Chitosan/PAMAM/Hydroxyapatite Engineered Drug Release Hydrogels with Tunable Rheological Properties

 ,
,  ,
,  ,
,  ,
,  ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
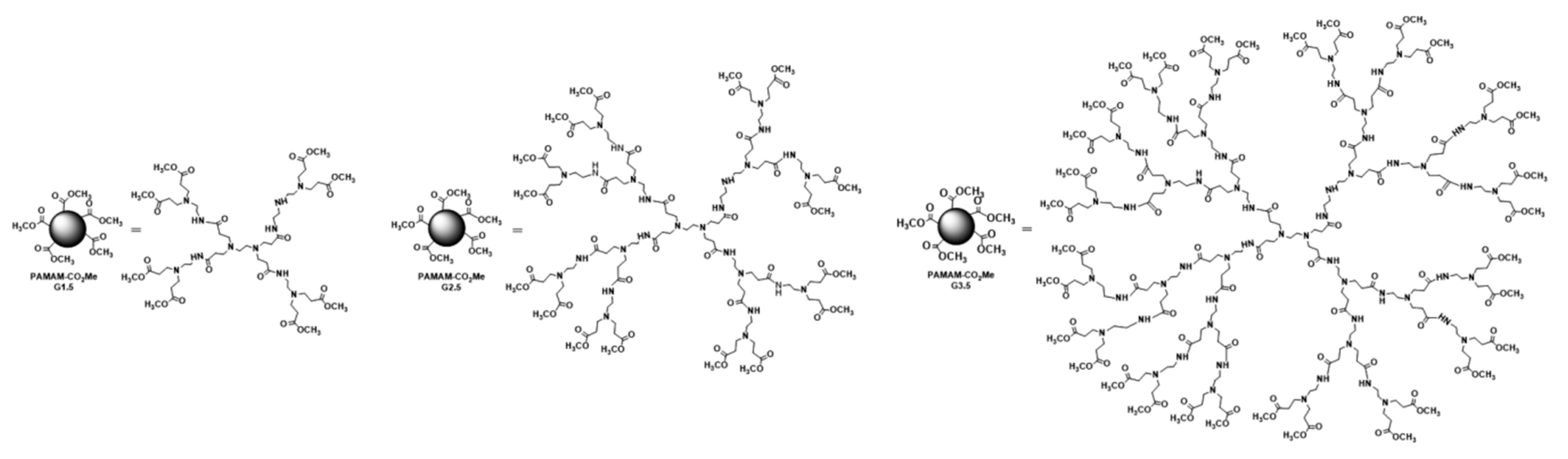
2.2.1. Synthesis of PAMAM Dendrimers and Chitosan–PAMAM Chains
2.2.2. Synthesis of Chitosan Hydrogel (CS Sample)
2.2.3. Synthesis of Chitosan-Hydroxyapatite Hydrogel (CS-HA Sample)
2.2.4. Synthesis of CS-PAMAM-HA Hydrogel (CS-D1.5-HA, CS-D2.5-HA, CS-D3.5-HA Samples)
2.2.5. Synthesis of Ketoprofen-Doped Hydrogel (CS-HA-Keto and CS-D1.5-HA-Keto, CS-D2.5-HA-Keto, CS-D3.5-HA-Keto Samples)
2.2.6. Drug Release Studies
2.2.7. Rheological Studies
3. Results and Discussion
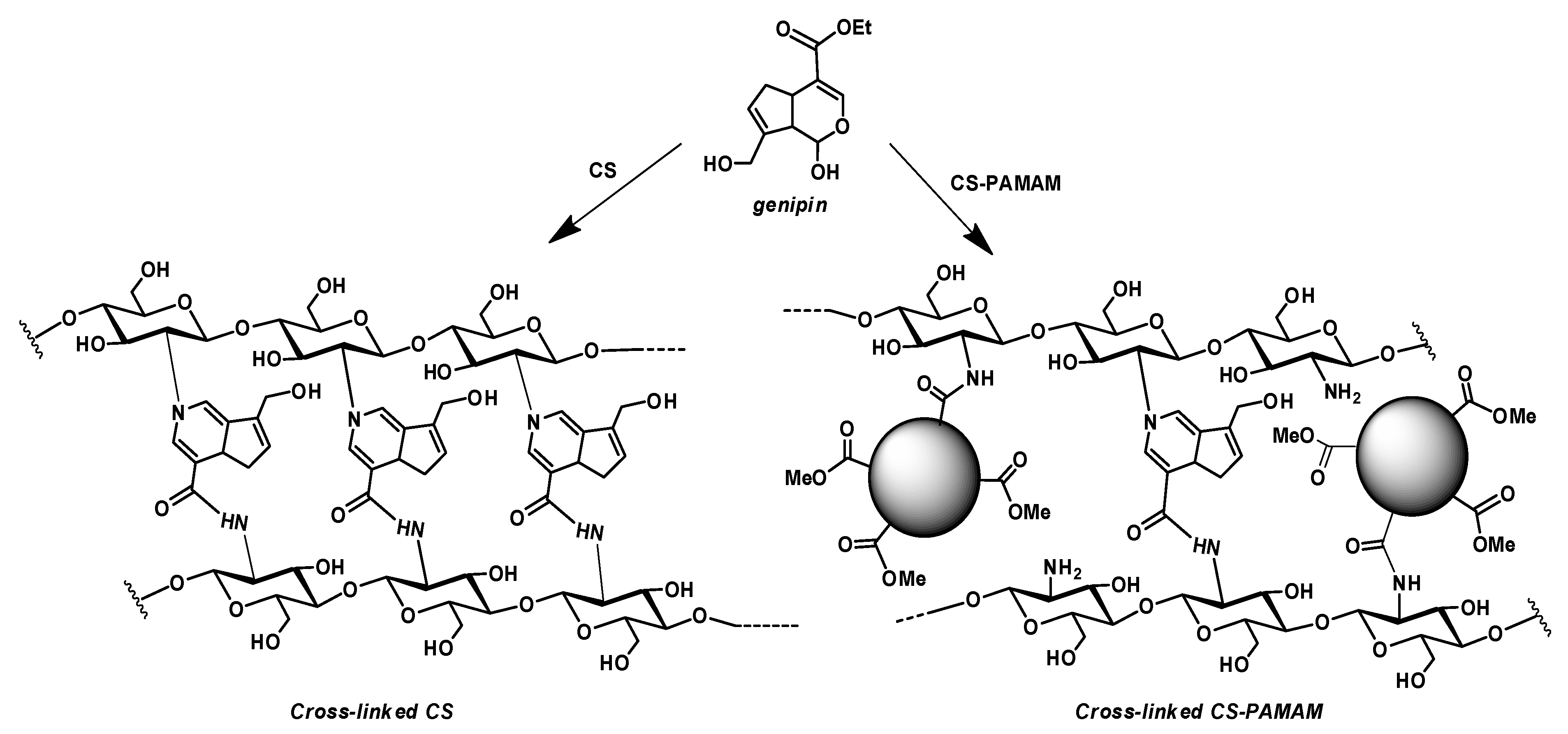
3.1. Synthesis of Chitosan-Based Hydrogels
3.1.1. Synthesis of Chitosan–PAMAM Chains
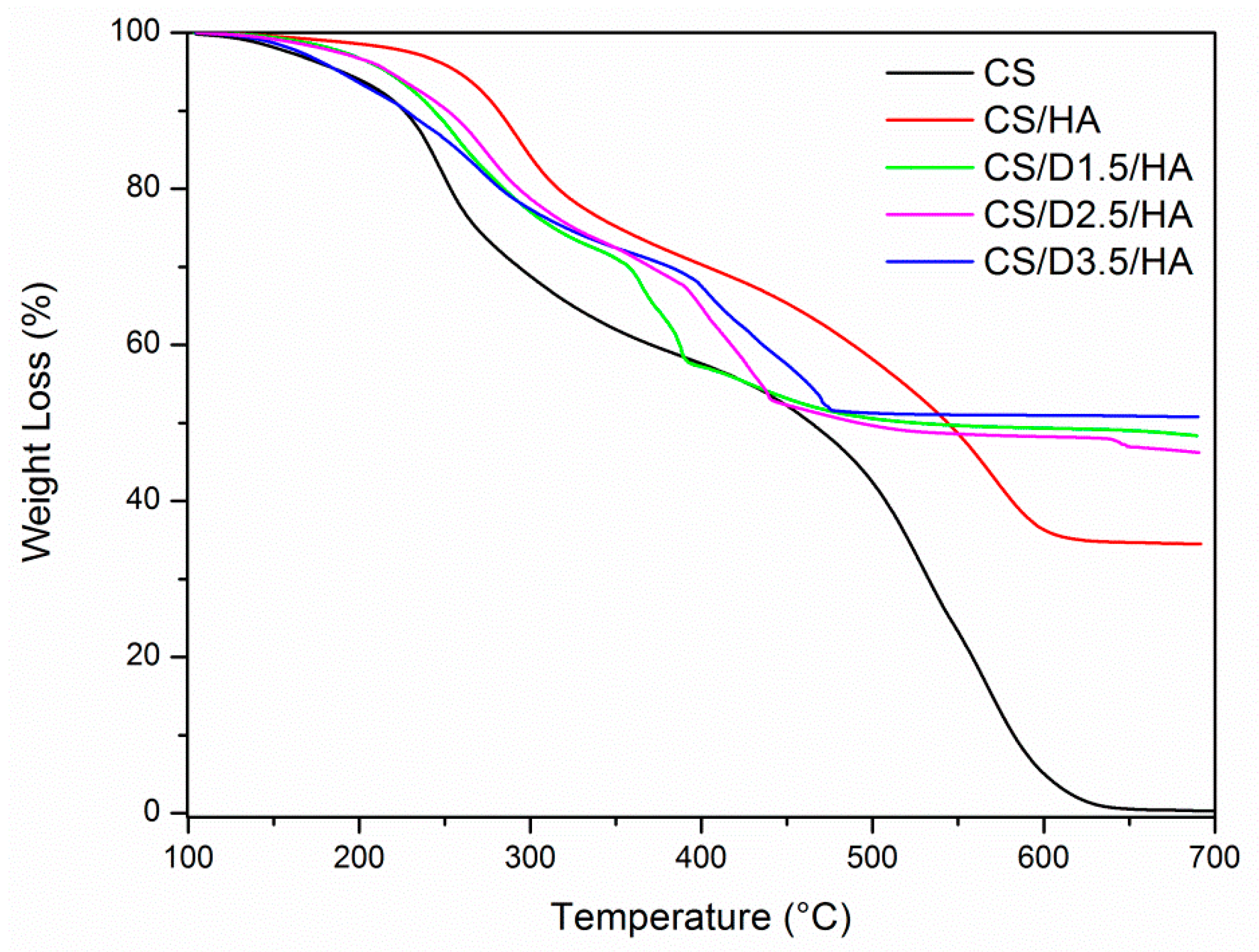
3.1.2. Synthesis of CS-D-HA Hydrogels
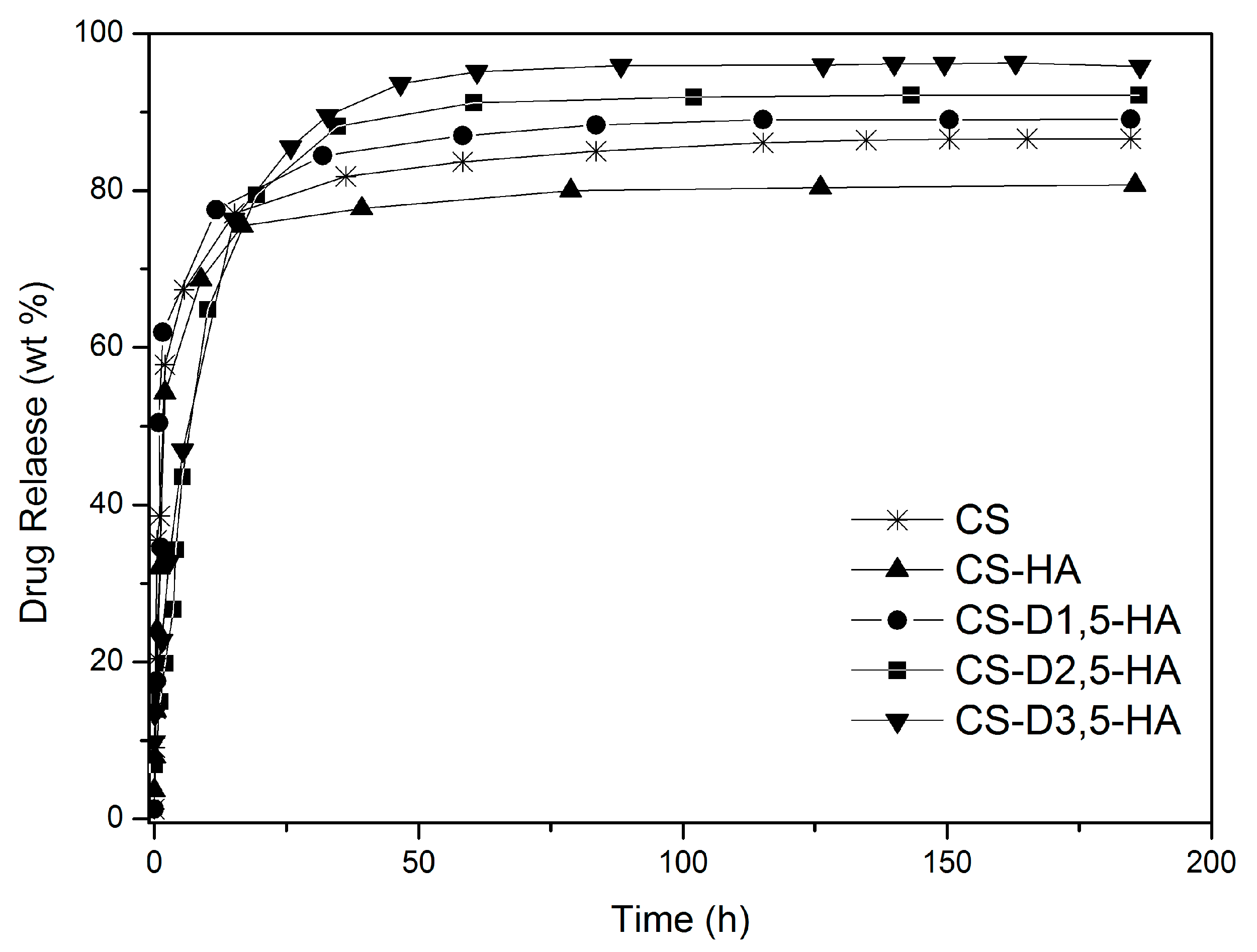
3.2. Drug Release Studies
3.3. Rheological Studies
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Albertsson, A.-C.; Varma, I.K. Aliphatic polyesters: Synthesis, properties and applications. In Degradable Aliphatic Polyesters; Springer: Berlin/Heidelberg, Germany, 2002; Volume 157, pp. 1–40. ISBN 978-3-540-45734-3. [Google Scholar]
- Visco, A.; Scolaro, C.; Giamporcaro, A.; De Caro, S.; Tranquillo, E.; Catauro, M. Threads made with blended biopolymers: Mechanical, physical and biological features. Polymers 2019, 11, 901. [Google Scholar] [CrossRef] [PubMed]
