Theoretical Insights into the Structures and Capacitive Performances of Confined Ionic Liquids


Abstract
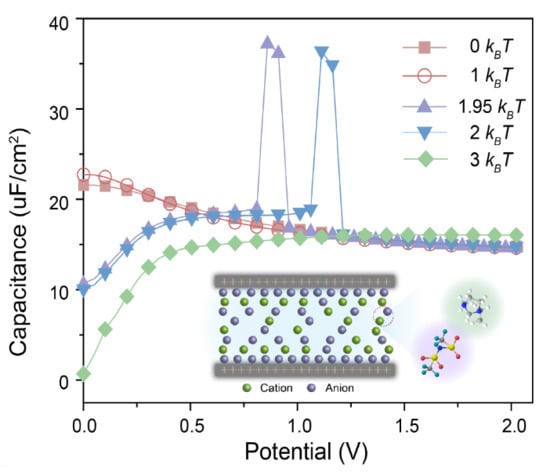
{kind=link}
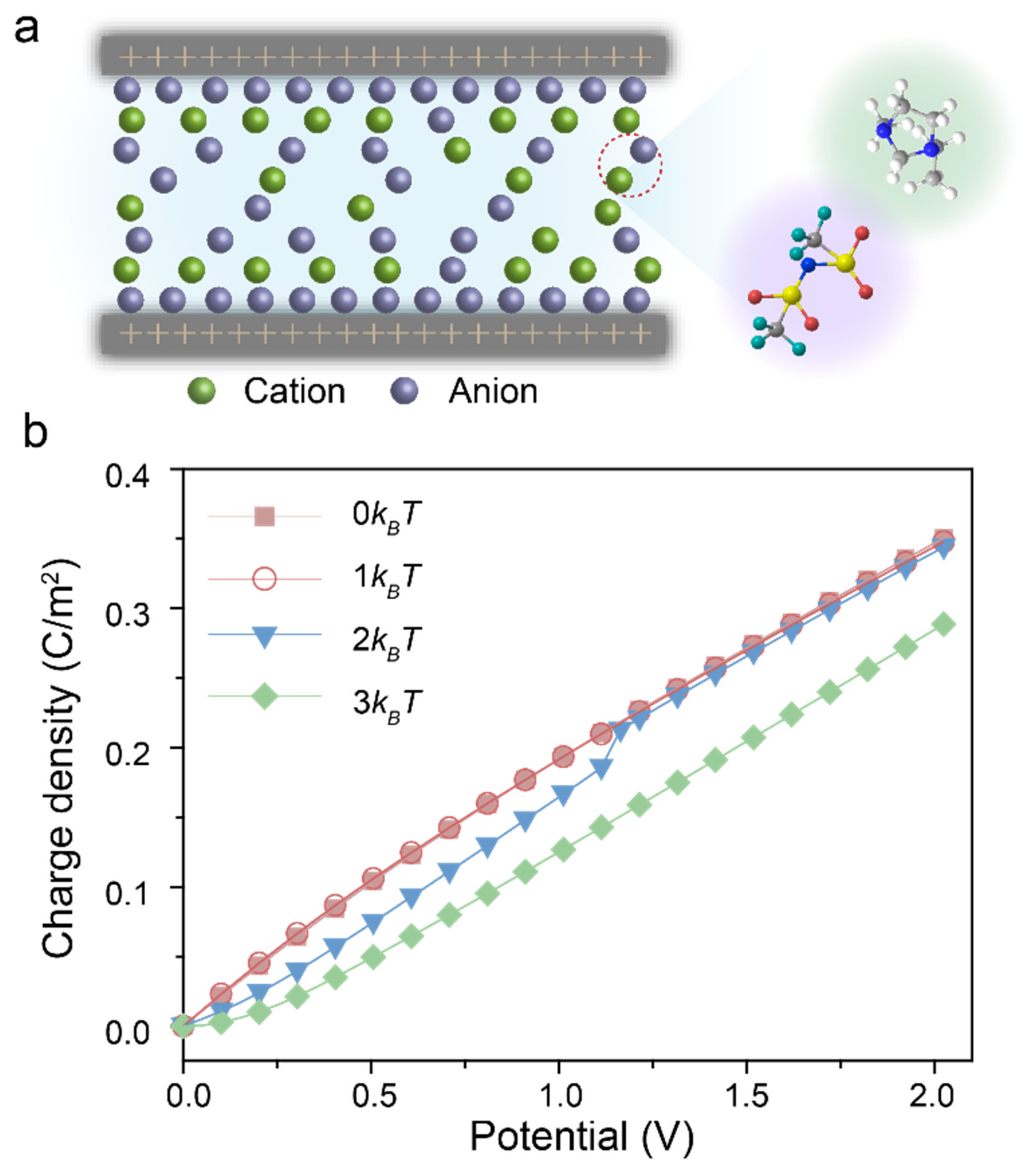{kind=link}
{kind=link}
{kind=link}
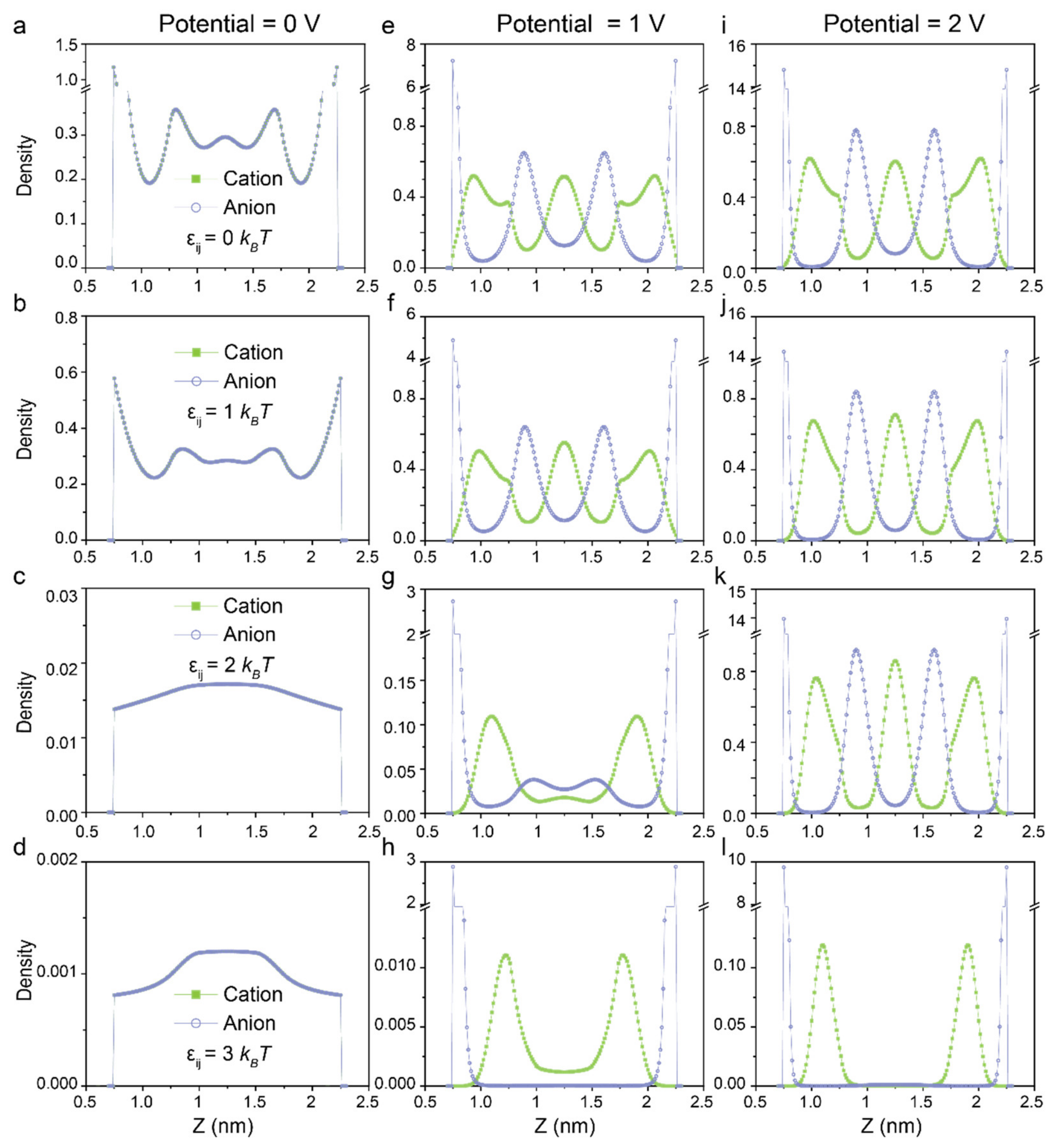{kind=link}
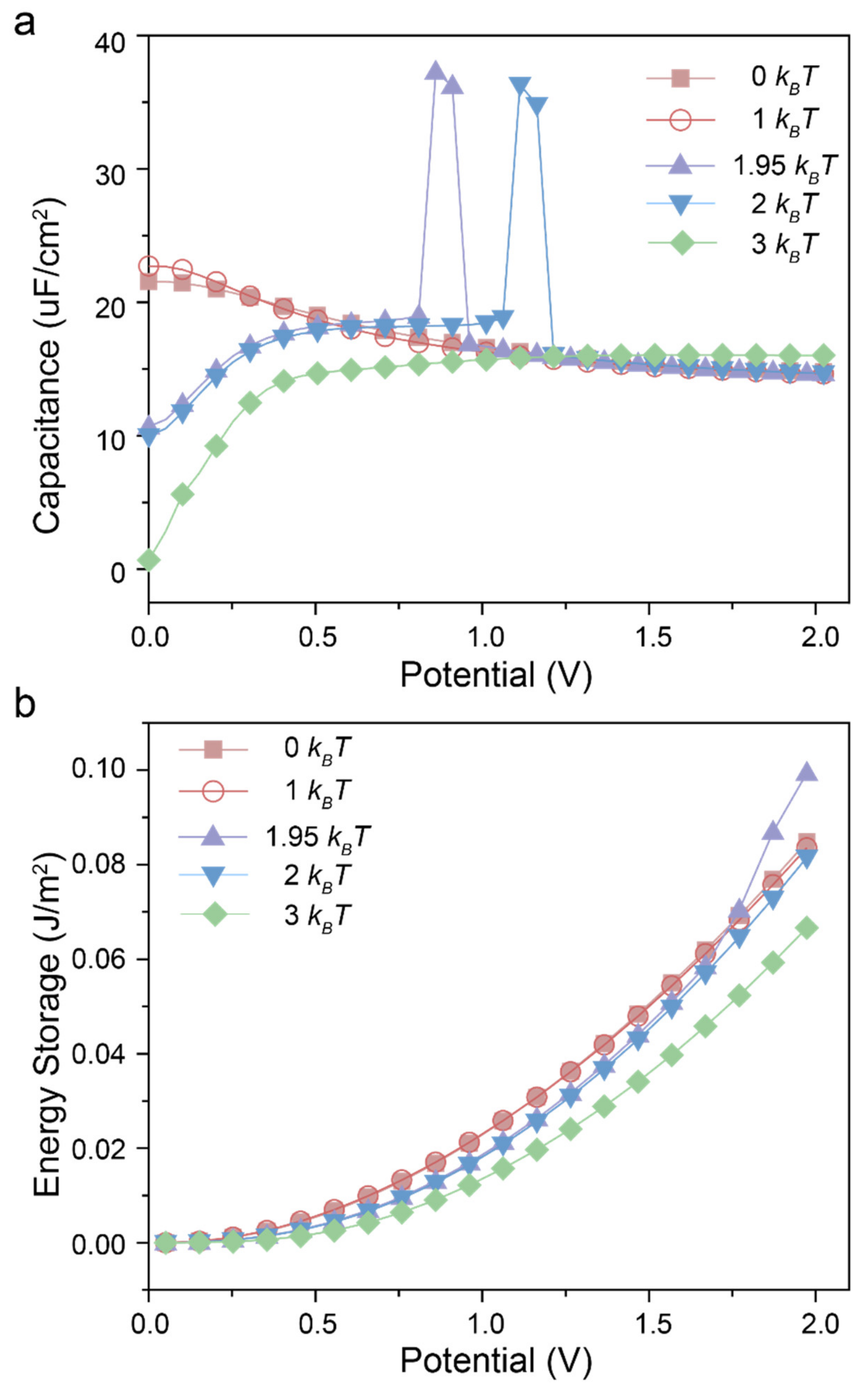{kind=link}
1. Introduction
2. Models and Methods
3. Results and Discussions
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chmiola, J.; Yushin, G.; Gogotsi, Y.; Portet, C.; Simon, P.; Taberna, P.L. Anomalous increase in carbon capacitance at pore sizes less than 1 nanometer. Science 2006, 313, 1760–1763. [Google Scholar] [CrossRef] [PubMed]
- Ali, B.A.; Allam, N.K. First-principles roadmap and limits to design efficient supercapacitor electrode materials. Phys. Chem. Chem. Phys. 2019, 21, 17494–17511. [Google Scholar] [CrossRef] [PubMed]
- Burt, R.; Birkett, G.; Zhao, X.S. A review of molecular modelling of electric double layer capacitors. Phys. Chem. Chem. Phys. 2014, 16, 6519–6538. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Deng, Y.; Hu, W.; Qiao, J.; Zhang, L.; Zhang, J. A review of electrolyte materials and compositions for electrochemical supercapacitors. Chem. Soc. Rev. 2015, 44, 7484–7539. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.E.; Wu, J. Microscopic insights into the electrochemical behavior of nonaqueous electrolytes in electric double-layer capacitors. J. Phys. Chem. Lett. 2013, 4, 1260–1267. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; El-Kady, M.F.; Wang, L.J.; Zhang, Q.; Li, Y.; Wang, H.; Mousavi, M.F.; Kaner, R.B. Graphene-based materials for flexible supercapacitors. Chem. Soc. Rev. 2015, 44, 3639–3665. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, M.V.; Kornyshev, A.A. Ionic liquids at electrified interfaces. Chem. Rev. 2014, 114, 2978–3036. [Google Scholar] [CrossRef]
