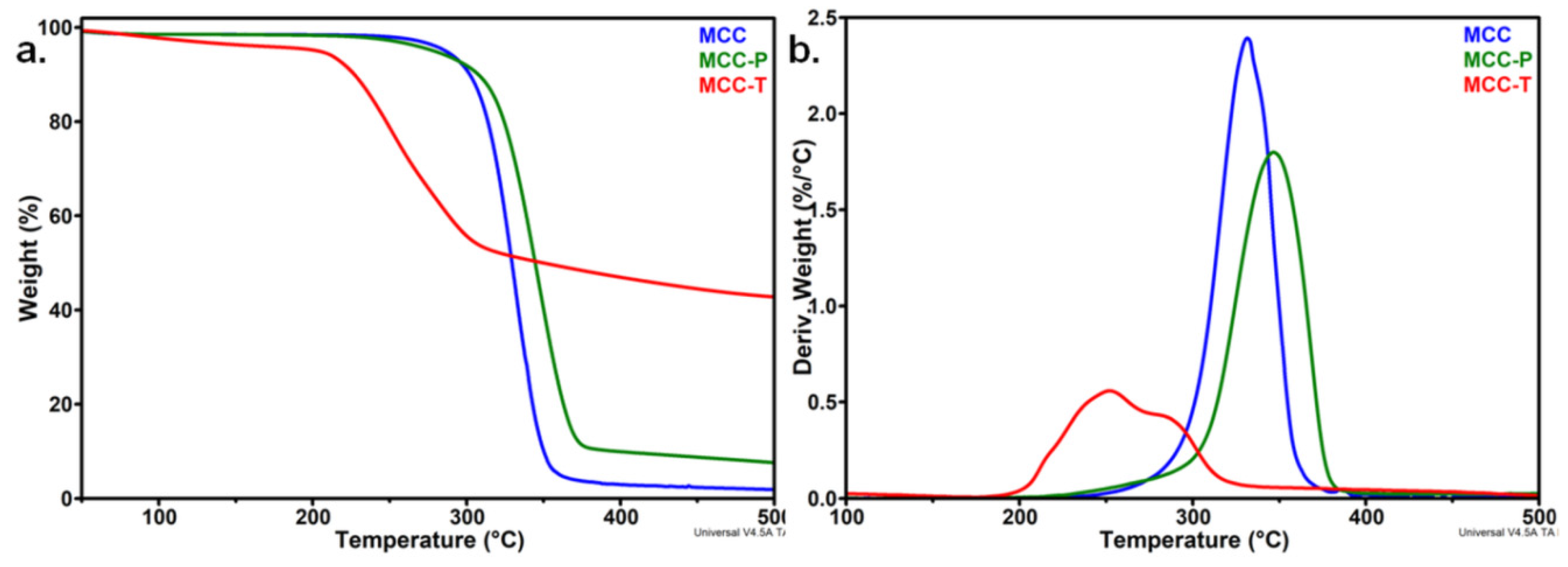
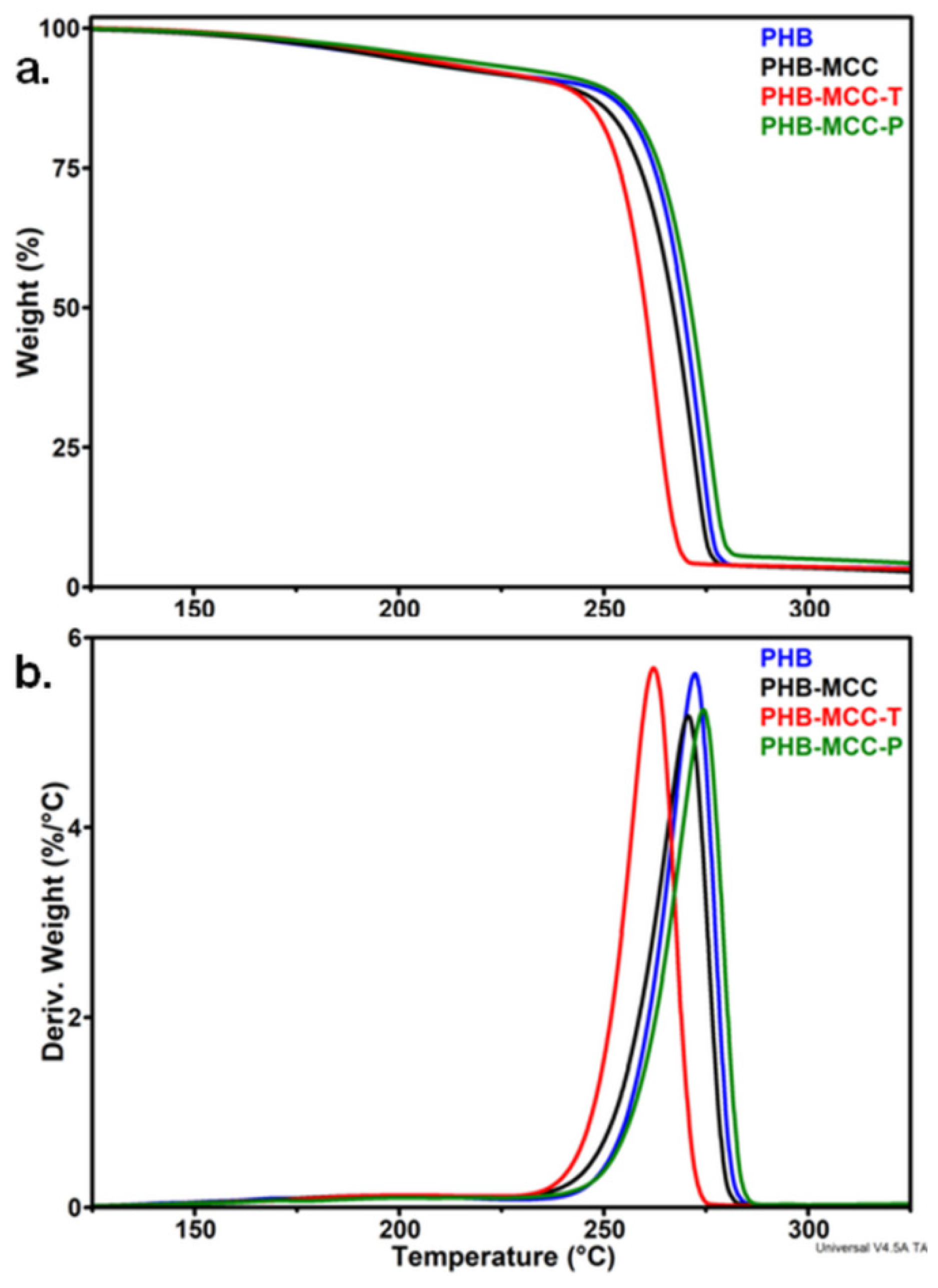
3.1.1. Effect of the Treatments on the Thermal Stability of MCC
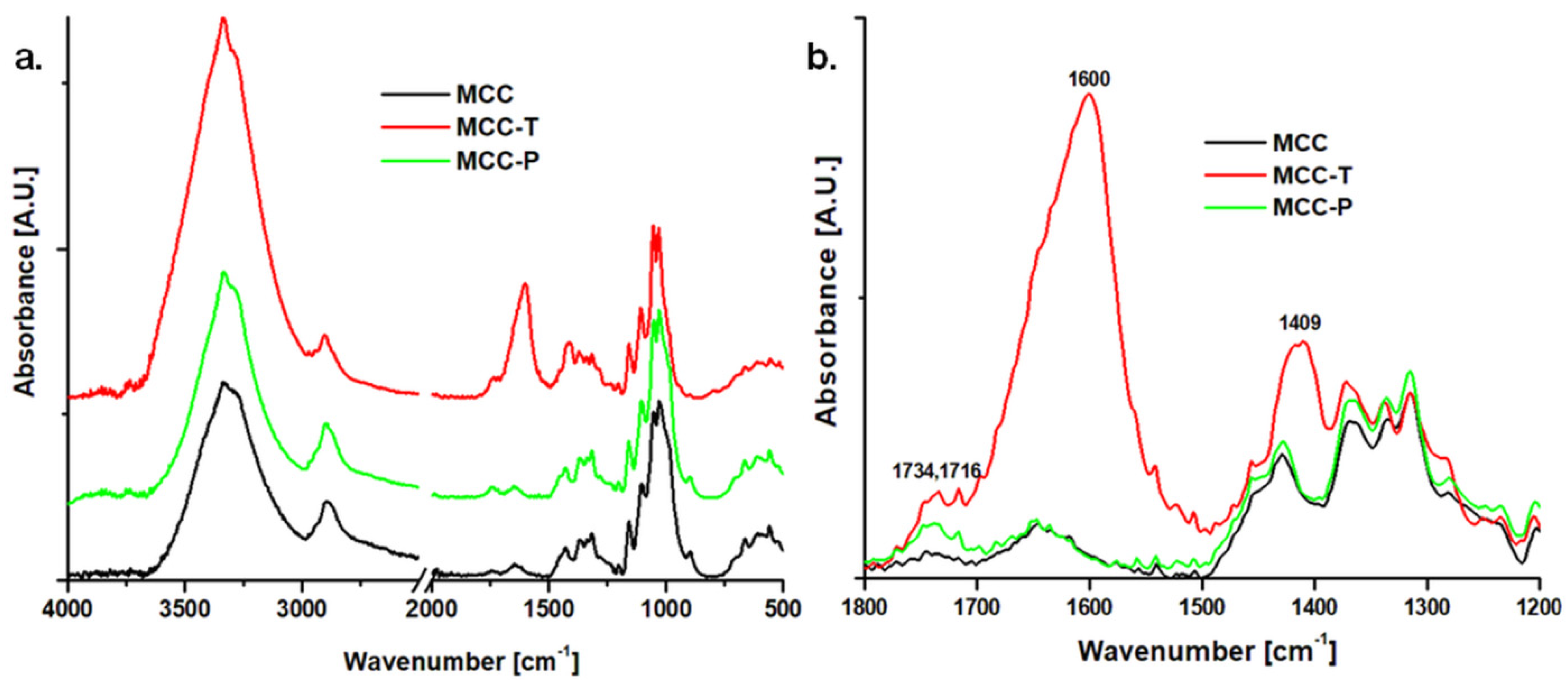
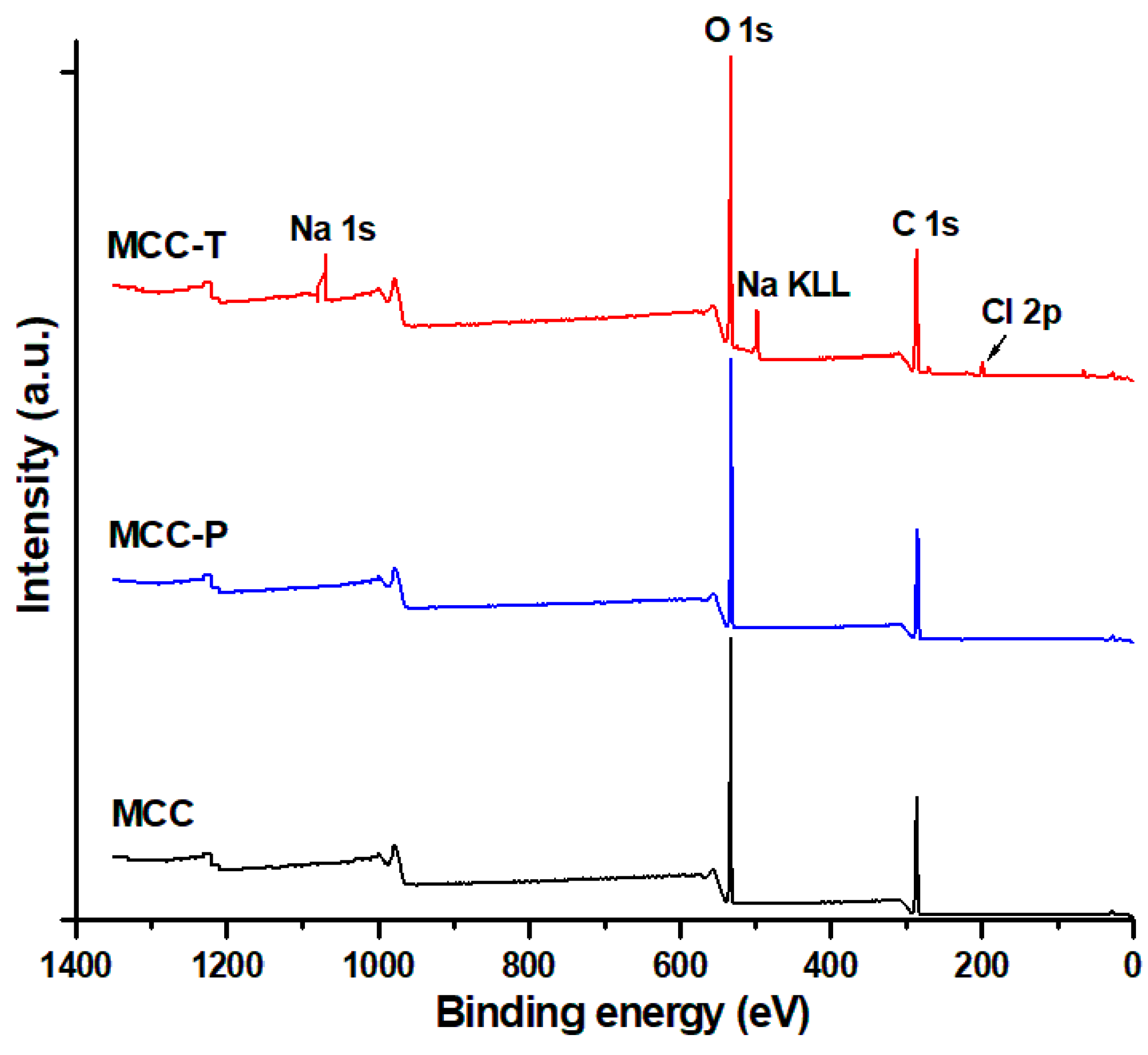
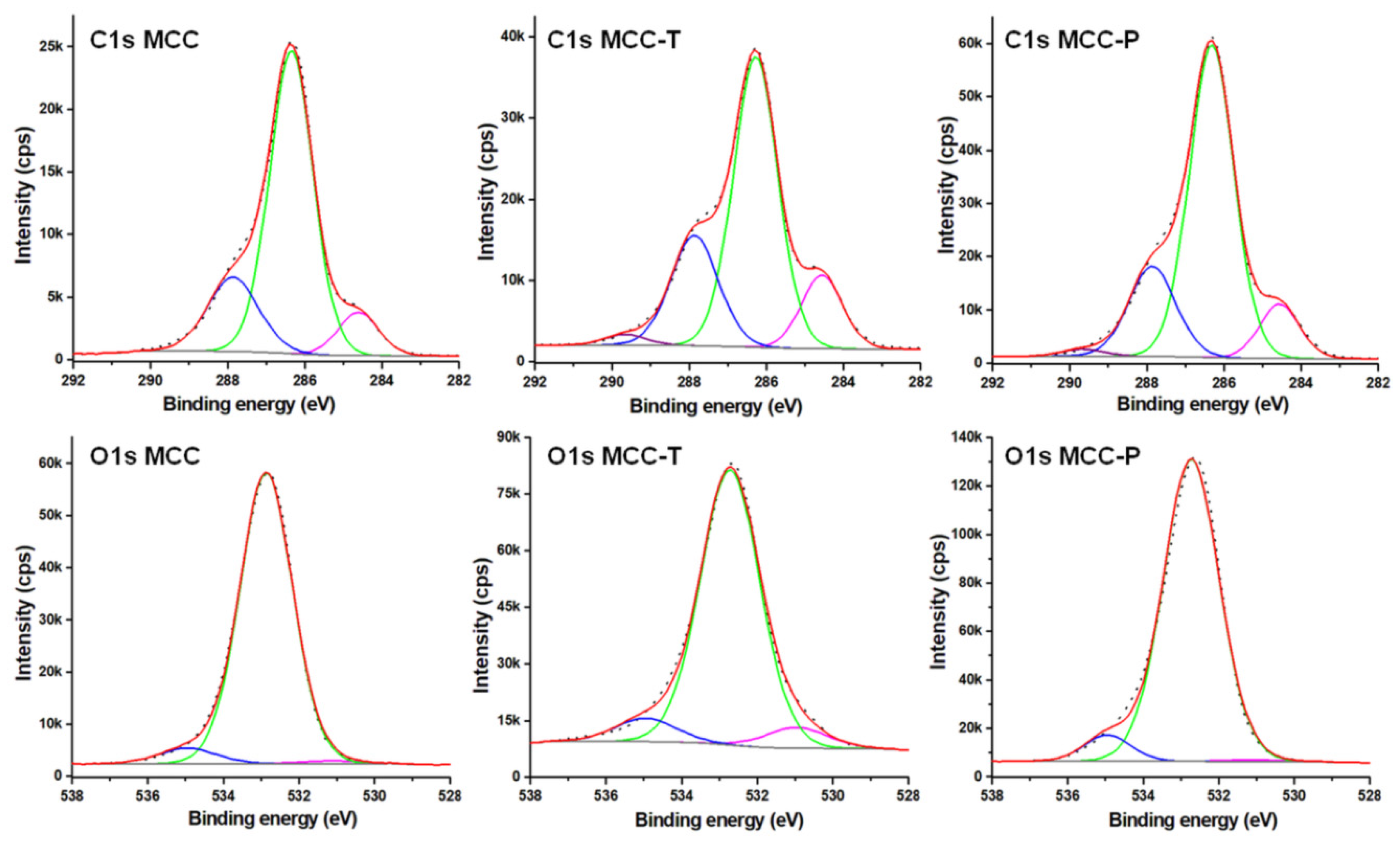
3.1.2. Surface Chemical Properties of Modified Celluloses
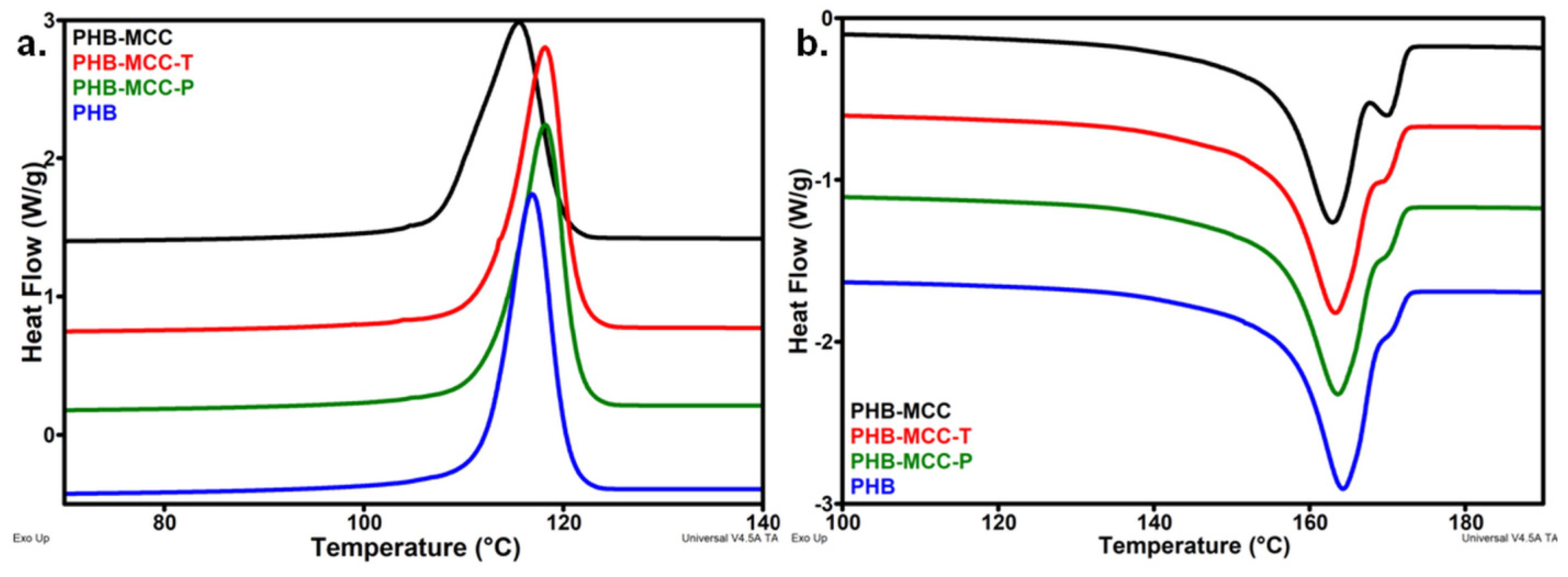
3.2. Thermal Properties of PHB Composites with Different Treated MCC
3.3. SEM Images of PHB Composites with Treated MCC
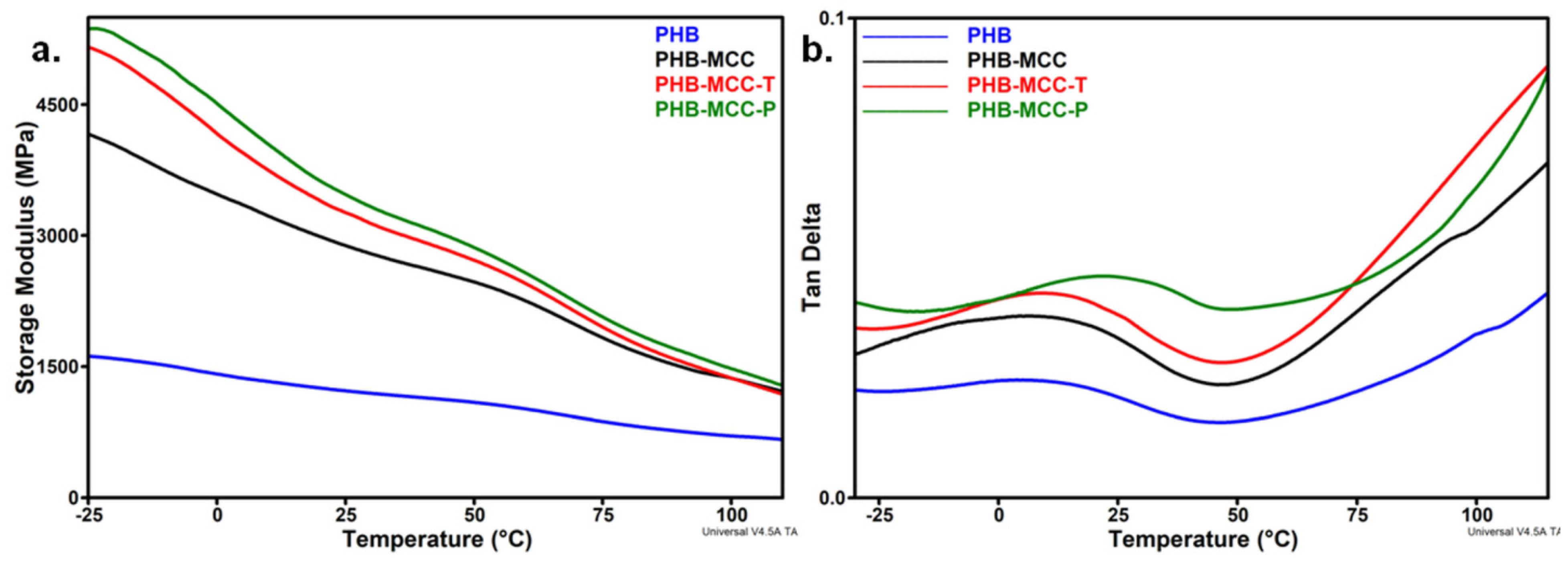
3.4. DMA Analysis of PHB Composites with Treated MCC
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gandini, A.; Belgacem, M.N. The state of the art of polymers from renewable resources. In Handbook of Biopolymers and Biodegradable Plastics: Properties, Processing and Applications, 1st ed.; Ebnesajjad, S., Ed.; Elsevier Inc.: Kidlington, Oxford, UK, 2013; Chapter 4; pp. 71–85. [Google Scholar]
- Kargarzadeh, H.; Huang, J.; Lin, N.; Ahmad, I.; Marino, M.; Dufresne, A.; Thomas, S.; Gałęski, A. Recent developments in nanocellulose-based biodegradable polymers, thermoplastic polymers, and porous nanocomposites. Prog. Polym. Sci. 2018, 87, 197–227. [Google Scholar] [CrossRef]
- Wróblewska-Krepsztul, J.; Rydzkowski, T.; Michalska-Pożoga, I.; Thakur, V.K. Biopolymers for Biomedical and Pharmaceutical Applications: Recent Advances and Overview of Alginate Electrospinning. Nanomaterials (Basel) 2019, 9, 404. [Google Scholar]
- Albuquerque, P.B.S.; Malafaia, C.B. Perspectives on the production, structural characteristics and potential applications of bioplastics derived from polyhydroxyalkanoates. Int. J. Biol. Macromol. 2018, 107, 615–625. [Google Scholar] [CrossRef]