- Piskin, E. Biodegradable polymers as biomaterials. J. Biomater. Sci. Polym. Ed. 1995, 6, 775–795. [Google Scholar] [CrossRef] [PubMed]
- Sultankulov, B.; Berillo, D.; Sultankulova, K.; Tokay, T.; Saparov, A. Progress in the development of chitosan-based biomaterials for tissue engineering and regenerative medicine. Biomolecules 2019, 9, 470. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.F. On the nature of biomaterials. Biomaterials 2009, 30, 5897–5909. [Google Scholar] [CrossRef]
- Ulery, B.D.; Nair, L.S.; Laurencin, C.T. Biomedical applications of biodegradable polymers. J. Polym. Sci. Part B Polym. Phys. 2011, 49, 832–864. [Google Scholar] [CrossRef] [PubMed]
- Jianghua, L.; Chao, C.; Jiarui, L.; Jun, L.; Jia, L.; Tiantian, S.; Lihao, W.; Haotian, W.; Guangli, Y. Chitosan-based nanomaterials for drug delivery. Molecules 2018, 23, 2661. [Google Scholar]
- Muzzarelli, R.A.A.; El Mehtedi, M.; Bottegoni, C.; Aquili, A.; Gigante, A. Genipin-Crosslinked Chitosan Gels and Scaffolds for Tissue Engineering and Regeneration of Cartilage and Bone. Mar. Drugs 2015, 13, 7314–7338. [Google Scholar] [CrossRef]
- Song, R.; Murphy, M.; Li, C.; Ting, K.; Soo, C.; Zheng, Z. Current development of biodegradable polymeric materials for biomedical applications. Drug Des. Dev. Ther. 2018, 12, 3117–3145. [Google Scholar] [CrossRef]
- Tiwari, S.; Patil, R.; Bahadur, P. Polysaccharide based scaffolds for soft tissue engineering applications. Polymers 2019, 11, 1. [Google Scholar] [CrossRef]