- Forse, A.C.; Merlet, C.; Griffin, J.M.; Grey, C.P. New perspectives on the charging mechanisms of supercapacitors. J. Am. Chem. Soc. 2016, 138, 5731–5744. [Google Scholar] [CrossRef]
- Largeot, C.; Portet, C.; Chmiola, J.; Taberna, P.L.; Gogotsi, Y.; Simon, P. Relation between the ion size and pore size for an electric double-layer capacitor. J. Am. Chem. Soc. 2008, 130, 2730–2731. [Google Scholar] [CrossRef]
- Jiang, D.E.; Jin, Z.; Henderson, D.; Wu, J. Solvent effect on the pore-size dependence of an organic electrolyte supercapacito. J. Phys. Chem. Lett. 2012, 3, 1727–1731. [Google Scholar] [CrossRef]
- Kondrat, S.; Perez, C.R.; Presser, V.; Gogotsi, Y.; Kornyshev, A.A. Effect of pore size and its dispersity on the energy storage in nanoporous supercapacitors. Energy Environ. Sci. 2012, 5, 6474–6479. [Google Scholar] [CrossRef]
- Seddon, K.R. Ionic liquids for clean technology. J. Chem. Technol. Biotechnol. 1997, 68, 351–356. [Google Scholar] [CrossRef]
- Simon, P.; Gogotsi, Y. Capacitive energy storage in nanostructured carbon–electrolyte systems. Acc. Chem. Res. 2013, 46, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Dong, K.; Liu, X.; Dong, H.; Zhang, X.; Zhang, S. Multiscale studies on ionic liquids. Chem. Rev. 2017, 117, 6636–6695. [Google Scholar] [CrossRef]
- Lian, C.; Liu, H.; Li, C.; Wu, J. Hunting ionic liquids with large electrochemical potential windows. AIChE J. 2019, 65, 804–810. [Google Scholar] [CrossRef]
- Wang, H.; Xu, Z.; Kohandehghan, A.; Li, Z.; Cui, K.; Tan, X.; Stephenson, T.J.; King’ondu, C.K.; Holt, C.M.B.; Olsen, B.C.; et al. Interconnected carbon nanosheets derived from hemp for ultrafast supercapacitors with high energy. ACS Nano 2013, 7, 5131–5141. [Google Scholar] [CrossRef]