- Panaitescu, D.M.; Nicolae, C.A.; Gabor, A.R.; Trusca, R. Thermal and mechanical properties of poly(3-hydroxybutyrate) reinforced with cellulose fibers from wood waste. Ind. Crops Prod. 2020, 145, 112071. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, W.; Wang, X.; Chen, X.; Chen, G.-Q.; Xu, K. Processability modifications of poly(3-hydroxybutyrate) by plasticizing, blending, and stabilizing. J. Appl. Polym. Sci. 2008, 107, 166–173. [Google Scholar] [CrossRef]
- Srithep, Y.; Ellingham, T.; Peng, J.; Sabo, R.; Clemons, C.; Turng, L.-S.; Pilla, S. Melt compounding of poly (3-hydroxybutyrate-co-3-hydroxyvalerate)/nanofibrillated cellulose nanocomposites. Polym. Degrad. Stab. 2013, 98, 1439–1449. [Google Scholar] [CrossRef]
- Panaitescu, D.M.; Nicolae, C.A.; Frone, A.N.; Chiulan, I.; Stanescu, P.O.; Draghici, C.; Iorga, M.; Mihailescu, M. Plasticized poly(3-hydroxybutyrate) with improved melt processing and balanced properties. J. Appl. Polym. Sci. 2017, 134, 44810. [Google Scholar] [CrossRef]
- Frone, A.N.; Panaitescu, D.M.; Chiulan, I.; Nicolae, C.A.; Vuluga, Z.; Vitelaru, C.; Damian, M.C. The effect of cellulose nanofibers on the crystallinity and nanostructure of poly (lactic acid) composites. J. Mater. Sci. 2016, 51, 9771–9791. [Google Scholar] [CrossRef]
- Garcia-Garcia, D.; Lopez-Martinez, J.; Balart, R.; Strömberg, E.; Moriana, R. Reinforcing capability of cellulose nanocrystals obtained from pine cones in a biodegradable poly(3-hydroxybutyrate)/poly(ε-caprolactone) (PHB/PCL) thermoplastic blend. Eur. Polym. J. 2018, 104, 10–18. [Google Scholar] [CrossRef]
- Panaitescu, D.M.; Frone, A.N.; Chiulan, I.; Gabor, R.A.; Spataru, I.C.; Căşărică, A. Biocomposites from polylactic acid and bacterial cellulose nanofibers obtained by mechanical treatment. BioResources 2017, 12, 662–672. [Google Scholar] [CrossRef]