- Galler, K.M.; D’Souza, R.N.; Hartgerink, J.D. Biomaterials and their potential applications for dental tissue engineering. J. Mater. Chem. 2010, 20, 8730–8746. [Google Scholar] [CrossRef]
- Rodríguez Vázquez, M.; Vega Ruiz, B.; Ramos Zúñiga, R.; Saldaña Koppel, D.A.; Quiñones Olvera, L.F. Chitosan and its potential use as a scaffold for tissue engineering in regenerative medicine. BioMed Res. Int. 2015, 2015, 821279. [Google Scholar] [CrossRef] [PubMed]
- Argüelles Monal, W.M.; Lizardi Mendoza, J.; Fernández Quiroz, D.; Recillas Mota, M.T.; Montiel Herrera, M. Chitosan derivatives: Introducing new functionalities with a controlled molecular architecture for innovative materials. Polymers 2018, 10, 342. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Kong, Y.; Zheng, H.; Ke, W.; Chen, X.; Yin, Y.; Yi, Y. Preparation and properties of poly(amidoamine) dendrimer/quaternary ammonium chitosan hydrogels. J. Wuhan Univ. Technol. 2018, 33, 736–743. [Google Scholar] [CrossRef]
- Sashiwa, H.; Yajima, H.; Aiba, S. Synthesis of a chitosan dendrimer hybrid and its biodegradation. Biomacromolecules 2003, 4, 1244–1249. [Google Scholar] [CrossRef]
- Xu, Q.; Wang, C.H.; Wayne Pack, D. Polymeric carriers for gene delivery: Chitosan and poly(amidoamine) dendrimers. Curr. Pharm. Des. 2010, 16, 2350–2368. [Google Scholar] [CrossRef]
- Abedi Gaballu, F.; Dehghan, G.; Ghaffari, M.; Yekta, R.; Abbaspour Ravasjani, S.; Baradaran, B.; Dolatabadi, J.E.N.; Hamblin, M.R. PAMAM dendrimers as efficient drug and gene delivery nanosystems for cancer therapy. Appl. Mater. Today 2018, 12, 177–190. [Google Scholar] [CrossRef]
