A Chitosan-Based Micellar System as Nanocarrier For the Delivery of Paclitaxel


Abstract
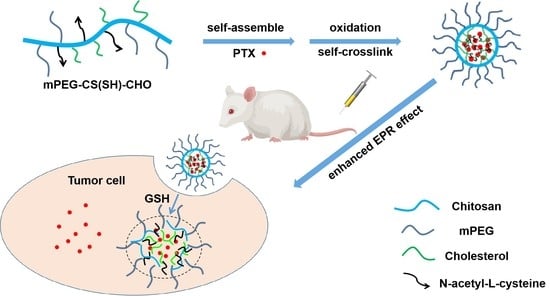
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
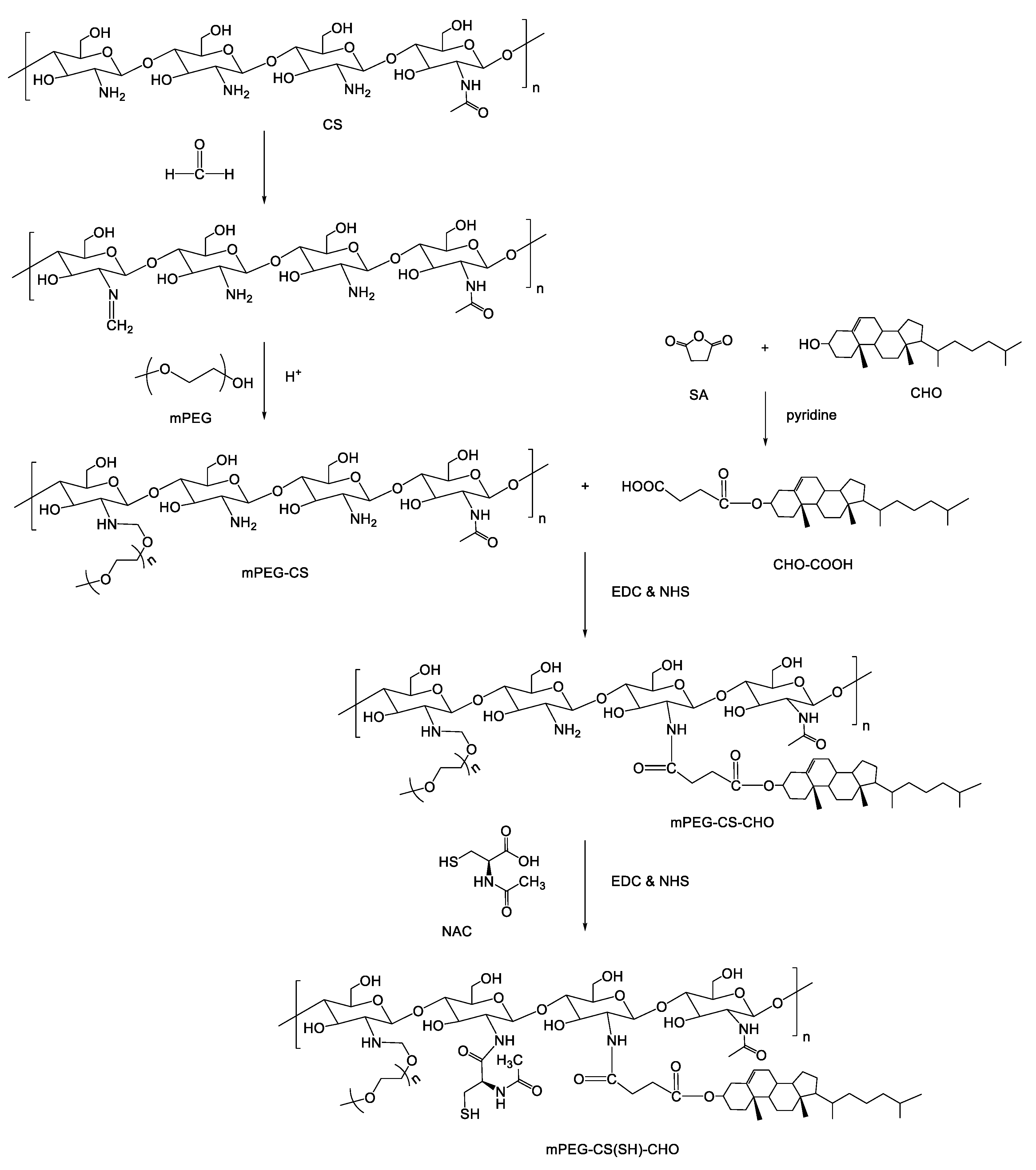
2.2. Synthesis of mPEG-CS(SH)-CHO
2.2.1. Synthesis of CHO-COOH
2.2.2. Synthesis of mPEG-CS
2.2.3. Synthesis of mPEG-CS-CHO
2.2.4. Synthesis of mPEG-CS(SH)-CHO
2.3. Characterization of mPEG-CS(SH)-CHO
2.4. Preparation and Characterization of the mPEG-CS(SH)-CHO Micelles
2.5. Protein Adsorption Tests
2.6. In Vitro Drug Release Kinetics
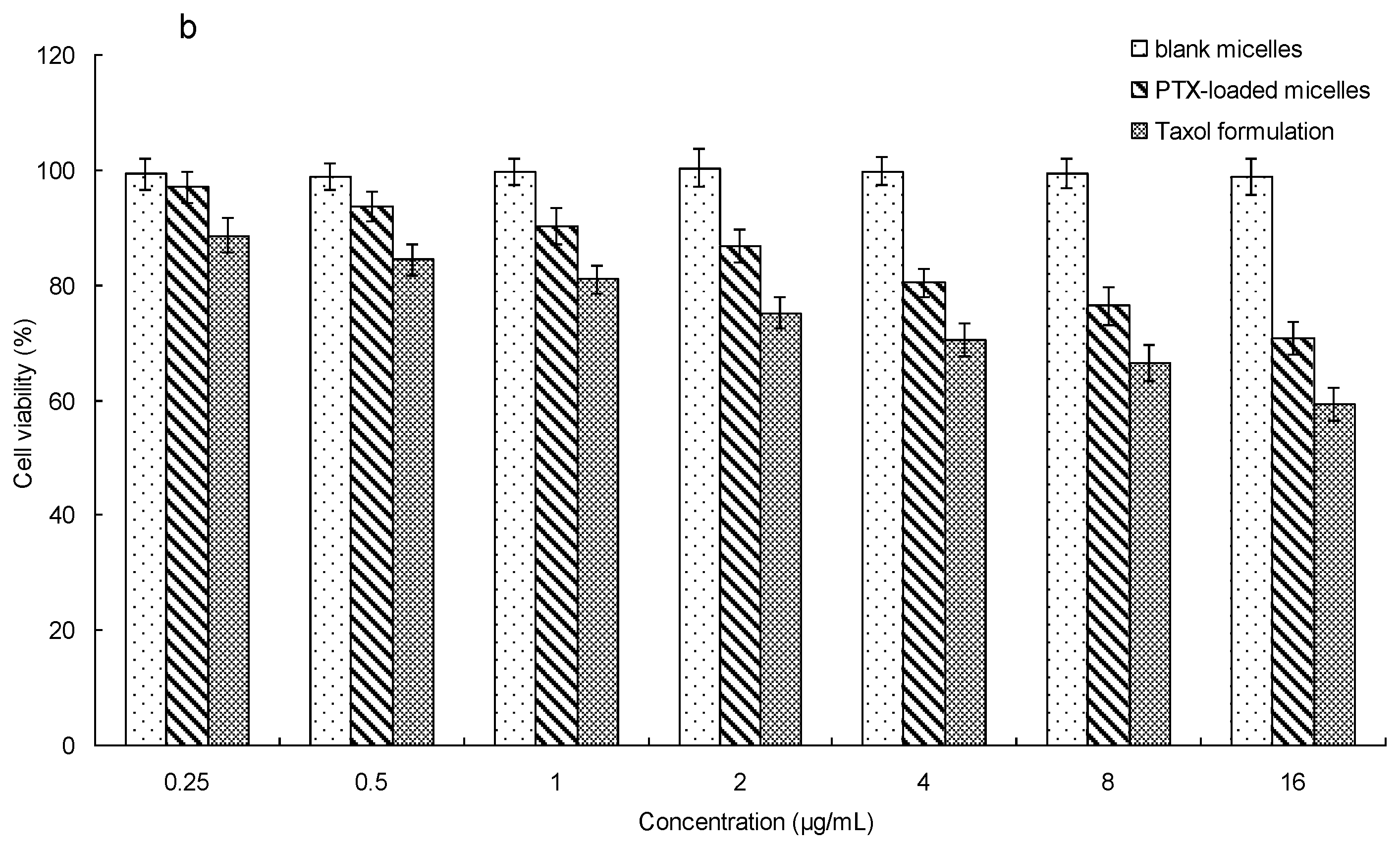
2.7. In Vitro Cytotoxicity
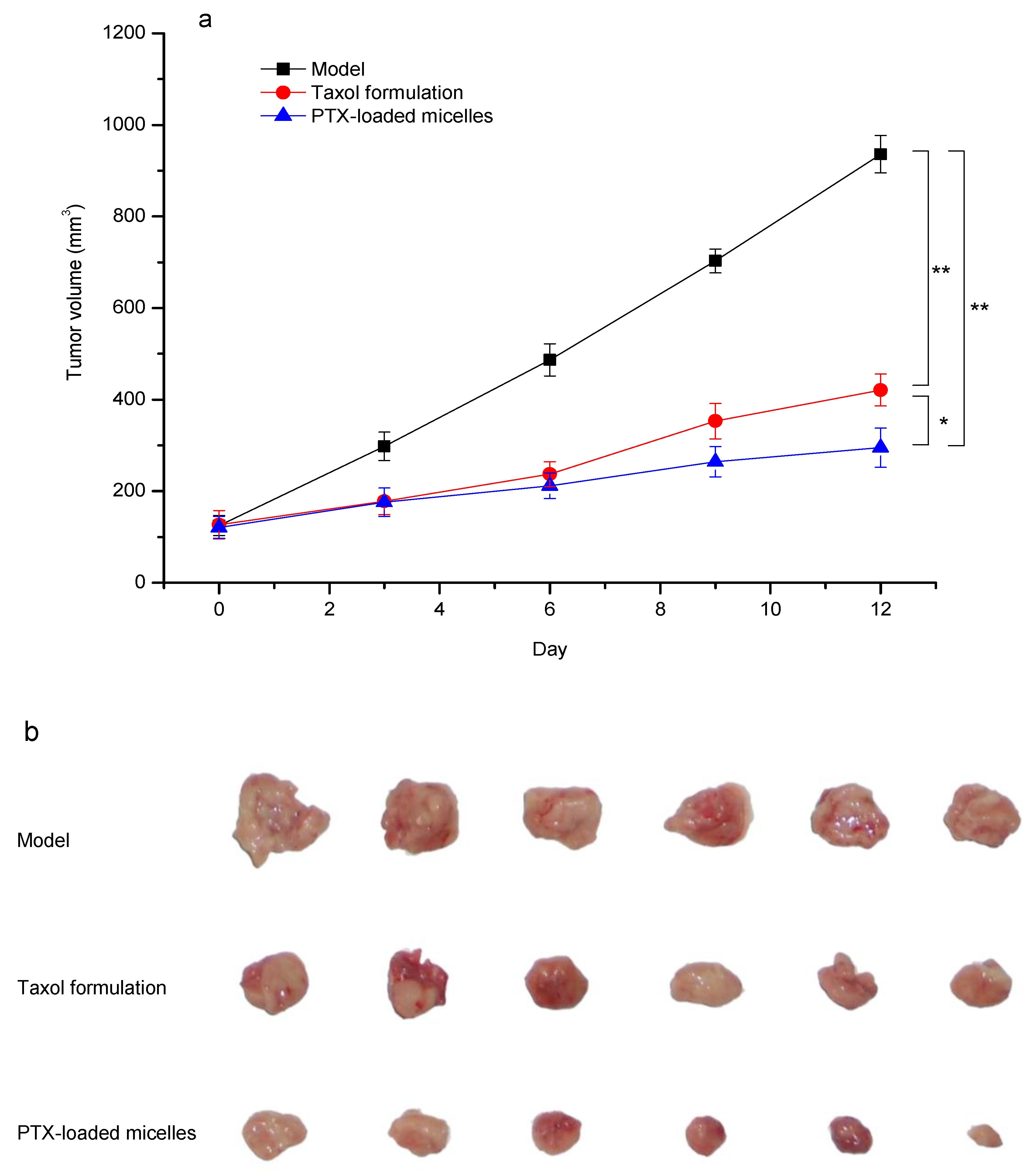
2.8. In Vivo Antitumor Efficacy
2.9. Statistical Analysis
3. Results and Discussion
3.1. Preparation of mPEG-CS(SH)-CHO
3.2. Characterization of mPEG-CS(SH)-CHO
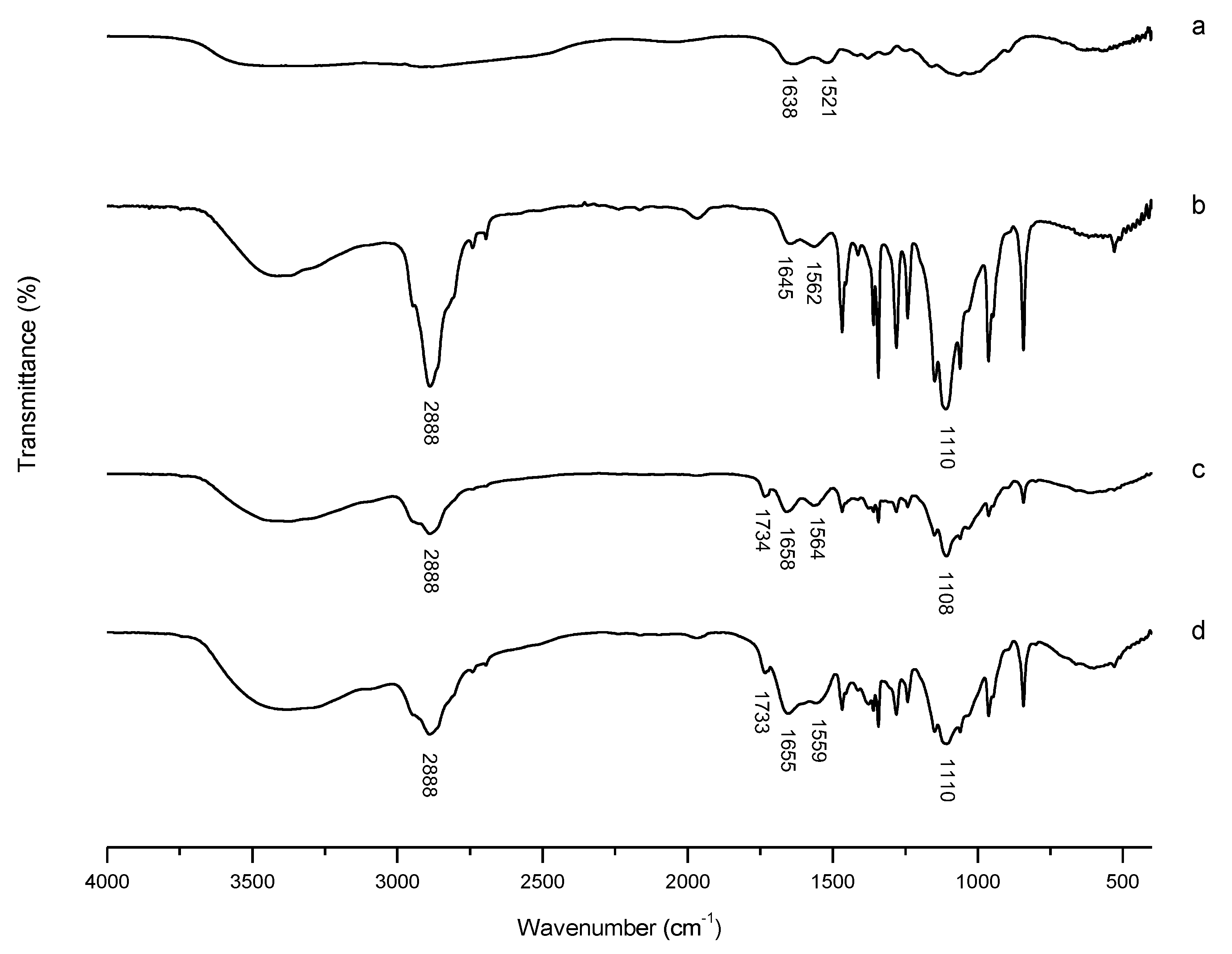
3.2.1. FT-IR Characterization
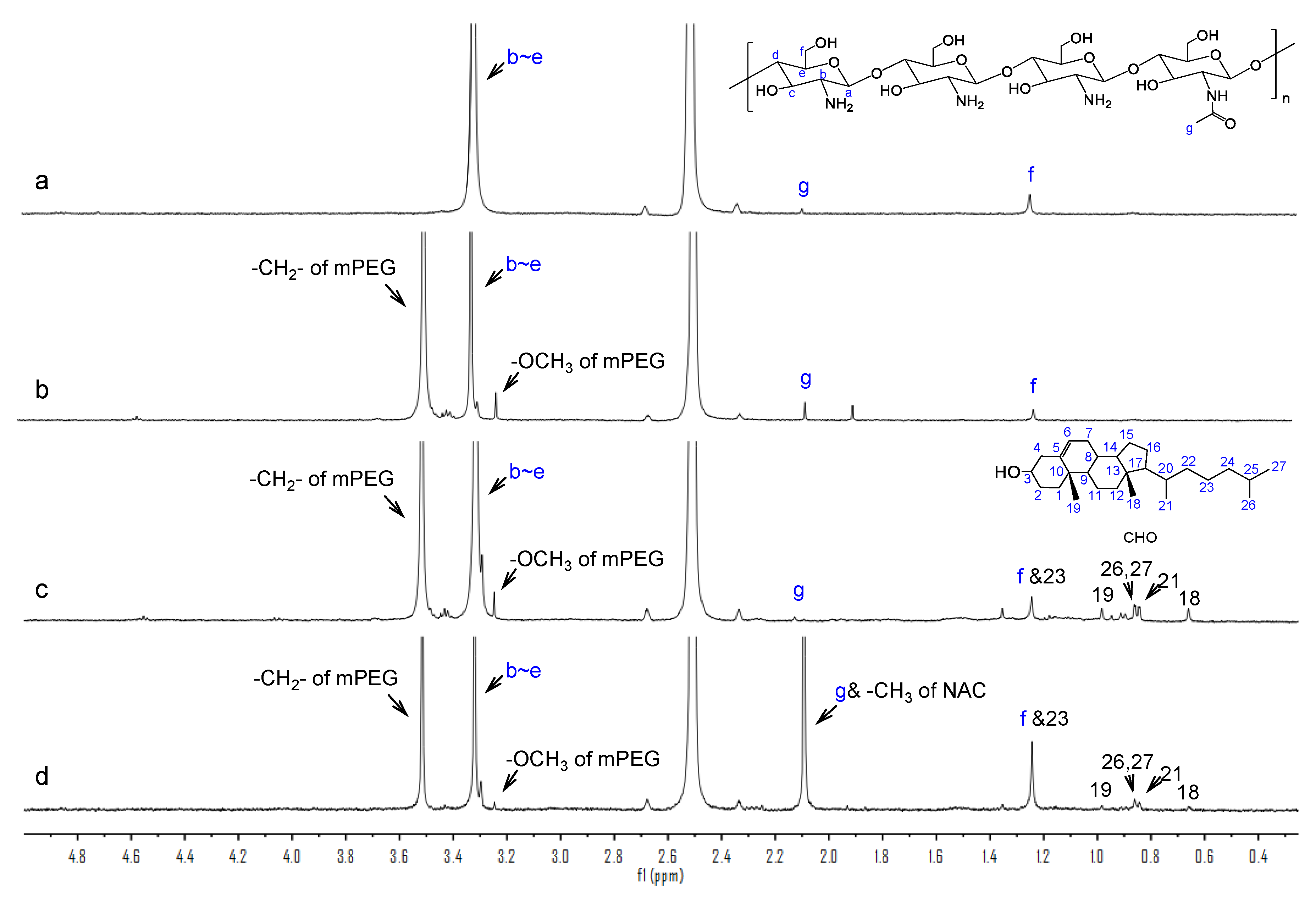
3.2.2. 1H-NMR Characterization
3.3. Preparation of mPEG-CS(SH)-CHO Micelles
3.4. Characterization of PTX-Loaded mPEG-CS(SH)-CHO Micelles
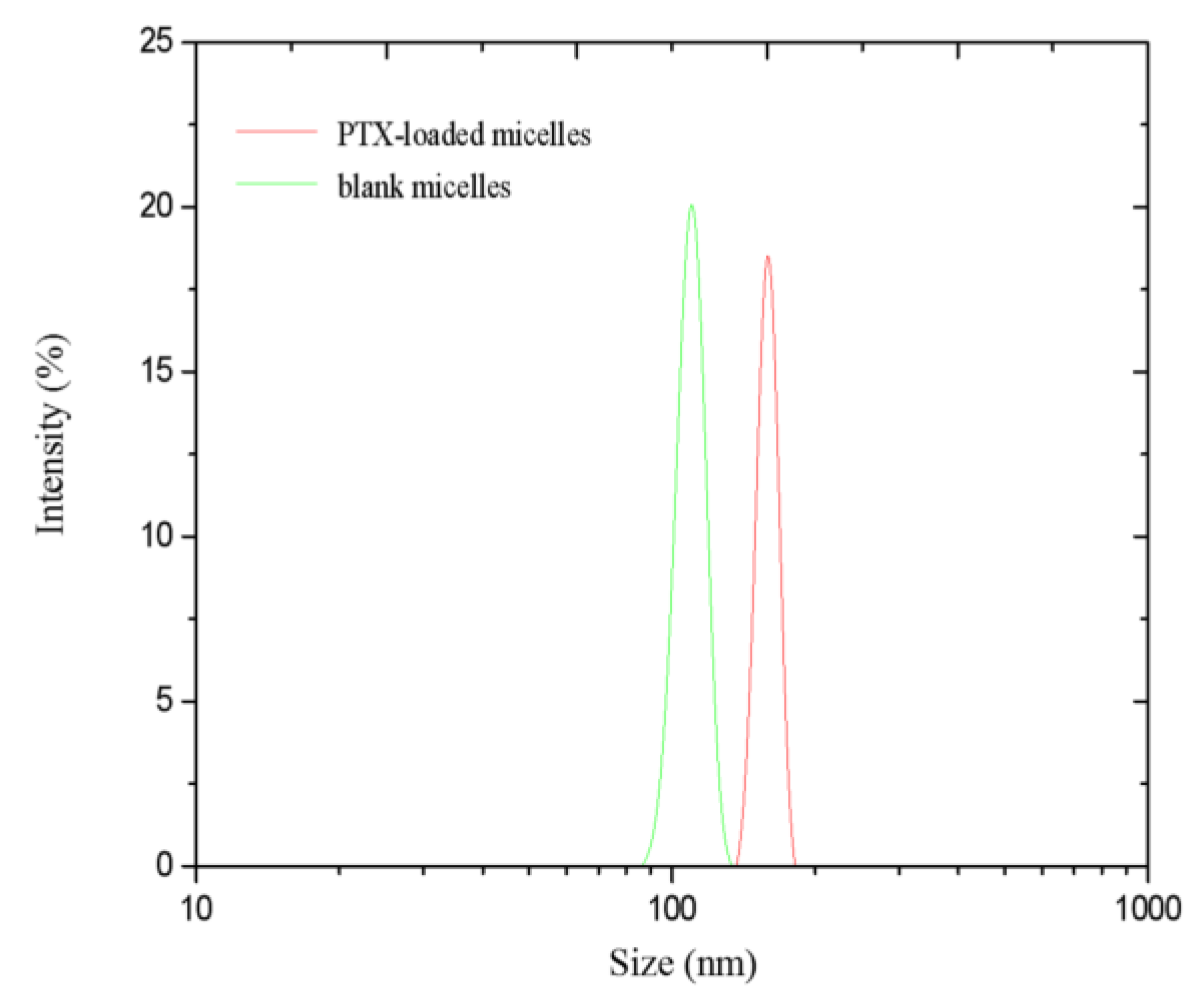
3.4.1. Hydrodynamic Diameter and Zeta Potential
3.4.2. TEM Observation
3.4.3. XRD Analysis
3.5. Protein Adsorption
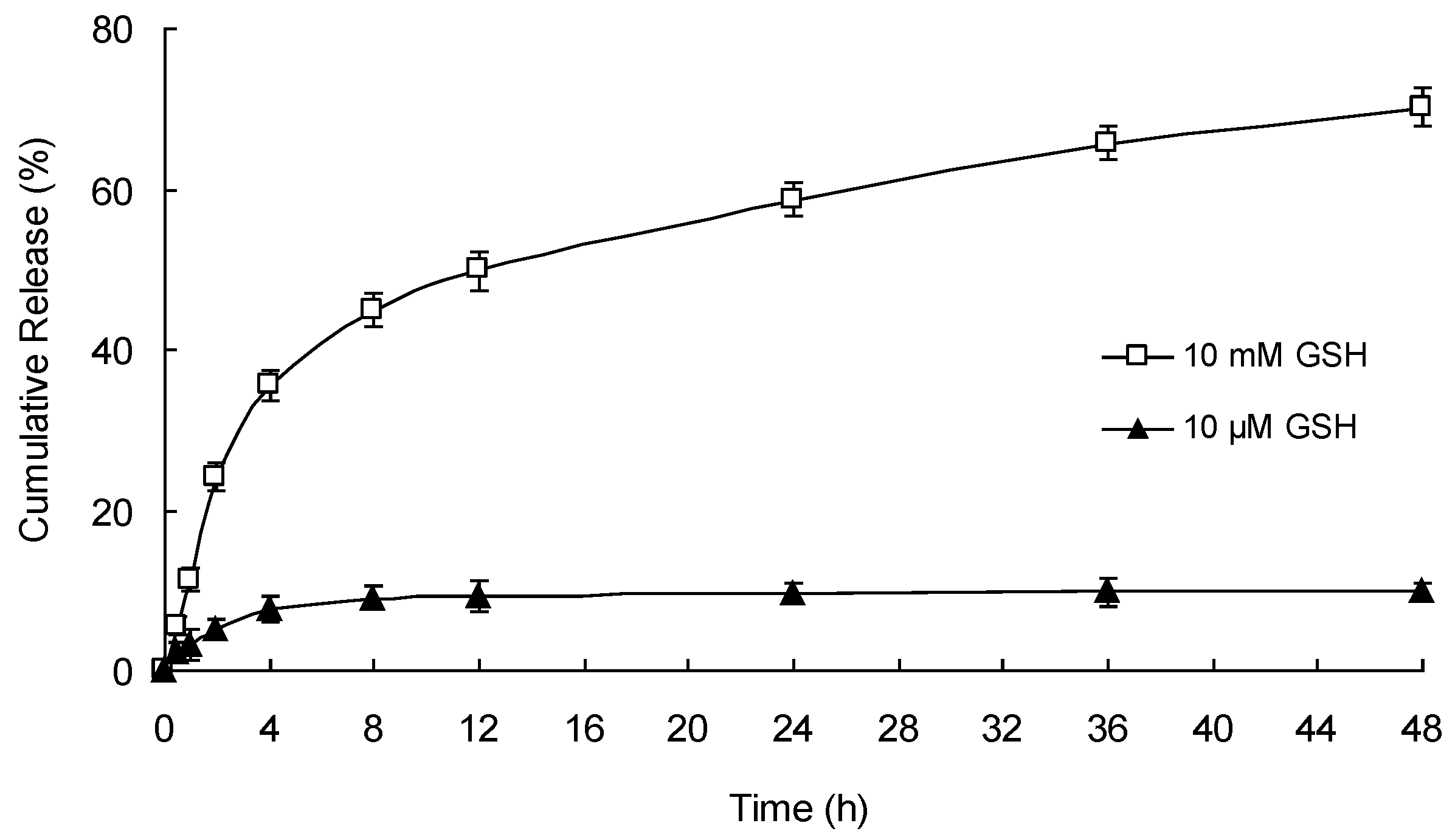
3.6. In Vitro Redox-Responsive Drug Release
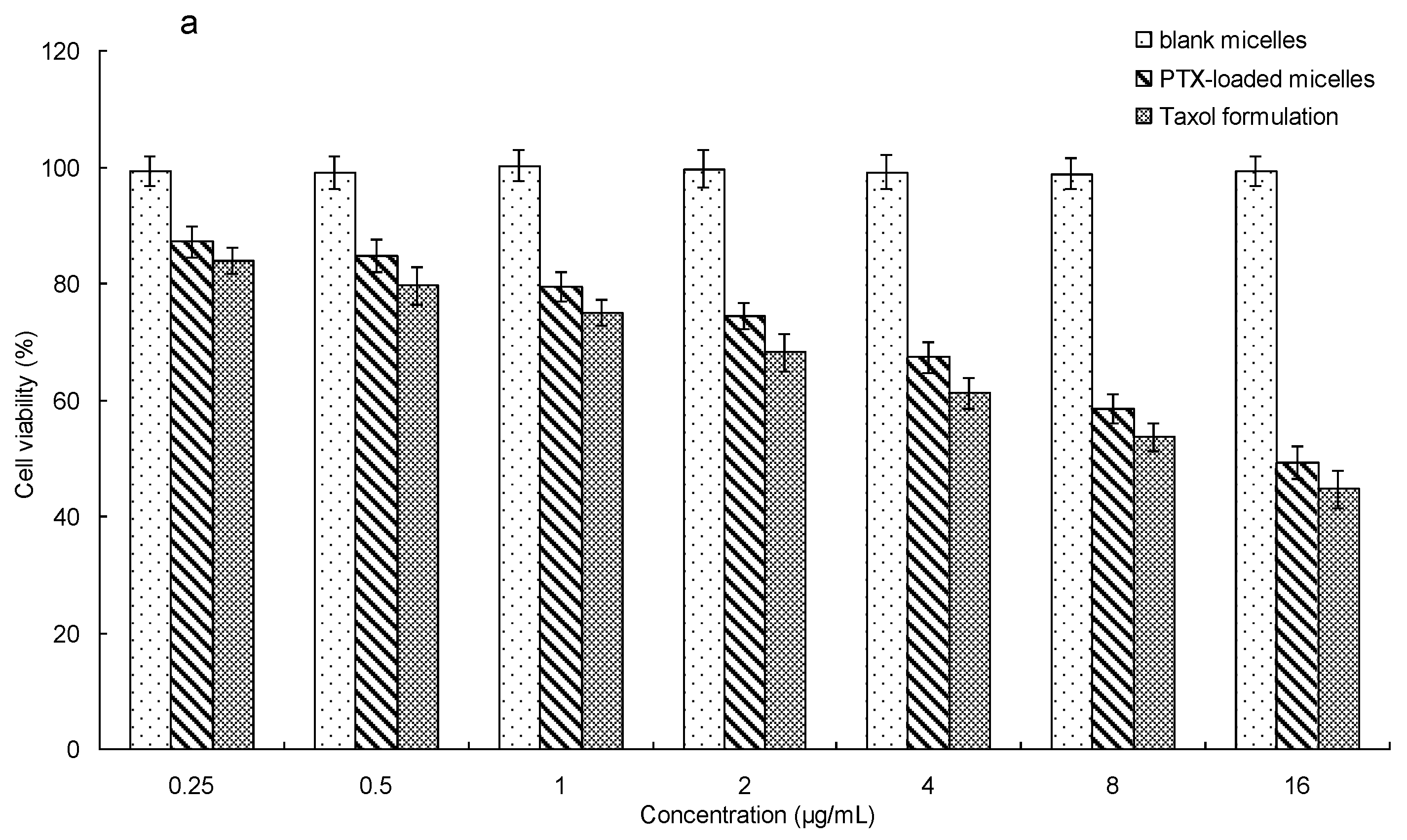
3.7. In Vitro Cytotoxicity
3.8. In Vivo Antitumor Efficacy