- Ambrosio-Martin, J.; Fabra, M.J.; Lopez-Rubio, A.; Gorrasi, G.; Sorrentino, A.; Lagaron, J.M. Assessment of ball milling as a compounding technique to develop nanocomposites of poly(3-hydroxybutyrate-co-3- hydroxyvalerate) and bacterial cellulose nanowhiskers. J. Polym. Environ. 2016, 24, 241–254. [Google Scholar] [CrossRef]
- Nechyporchuk, O.; Belgacem, M.N.; Bras, J. Production of cellulose nanofibrils: A review of recent advances. Ind. Crops Prod. 2016, 93, 2–25. [Google Scholar] [CrossRef]
- Agarwal, C.; Csoka, L. Surface-Modified Cellulose in Biomedical Engineering In Materials for Biomedical Engineering: Bioactive Materials, Properties, and Applications; Grumezescu, V., Grumezescu, A.M., Eds.; Elsevier Radarweg: Amsterdam, The Netherlands, 2019; Chapter 6; pp. 251–261. [Google Scholar]
- Frone, A.N.; Chiulan, I.; Panaitescu, D.M.; Cristian, A.N.; Ghiurea, M.; Galan, A.-M. Isolation of cellulose nanocrystals from plum seed shells, structural and morphological characterization. Mater. Lett. 2017, 194, 160–163. [Google Scholar] [CrossRef]
- Saito, T.; Isogai, A. TEMPO-mediated oxidation of native cellulose. The effect of oxidation conditions on chemical and crystal structures of the water-insoluble fractions. Biomacromolecules 2004, 5, 1983–1989. [Google Scholar] [CrossRef]
- Besbes, I.; Alila, S.; Boufi, S. Nanofibrillated cellulose from TEMPO-oxidized eucalyptus fibres: Effect of the carboxyl content. Carbohydr. Polym. 2011, 84, 975–983. [Google Scholar] [CrossRef]
- Lavoine, N.; Desloges, I.; Dufresne, A.; Bras, J. Microfibrillated cellulose—Its barrier properties and applications in cellulosic materials: A review. Carbohydr. Polym. 2012, 90, 735–764. [Google Scholar] [CrossRef]