- Yoo, J.J.; Balakrishnan, K.; Huang, J.; Meunier, V.; Sumpter, B.G.; Srivastava, A.; Conway, M.; Reddy, A.L.; Yu, J.; Vajtai, R.; et al. Ultrathin planar graphene supercapacitors. Nano Lett. 2011, 11, 1423–1427. [Google Scholar] [CrossRef]
- Zhu, Y.; Murali, S.; Stoller, M.D.; Ganesh, K.J.; Cai, W.; Ferreira, P.J.; Pirkle, A.; Wallace, R.M.; Cychosz, K.A.; Thommes, M.; et al. Carbon-based supercapacitors produced by activation of grapheme. Science 2011, 332, 1537. [Google Scholar] [CrossRef]
- Yang, X.; Cheng, C.; Wang, Y.; Qiu, L.; Li, D. Liquid-mediated dense integration of graphene materials for compact capacitive energy storage. Science 2013, 341, 534–537. [Google Scholar] [CrossRef]
- Lukatskaya, M.R.; Mashtalir, O.; Ren, C.E.; Dall’Agnese, Y.; Rozier, P.; Taberna, P.L.; Naguib, M.; Simon, P.; Barsoum, M.W.; Gogotsi, Y. Cation intercalation and high volumetric capacitance of two-dimensional titanium carbide. Science 2013, 341, 1502. [Google Scholar] [CrossRef]
- Feng, G.; Zhang, J.S.; Qiao, R. Microstructure and capacitance of the electrical double layers at the interface of ionic liquids and planar electrodes. J. Phys. Chem. C 2009, 113, 4549–4559. [Google Scholar] [CrossRef]
- Wu, P.; Huang, J.; Meunier, V.; Sumpter, B.G.; Qiao, R. Complex capacitance scaling in ionic liquids-filled nanopores. ACS Nano 2011, 5, 9044–9051. [Google Scholar] [CrossRef] [PubMed]
- Kondrat, S.; Georgi, N.; Fedorov, M.V.; Kornyshev, A.A. A superionic state in nano-porous double-layer capacitors: Insights from Monte Carlo simulations. Phys. Chem. Chem. Phys. 2011, 13, 11359–11366. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, L.; Zhang, J. A review of electrode materials for electrochemical supercapacitors. Chem. Soc. Rev. 2012, 41, 797–828. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Duay, J.; Lee, S.B. Heterogeneous nanostructured electrode materials for electrochemical energy storage. Chem. Commun. 2011, 47, 1384–1404. [Google Scholar] [CrossRef] [PubMed]
- Vatamanu, J.; Hu, Z.; Bedrov, D.; Perez, C.; Gogotsi, Y. Increasing energy storage in electrochemical capacitors with ionic liquid electrolytes and nanostructured carbon electrodes. J. Phys. Chem. Lett. 2013, 4, 2829–2837. [Google Scholar] [CrossRef]
- Hayes, R.; Warr, G.G.; Atkin, R. Structure and nanostructure in ionic liquids. Chem. Rev. 2015, 115, 6357–6426. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, C.; Zhang, Y.; Huo, F.; He, H.; Zhang, S. Molecular insights into the regulatable interfacial property and flow behavior of confined ionic liquids in graphene nanochannels. Small 2019, 15, e1804508. [Google Scholar] [CrossRef]
- Tong, J.; Xiao, X.; Liang, X.; von Solms, N.; Huo, F.; He, H.; Zhang, S. Insights into the solvation and dynamic behaviors of a lithium salt in organic- and ionic liquid-based electrolytes. Phys. Chem. Chem. Phys. 2019, 21, 19216–19225. [Google Scholar] [CrossRef]
- Trulsson, M.; Algotsson, J.; Forsman, J.; Woodward, C.E. Differential capacitance of room temperature Ionic liquids: The role of dispersion forces. J. Phys. Chem. Lett. 2010, 1, 1191–1195. [Google Scholar] [CrossRef]
- Forsman, J.; Woodward, C.E.; Trulsson, M. A classical density functional theory of ionic liquids. J. Phys. Chem. B 2011, 115, 4606–4612. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.T.; Islam, M.M.; Okajima, T.; Ohsaka, T. Measurements of differential capacitance at mercury/room-temperature ionic liquids interfaces. J. Phys. Chem. C 2007, 111, 18326–18333. [Google Scholar] [CrossRef]