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Singla, A.K.; Garg, A.; Aggarwal, D. Paclitaxel and its formulations. Int. J. Pharm. 2002, 235, 179–192. [Google Scholar] [CrossRef]
- Bernabeu, E.; Cagel, M.; Lagomarsino, E.; Moretton, M.; Chiappetta, D.A. Paclitaxel: What has been done and the challenges remain ahead. Int. J. Pharm. 2017, 526, 474–495. [Google Scholar] [CrossRef]
- Tian, Q.; Fei, C.; Yin, H.; Feng, Y. Stimuli-responsive polymer wormlike micelles. Prog. Polym. Sci. 2019, 89, 108–132. [Google Scholar] [CrossRef]
- Deshmukh, A.S.; Chauhan, P.N.; Noolvi, M.N.; Chaturvedi, K.; Ganguly, K.; Shukla, S.S.; Nadagouda, M.N.; Aminabhavi, T.M. Polymeric micelles: Basic research to clinical practice. Int. J. Pharm. 2017, 532, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Nakamura, H.; Fang, J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug Deliv. Rev. 2013, 65, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Chen, Y.; Zhang, Z.-H.; Han, X. Stimuli-responsive block copolymer-based assemblies for cargo delivery and theranostic applications. Polymers 2016, 8, 268. [Google Scholar] [CrossRef] [PubMed]
- Gulzar, A.; Xu, J.; Wang, C.; He, F.; Yang, D.; Gai, S.; Yang, P.; Lin, J.; Jin, D.; Xing, B. Tumour microenvironment responsive nanoconstructs for cancer theranostic. Nano Today 2019, 26, 16–56. [Google Scholar] [CrossRef]
- Hong, R.; Han, G.; Fernández, J.M.; Kim, B.-J.; Forbes, N.S.; Rotello, V.M. Glutathione-mediated delivery and release using monolayer protected nanoparticle carriers. J. Am. Chem. Soc. 2006, 128, 1078–1079. [Google Scholar] [CrossRef] [PubMed]