- Missoum, K.; Belgacem, M.N.; Bras, J. Nanofibrillated Cellulose Surface Modification: A Review. Materials (Basel) 2013, 6, 1745–1766. [Google Scholar] [CrossRef]
- Panaitescu, D.M.; Vizireanu, S.; Nicolae, C.A.; Frone, A.N.; Casarica, A.; Carpen, L.G.; Dinescu, G. Treatment of nanocellulose by submerged liquid plasma for surface functionalization. Nanomaterials (Basel) 2018, 8, 467. [Google Scholar] [CrossRef]
- Vizireanu, S.; Panaitescu, D.M.; Nicolae, C.A.; Frone, A.N.; Chiulan, I.; Ionita, M.D.; Satulu, V.; Carpen, L.G.; Petrescu, S.; Birjega, R.; et al. Cellulose defibrillation and functionalization by plasma in liquid treatment. Sci. Rep. 2018, 8, 15473. [Google Scholar] [CrossRef]
- International Organisation for Standardization (ISO). ISO 527-2:2012: Plastics—Determination of Tensile Properties—Part. 2: Test. Conditions for Moulding and Extrusion Plastics; International Organization for Standardization: Geneva, Switzeland, 2012. [Google Scholar]
- Teodorescu, M.; Bazavan, M.; Ionita, E.R.; Dinescu, G. Characteristics of a long and stable filamentary argon plasma jet generated in ambient atmosphere. Plasma Sources Sci. Technol. 2015, 24, 25033. [Google Scholar] [CrossRef]
- Carpen, L.G.; Chireceanu, C.; Teodorescu, M.; Chiriloaie, A.; Teodoru, A.; Dinescu, G. The effect of argon/oxygen and argon/nitrogen atmospheric plasma jet on stored products pests. Rom. J. Phys. 2019, 64, 503. [Google Scholar]
- Barham, P.J.; Keller, A.; Otun, E.L.; Holmer, P.A. Crystallization and morphology of a bacterial thermoplastic: Poly-3-hydroxybutyrate. J. Mater. Sci. 1984, 19, 2781–2794. [Google Scholar] [CrossRef]