- Alam, M.T.; Islam, M.M.; Okajima, T.; Ohsaka, T. Capacitance measurements in a series of room-temperature ionic liquids at glassy carbon and gold electrode interfaces. J. Phys. Chem. C 2008, 112, 16600–16608. [Google Scholar] [CrossRef]
- Lockett, V.; Sedev, R.; Ralston, J.; Horne, M.; Rodopoulos, T. Differential capacitance of the electrical Double layer in imidazolium-based ionic liquids: Influence of potential, cation size, and temperature. J. Phys. Chem. C 2008, 112, 7486–7495. [Google Scholar] [CrossRef]
- Shrivastav, G.; Remsing, R.C.; Kashyap, H.K. Capillary evaporation of the ionic liquid [EMIM][BF4] in nanoscale solvophobic confinement. J. Chem. Phys. 2018, 148, 193810. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Zhang, P.; Wu, J. Does capillary evaporation limit the accessibility of nonaqueous electrolytes to the ultrasmall pores of carbon electrodes? J. Chem. Phys. 2018, 149, 234708. [Google Scholar] [CrossRef]
- Szparaga, R.; Woodward, C.E.; Forsman, J. Theoretical prediction of the capacitance of ionic liquid films. J. Phys. Chem. C 2012, 116, 15946–15951. [Google Scholar] [CrossRef]
- Szparaga, R.; Woodward, C.E.; Forsman, J. Capillary condensation of ionic liquid solutions in porous electrodes. J. Phys. Chem. C 2013, 117, 1728–1734. [Google Scholar] [CrossRef][Green Version]
- Lian, C.; Liu, K.; Liu, H.; Wu, J. Impurity effects on charging mechanism and energy storage of nanoporous supercapacitors. J. Phys. Chem. C 2017, 121, 14066–14072. [Google Scholar] [CrossRef]
- Jiang, D.E.; Jin, Z.; Wu, J.Z. Oscillation of capacitance inside nanopores. Nano Lett. 2011, 11, 5373–5377. [Google Scholar] [CrossRef]