- Brülisauer, L.; Gauthier, M.A.; Leroux, J.C. Disulfide-containing parenteral delivery systems and their redox-biological fate. J. Control. Release 2014, 195, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Cort, A.; Ozdemir, E.; Timur, M.; Ozben, T. Effects of N-acetyl-l-cysteine on bleomycin induced oxidative stress in malignant testicular germ cell tumors. Biochimie 2012, 94, 2734–2739. [Google Scholar] [CrossRef]
- Harris, J.M.; Martin, N.E.; Modi, M. Pegylation: A novel process for modifying pharmacokinetics. Clin. Pharm. 2001, 40, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Nayak, B.; Dey, R.K. PEGylation in anti-cancer therapy: An overview. Asian J. Pharm. Sci. 2016, 11, 337–348. [Google Scholar] [CrossRef]
- Huang, S.; Yu, X.; Yang, L.; Song, F.; Chen, G.; Lv, Z.; Li, T.; Chen, D.; Zhu, W.; Yu, A.; et al. The efficacy of nimodipine drug delivery using mPEG-PLA micelles and mPEG-PLA/TPGS mixed micelles. Eur. J. Pharm. Sci. 2014, 63, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Kumari, P.; Lakhani, P.M.; Ghosh, B. Recent advances in polymeric micelles for anti-cancer drug delivery. Eur. J. Pharm. Sci. 2016, 83, 184–202. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, S.M.; Thomas, M.; Reddy, K.K.; Sooraparaju, S.G.; Asthana, A.; Bhatnagar, I. Chitosan as biomaterial in drug delivery and tissue engineering. Int. J. Biol. Macromol. 