- Uswatta, S.P.; Okeke, I.U.; Jayasuriya, A.C. Injectable porous nano hydroxyapatite/chitosan/tripolyphosphate scaffolds with improved compressive strength for bone regeneration. Mater. Sci. Eng. C 2016, 69, 505–512. [Google Scholar] [CrossRef]
- Reza Mahdavinia, G.; Karimi, M.H.; Soltaniniya, M.; Massoumi, B. In vitro evaluation of sustained ciprofloxacin release from κ-carrageenan-crosslinked chitosan/hydroxyapatite hydrogel nanocomposites. Int. J. Biol. Macromol. 2019, 126, 443–453. [Google Scholar] [CrossRef]
- Frohbergh, M.E.; Katsman, A.; Botta, G.P.; Lazarovici, P.; Schauer, C.L.; Wegst, U.G.K.; Lelkes, P.I. Electrospun hydroxyapatite-containing chitosan nanofibers crosslinkedwith genipin for bone tissue engineering. Biomaterials 2012, 33, 9167–9178. [Google Scholar] [CrossRef]
- Pistone, A.; Iannazzo, D.; Celesti, C.; Piperopoulos, E.; Ashok, D.; Cembran, A.; Tricoli, A.; Nisbet, D. Engineering of chitosan-hydroxyapatite-magnetite hierarchical scaffolds for guided bone growth. Materials 2019, 12, 2321. [Google Scholar] [CrossRef]
- Yuan, Q.; Shah, J.; Hein, S.; Misra, R.D.K. Controlled and extended drug release behaviorof chitosan-based nanoparticle carrier. Acta Biomater. 2010, 6, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Pistone, A.; Iannazzo, D.; Espro, C.; Galvagno, S.; Tampieri, A.; Montesi, M.; Panseri, S.; Sandri, M. Tethering of gly-arg-gly-asp-ser-pro-lyspeptides on mg-doped hydroxyapatite. Engineering 2017, 3, 55–59. [Google Scholar] [CrossRef]