- Baruah, J.; Deka, R.C.; Kalita, E. Greener production of microcrystalline cellulose (MCC) from Saccharum spontaneum (Kans grass): Statistical optimization. Int. J. Biol. Macromol. 2020, 154, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Fukuzumi, H.; Saito, T.; Iwata, T.; Kumamoto, Y.; Isogai, A. Transparent and high gas barrier films of cellulose nanofibers prepared by TEMPO-mediated oxidation. Biomacromolecules 2009, 10, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Fukuzumi, H.; Saito, T.; Okita, Y.; Isogai, A. Thermal stabilization of TEMPO-oxidized cellulose. Polym. Degrad. Stab. 2010, 95, 1502–1508. [Google Scholar] [CrossRef]
- Loof, D.; Hiller, M.; Oschkinat, H.; Koschek, K. Quantitative and qualitative analysis of surface modified cellulose utilizing TGA-MS. Materials (Basel) 2016, 9, 415. [Google Scholar] [CrossRef]
- Peng, Y.; Gardner, D.J.; Han, Y.; Kiziltas, A.; Cai, Z.; Tshabalala, M.A. Influence of drying method on the material properties of nanocellulose I: Thermostability and crystallinity. Cellulose 2013, 20, 2379–2392. [Google Scholar] [CrossRef]
- Piskorz, J.; Radlein, D.S.A.G.; Donald, S.S.; Czernik, S. Pretreatment of wood and cellulose for production of sugars by fast pyrolysis. J. Anal. Appl. Pyrolysis 1989, 16, 127–142. [Google Scholar] [CrossRef]
- Oh, S.Y.; Yoo, D.I.; Shin, Y.; Kim, H.C.; Kim, H.Y.; Chung, Y.S.; Park, W.H.; Youk, J.H. Crystalline structure analysis of cellulose treated with sodium hydroxide and carbon dioxide by means of X-ray diffraction and FTIR spectroscopy. Carbohydr. Res. 2005, 340, 2376–2391. [Google Scholar] [CrossRef]
- Gwon, J.G.; Lee, S.Y.; Doh, G.H.; Kim, J.H. Characterization of chemically modified wood fibers using ftir spectroscopy for biocomposites. J. Appl. Polym. Sci. 2010, 116, 3212–3219. [Google Scholar] [CrossRef]