2018, 110, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Tan, Y.F.; Wong, Y.S.; Liew, M.W.J.; Venkatraman, S. Recent advances in chitosan-based carriers for Gene delivery. Mar. Drugs 2019, 17, 381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liang, N.; Gong, X.; Kawashima, Y.; Cui, F.; Sun, S. Tumor-targeting micelles based on folic acid and α-tocopherol succinate conjugated hyaluronic acid for paclitaxel delivery. Colloids Surf. B Biointerfaces 2019, 177, 11–18. [Google Scholar] [CrossRef]
- Ermilova, I.; Lyubartsev, A.P. Cholesterol in phospholipid bilayers: Positions and orientations inside membranes with different unsaturation degrees. Soft Matter 2019, 15, 78–93. [Google Scholar] [CrossRef]
- Chou, T.-H.; Chen, C.-W.; Liang, C.-H.; Yeh, L.-H.; Qian, S. Simple synthesis, self-assembly, and cytotoxicity of novel dimeric cholesterol derivatives. Colloids Surf. B Biointerfaces 2014, 116, 153–159. [Google Scholar] [CrossRef]
- Chang, K.; Chang, F.-H.; Chen, M.-H. Developing a novel cholesterol-based nanocarrier with high transfection efficiency and serum compatibility for gene therapy. J. Formos. Med Assoc. 2019, 118, 766–775. [Google Scholar] [CrossRef]
- Maletínská, L.; A Blakely, E.; A Bjornstad, K.; Deen, D.F.; Knoff, L.J.; Forte, T.M. Human glioblastoma cell lines: Levels of low-density lipoprotein receptor and low-density lipoprotein receptor-related protein. Cancer Res. 2000, 60, 2300–2303. [Google Scholar] [PubMed]