- Lian, C.; Jiang, D.E.; Liu, H.; Wu, J.A. Generic model for electric double layers in porous electrodes. J. Phys. Chem. C 2016, 120, 8704–8710. [Google Scholar] [CrossRef]
- Jiang, D.E.; Wu, J. Unusual effects of solvent polarity on capacitance for organic electrolytes in a nanoporous electrode. Nanoscale 2014, 6, 5545–5550. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, Z. Density-functional theory for complex fluids. Annu. Rev. Phys. Chem. 2007, 58, 85–112. [Google Scholar] [CrossRef]
- Wu, J.; Jiang, T.; Jiang, D.E.; Jin, Z.; Henderson, D. A classical density functional theory for interfacial layering of ionic liquids. Soft Matter 2011, 7, 11222–11231. [Google Scholar] [CrossRef]
- Lian, C.; Liu, K.; Van Aken, K.L.; Gogotsi, Y.; Wesolowski, D.J.; Liu, H.L.; Jiang, D.E.; Wu, J.Z. Enhancing the capacitive performance of electric double-layer capacitors with ionic liquid mixtures. ACS Energy Lett. 2016, 1, 21–26. [Google Scholar] [CrossRef]
- Boda, D.; Fawcett, W.R.; Henderson, D.; Sokołowski, S. Monte Carlo, density functional theory, and Poisson-Boltzmann theory study of the structure of an electrolyte near an electrode. J. Chem. Phys. 2002, 116, 7170–7176. [Google Scholar] [CrossRef]
- Gillespie, D.; Valiskó, M.; Boda, D. Density functional theory of the electrical double layer: The RFD functional. J. Phys. Condens. Matter 2005, 17, 6609–6626. [Google Scholar] [CrossRef]
- Yu, Y.X.; Wu, J.; Gao, G.H. Density-functional theory of spherical electric double layers and zeta potentials of colloidal particles in restricted-primitive-model electrolyte solutions. J. Chem. Phys. 2004, 120, 7223–7233. [Google Scholar] [CrossRef]
- Li, Z.; Wu, J. Density functional theory for planar electric double layers: Closing the gap between simple and polyelectrolytes. J. Phys. Chem. B 2006, 110, 7473–7484. [Google Scholar] [CrossRef]