- Sehaqui, H.; de Larraya, U.P.; Liu, P.; Pfenninger, N.; Mathew, A.P.; Zimmermann, T.; Tingaut, P. Enhancing adsorption of heavy metal ions onto biobased nanofibers from waste pulp residues for application in wastewater treatment. Cellulose 2014, 21, 2831–2844. [Google Scholar] [CrossRef]
- Zhu, C.; Soldatov, A.; Mathew, A.P. Advanced microscopy and spectroscopy reveal the adsorption and clustering of Cu(II) onto TEMPO oxidized cellulose nanofibers. Nanoscale 2017, 9, 7419–7428. [Google Scholar] [CrossRef]
- Habibi, Y.; Chanzy, H.; Vignon, M.R. TEMPO-mediated surface oxidation of cellulose whiskers. Cellulose 2006, 13, 679–687. [Google Scholar] [CrossRef]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies, 3rd ed.; John Wiley & Sons Ltd.: Chichester, UK, 2001; pp. 50–67. [Google Scholar]
- Coseri, S.; Biliuta, G.; Zemljič, L.F.; Srndovic, J.S.; Larsson, P.T.; Strnad, S.; Kreže, T.; Naderi, A.; Lindstrom, T. One-shot carboxylation of microcrystalline cellulose in the presence of nitroxyl radicals and sodium periodate. RSC Adv. 2015, 5, 85889–85897. [Google Scholar] [CrossRef]
- Clark, D.T.; Cromarty, B.J.; Dilks, A. A theoretical investigation of molecular core binding and relaxation energies in a series of oxygen-containing organic molecules of interest in the study of surface oxidation of polymers. J. Polym. Sci. Polym. Chem. Ed. 1978, 16, 3173–3184. [Google Scholar] [CrossRef]
- Zhou, J.-H.; Sui, Z.-J.; Zhu, J.; Li, P.; Chen, D.; Dai, Y.-C.; Yuan, W.-K. Characterization of surface oxygen complexes on carbon nanofibers by TPD, XPS and FT-IR. Carbon 2007, 45, 785–796. [Google Scholar] [CrossRef]