- Lee, J.-J.; Park, J.-H.; Kim, D.-D.; Cho, H.-J. Cholesterol-modified poly(lactide-co-glycolide) nanoparticles for tumor-targeted drug delivery. Int. J. Pharm. 2016, 509, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.-K.; Jeong, Y.-I.; Nah, J.-W. Characterization and preparation of core–shell type nanoparticle for encapsulation of anticancer drug. Colloids Surf. B Biointerfaces 2010, 81, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Qu, G.; Yao, Z.; Zhang, C.; Wu, X.; Ping, Q. PEG conjugated N-octyl-O-sulfate chitosan micelles for delivery of paclitaxel: In vitro characterization and in vivo evaluation. Eur. J. Pharm. Sci. 2009, 37, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Liu, Y.; Yang, W.; Li, X.; Liu, L.; Zhou, Z.; Wang, Y.; Li, R.; Zhang, Q. Preparation and characterization of self-assembled nanoparticles of 6-O-cholesterol-modified chitosan for drug delivery. Carbohydr. Polym. 2011, 84, 1244–1251. [Google Scholar] [CrossRef]
- Li, L.; Liang, N.; Wang, D.; Yan, P.; Kawashima, Y.; Cui, F.; Sun, S. Amphiphilic polymeric micelles based on deoxycholic acid and folic acid modified chitosan for the delivery of paclitaxel. Int. J. Mol. Sci. 2018, 19, 3132. [Google Scholar] [CrossRef]
- Liang, N.; Sun, S.; Li, X.; Piao, H.; Piao, H.; Cui, F.; Fang, L. α-Tocopherol succinate-modified chitosan as a micellar delivery system for paclitaxel: Preparation, characterization and in vitro/in vivo evaluations. Int. J. Pharm. 2012, 423, 480–488. [Google Scholar] [CrossRef]