- Iannazzo, D.; Pistone, A.; Espro, C.; Galvagno, S. Drug delivery strategies for bone tissue regeneration. In Biomimetic Approaches for Tissue Healing; Panseri, S., Taraballi, F., Cunha, C., Eds.; OMICS Group eBooks: Foster City, CA, USA, 2015; pp. 1–39. [Google Scholar]
- Tsai, C.C.; Huang, R.N.; Sung, H.W.; Liang, H.C. In vitro evaluation of the genotoxicity of a naturally occurring crosslinking agent (genipin) for biologic tissue fixation. J. Biomed. Mater. Res. 2000, 52, 58–65. [Google Scholar] [CrossRef]
- Gao, L.; Gan, H.; Meng, Z.; Gu, R.; Wu, Z.; Zhang, L.; Zhu, X.; Sun, W.; Li, J.; Zheng, Y.; et al. Effects of genipin cross-linking of chitosan hydrogels on cellular adhesion and viability. Colloids Surf. B 2014, 117, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Tan, Z.; Sun, F.; Sheng, L.; Zhang, X.; Yao, F. Synthesis and characterization of quaternizedcarboxymethyl chitosan/poly(amidoamine) dendrimer core–shell nanoparticles. Mater. Sci. Eng. C 2012, 32, 2026–2036. [Google Scholar] [CrossRef]
- Arguelles Monal, W.; Goycoolea, F.M.; Peniche, C.; Higuera Ciapara, I. Polymer gels and networks. Rheological study of the chitosan/glutaraldehyde chemical gel system. Polym. Gels Netw. 1998, 6, 429–440. [Google Scholar] [CrossRef]
- Kim, B.R.; Lee, H.G.; Kang, S.B.; Sung, G.H.; Kim, J.J.; Park, J.K.; Lee, S.G.; Yoon, Y.J. tert-Butoxide-Assisted amidation of esters under green conditions. Synthesis 2012, 44, 42–50. [Google Scholar]