- Li, S.-S.; Wang, X.-L.; An, Q.-D.; Xiao, Z.-Y.; Zhai, S.-R.; Cui, L.; Li, Z.-C. Upon designing carboxyl methylcellulose and chitosan-derived nanostructured sorbents for efficient removal of Cd(II) and Cr(VI) from water. Int. J. Biol. Macromol. 2020, 143, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; Scudiero, L.; Espinal, J.; McEwen, J.-S.; Garcia-Perez, M. Improving the deconvolution and interpretation of XPS spectra from chars by ab initio calculations. Carbon 2016, 110, 155–171. [Google Scholar] [CrossRef]
- Ariffin, H.; Nishida, H.; Shirai, Y.; Hassan, M.A. Highly selective transformation of poly[(R)-3-hydroxybutyric acid] into trans-crotonic acid by catalytic thermal degradation. Polym. Degrad. Stab. 2010, 95, 1375–1381. [Google Scholar] [CrossRef]
- Kaiser, M.R.; Anuar, H.; Razak, S.B.A. Ductile–brittle transition temperature of polylactic acid-based biocomposite. J. Thermoplast. Compos. Mater. 2011, 26, 216–226. [Google Scholar] [CrossRef]
- Kim, K.J.; Doi, Y.; Abe, H. Effect of metal compounds on thermal degradation behavior of aliphatic poly(hydroxyalkanoic acid)s. Polym. Degrad. Stab. 2008, 93, 776–785. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | C1s (%) | O1s (%) | Na1s (%) | Cl 2p (%) | O/C |
|---|---|---|---|---|---|
| MCC | 57.3 | 42.7 | - | - | 0.75 |
| MCC-T | 52.8 | 39.4 | 5.3 | 2.5 | 0.75 |
| MCC-P | 56.9 | 43.1 | - | - | 0.76 |
| Samples | C1s | O1s | |||||
|---|---|---|---|---|---|---|---|
| C1 (%) | C2 (%) | C3 (%) | C4 (%) | O1 (%) | O2 (%) | O3 (%) | |
| MCC | 9.4 | 70.3 | 20.3 | - | 1.1 | 93.4 | 5.5 |
| MCC-T | 14.0 | 59.6 | 24.3 | 2.1 | 6.7 | 85.7 | 7.6 |
| MCC-P | 10.5 | 67.4 | 20.6 | 1.4 | 0.6 | 92.7 | 6.8 |
| Characteristic Temperatures | PHB | PHB-MCC | PHB-MCC-T | PHB-MCC-P |
|---|---|---|---|---|
| T5%, °C | 195.6 | 195.7 | 200.9 | 206.8 |
| Ton, °C | 262.6 | 259.4 | 253.7 | 263.6 |
| Td, °C | 272.3 | 270.6 | 262.1 | 274.3 |