- Fu, D.-J.; Jin, Y.; Xie, M.-Q.; Ye, Y.-J.; Qin, D.-D.; Lou, K.-Y.; Chen, Y.-Z.; Gao, F. Preparation and characterization of mPEG grafted chitosan micelles as 5-fluorouracil carriers for effective anti-tumor activity. Chin. Chem. Lett. 2014, 26, 1435–1440. [Google Scholar] [CrossRef]
- Kasaai, M.R. Determination of the degree of N-acetylation for chitin and chitosan by various NMR spectroscopy techniques: A review. Carbohydr. Polym. 2010, 79, 801–810. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, Q.; Huang, K.; Li, J.; Wang, K.; Zhang, K.; Tang, X. Preparation and characterization of carboxymethyl cellulose containing quaternized chitosan for potential drug carrier. Int. J. Biol. Macromol. 2019. [Google Scholar] [CrossRef]
- Zipser, B.; Bradford, J.J.; Hollingsworth, R.I. Cholesterol and its derivatives, are the principal steroids isolated from the leech species Hirudo medicinalis. Comp. Biochem. Physiol. Part C Pharmacol. Toxicol. Endocrinol. 1998, 120, 269–282. [Google Scholar] [CrossRef]
- Sang, X.; Yang, Q.; Shi, G.; Zhang, L.; Wang, D.; Ni, C. Preparation of pH/redox dual responsive polymeric micelles with enhanced stability and drug controlled release. Mater. Sci. Eng. C 2018, 91, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Albanese, A.; Tang, P.S.; Chan, W.C.W. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu. Rev. Biomed. Eng. 2012, 14, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-X.; Zuo, Z.-Q.; Du, J.-Z.; Wang, Y.-C.; Sun, R.; Cao, Z.-T.; Ye, X.-D.; Wang, J.