- Fathi, M.H.; Hanifi, A.; Mortazavi, V. Preparation and bioactivity evaluation of bone-like hydroxyapatite nanopowder. J. Mater. Process. Technol. 2008, 202, 536–542. [Google Scholar] [CrossRef]
- Nasiri, N.; Mukherjee, S.; Panneerselvan, A.; Nisbet, D.R.; Tricoli, A. Optimally hierarchical nanostructured hydroxyapatite coatings for superior prosthesis biointegration. ACS Appl. Mater. Interfaces 2018, 10, 24840–24849. [Google Scholar] [CrossRef]
- Dong, Q.X.; Chen, Q.J.; Yang, W.; Zheng, Y.L.; Liu, X.; Li, Y.L.; Yang, M.B. Thermal properties and flame retardancy of polycarbonate/hydroxyapatite nanocomposite. J. Appl. Polym. Sci. 2008, 109, 659–663. [Google Scholar] [CrossRef]
- Branca, C.; Crupi, C.; D’Angelo, G.; Khouzami, K.; Rifici, S.; Visco, A.; Wanderlingh, U. Effect of montmorillonite on the rheological properties of dually crosslinked guar gum-based hydrogels. J. Appl. Polym. Sci. 2015, 132, 41373. [Google Scholar] [CrossRef]
- Moura, M.J.; Figueiredo, M.M.; Gil, M.H. Rheological study of genipin cross-linked chitosan hydrogels. Biomolecules 2007, 8, 3823–3829. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Code | Sample Composition | Gelling Time (Figure 6) (h:m) | Freq. 0.1 rad/s | Freq. 10 rad/s | ||
|---|---|---|---|---|---|---|
| G’ (Pa) | η* (106 Pa∙s) | G’ (Pa) | η* (106 Pa∙s) | |||
| CS | 100 wt% pure chitosan | 1:05 | 112,752 | 1.128 | 114,072 | 0.011409 |
| CS-HA | 65 wt% pure chitosan + 35 wt% hydroxyapatite | 1:05 | 151,501 | 1.519 | 171,425 | 0.017144 |