- Lian, C.; Zhao, S.; Liu, H.; Wu, J. Time-dependent density functional theory for the charging kinetics of electric double layer containing room-temperature ionic liquids. J. Chem. Phys. 2016, 145, 204707. [Google Scholar] [CrossRef]
- Yu, Y.X.; Wu, J.Z. Structures of hard-sphere fluids from a modified fundamental-measure theory. J. Chem. Phys. 2002, 117, 10156–10164. [Google Scholar] [CrossRef]
- Fouad, W.A.; Haghmoradi, A.; Wang, L.; Bansal, A.; Hammadi, A.; Asthagiri, A.; Asthagiri, D.; Djamali, E.; Cox, K.R.; Chapman, W.G. Extensions of the SAFT model for complex association in the bulk and interface. Fluid Phase Equilib. 2016, 416, 62–71. [Google Scholar] [CrossRef]
- Lian, C.; Kong, X.; Liu, H.; Wu, J. On the hydrophilicity of electrodes for capacitive energy extraction. J. Phys. Condens. Matter 2016, 28, 464008. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Ding, Y.; Lian, C.; Ying, S.; Liu, H. Theoretical Insights into the Structures and Capacitive Performances of Confined Ionic Liquids. Polymers 2020, 12, 722. https://doi.org/10.3390/polym12030722
Yang J, Ding Y, Lian C, Ying S, Liu H. Theoretical Insights into the Structures and Capacitive Performances of Confined Ionic Liquids. Polymers. 2020; 12(3):722. https://doi.org/10.3390/polym12030722
Chicago/Turabian StyleYang, Jie, Yajun Ding, Cheng Lian, Sanjiu Ying, and Honglai Liu. 2020. "Theoretical Insights into the Structures and Capacitive Performances of Confined Ionic Liquids" Polymers 12, no. 3: 722. https://doi.org/10.3390/polym12030722
APA StyleYang, J., Ding, Y., Lian, C., Ying, S., & Liu, H. (2020). Theoretical Insights into the Structures and Capacitive Performances of Confined Ionic Liquids. Polymers, 12(3), 722. https://doi.org/10.3390/polym12030722