-L.; Leong, K.W.; Wang, J. Surface charge critically affects tumor penetration and therapeutic efficacy of cancer nanomedicines. Nano Today 2016, 11, 133–144. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, F.; Chen, Y.; Zhang, Q.; Zheng, D.; Hao, L.; Liu, Y.; Duan, C.; Jia, L.; Liu, G. Folate-mediated targeted and intracellular delivery of paclitaxel using a novel deoxycholic acid-O-carboxymethylated chitosan-folic acid micelles. Int. J. Nanomed. 2012, 7, 325–337. [Google Scholar] [CrossRef]
- Kumar, P.; Dehiya, B.S.; Sindhu, A. Synthesis and characterization of nHA-PEG and nBG-PEG scaffolds for hard tissue engineering applications. Ceram. Int. 2019, 45, 8370–8379. [Google Scholar] [CrossRef]
- Rabe, M.; Verdes, D.; Seeger, S. Understanding protein adsorption phenomena at solid surfaces. Adv. Colloid Interface Sci. 2011, 162, 87–106. [Google Scholar] [CrossRef]
- Chen, S.; Li, L.; Zhao, C.; Zheng, J. Surface hydration: Principles and applications toward low-fouling/nonfouling biomaterials. Polymer 2010, 51, 5283–5293. [Google Scholar] [CrossRef]
- Rahmati, M.; Mozafari, M. Protein adsorption on polymers. Mater. Today Commun. 2018, 17, 527–540. [Google Scholar] [CrossRef]
- Fleischer, C.C.; Payne, C.K. Nanoparticle–cell interactions: Molecular structure of the protein corona and cellular outcomes. Acc. Chem. Res. 2014, 47, 2651–2659. [Google Scholar] [CrossRef]