| CS-D1.5-HA | 50 wt% dendrimer modified chitosan + 50 wt% hydroxyapatite | 1:35 | 424,121 | 4.401 | 428347.7 | 0.033921 |
| CS-D2.5-HA | 50 wt% dendrimer modified chitosan + 50 wt% hydroxyapatite | 2:05 | 838,935 | 10.665 | 808,642 | 0.081778 |
| CS-D3.5-HA | 50 wt% dendrimer modified chitosan + 50 wt% hydroxyapatite | 2:40 | 1.36962 × 106 | 18.821 | 1.8724 × 106 | 0.191665 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pistone, A.; Iannazzo, D.; Celesti, C.; Scolaro, C.; Giofré, S.V.; Romeo, R.; Visco, A. Chitosan/PAMAM/Hydroxyapatite Engineered Drug Release Hydrogels with Tunable Rheological Properties. Polymers 2020, 12, 754. https://doi.org/10.3390/polym12040754
Pistone A, Iannazzo D, Celesti C, Scolaro C, Giofré SV, Romeo R, Visco A. Chitosan/PAMAM/Hydroxyapatite Engineered Drug Release Hydrogels with Tunable Rheological Properties. Polymers. 2020; 12(4):754. https://doi.org/10.3390/polym12040754
Chicago/Turabian StylePistone, Alessandro, Daniela Iannazzo, Consuelo Celesti, Cristina Scolaro, Salvatore V. Giofré, Roberto Romeo, and Annamaria Visco. 2020. "Chitosan/PAMAM/Hydroxyapatite Engineered Drug Release Hydrogels with Tunable Rheological Properties" Polymers 12, no. 4: 754. https://doi.org/10.3390/polym12040754
APA StylePistone, A., Iannazzo, D., Celesti, C., Scolaro, C., Giofré, S. V., Romeo, R., & Visco, A. (2020). Chitosan/PAMAM/Hydroxyapatite Engineered Drug Release Hydrogels with Tunable Rheological Properties. Polymers, 12(4), 754. https://doi.org/10.3390/polym12040754