| Composites | PHB | PHB-MCC | PHB-MCC-T | PHB-MCC-P |
|---|---|---|---|---|
| Tc, °C | 116.9 | 115.5 | 118.2 | 118.2 |
| ∆Hc | 76.6 | 74.0 | 77.1 | 76.9 |
| Tm, °C | 164.2 | 163.0 | 163.3 | 163.6 |
| ∆Hm | 81.1 | 78.4 | 81.5 | 81.5 |
| Xc, % | 55.5 | 54.8 | 57.0 | 57.0 |
| Composites | PHB | PHB-MCC | PHB-MCC-T | PHB-MCC-P |
|---|---|---|---|---|
| Tg, °C | 3.8 | 5.8 | 9.2 | 21.8 |
| E’ (−25 °C), MPa | 1617 | 4164 | 5160 | 5370 |
| E’ (25 °C), MPa | 1221 | 2885 | 3264 | 3470 |
| E’ (75 °C), MPa | 871 | 1830 | 1952 | 2068 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panaitescu, D.M.; Vizireanu, S.; Stoian, S.A.; Nicolae, C.-A.; Gabor, A.R.; Damian, C.M.; Trusca, R.; Carpen, L.G.; Dinescu, G. Poly(3-hydroxybutyrate) Modified by Plasma and TEMPO-Oxidized Celluloses. Polymers 2020, 12, 1510. https://doi.org/10.3390/polym12071510
Panaitescu DM, Vizireanu S, Stoian SA, Nicolae C-A, Gabor AR, Damian CM, Trusca R, Carpen LG, Dinescu G. Poly(3-hydroxybutyrate) Modified by Plasma and TEMPO-Oxidized Celluloses. Polymers. 2020; 12(7):1510. https://doi.org/10.3390/polym12071510
Chicago/Turabian StylePanaitescu, Denis Mihaela, Sorin Vizireanu, Sergiu Alexandru Stoian, Cristian-Andi Nicolae, Augusta Raluca Gabor, Celina Maria Damian, Roxana Trusca, Lavinia Gabriela Carpen, and Gheorghe Dinescu. 2020. "Poly(3-hydroxybutyrate) Modified by Plasma and TEMPO-Oxidized Celluloses" Polymers 12, no. 7: 1510. https://doi.org/10.3390/polym12071510
APA StylePanaitescu, D. M., Vizireanu, S., Stoian, S. A., Nicolae, C.-A., Gabor, A. R., Damian, C. M., Trusca, R., Carpen, L. G., & Dinescu, G. (2020). Poly(3-hydroxybutyrate) Modified by Plasma and TEMPO-Oxidized Celluloses. Polymers, 12(7), 1510. https://doi.org/10.3390/polym12071510