- Cui, C.; Xue, Y.-N.; Wu, M.; Zhang, Y.; Yu, P.; Liu, L.; Zhuo, R.-X.; Huang, S.-W. Poly(L-aspartamide)-based reduction-sensitive micelles as nanocarriers to improve doxorubicin content in cell nuclei and to enhance antitumor activity. Macromol. Biosci. 2013, 13, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Qin, B.; Liu, L.; Wu, X.; Liang, F.; Hou, T.; Pan, Y.; Song, S. mPEGylated solanesol micelles as redox-responsive nanocarriers with synergistic anticancer effect. Acta Biomater. 2017, 64, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Yen, H.-J.; Young, Y.-A.; Tsai, T.-N.; Cheng, K.-M.; Chen, X.-A.; Chen, Y.-C.; Chen, C.-C.; Young, J.-J.; Hong, P.-D. Positively charged gold nanoparticles capped with folate quaternary chitosan: Synthesis, cytotoxicity, and uptake by cancer cells. Carbohydr. Polym. 2018, 183, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomed. 2012, 7, 5577–5591. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-F.; Chang, C.-H.; Lin, C.-Y.; Lin, L.-F.; Yeh, M.-L.; Jan, J.-S. Disulfide-cross-linked PEG-block-polypeptide nanoparticles with high drug loading content as glutathione-triggered anticancer drug nanocarriers. Colloids Surf. B Biointerfaces 2018, 165, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, S.P.H.; Yazdimamaghani, M.; Ghandehari, H. Glutathione-sensitive hollow mesoporous silica nanoparticles for controlled drug delivery. J. Control. Release 2018, 282, 62–75. [Google Scholar] [CrossRef] [PubMed]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, Y.; Liang, N.; Yan, P.; Kawashima, Y.; Cui, F.; Sun, S. A Chitosan-Based Micellar System as Nanocarrier For the Delivery of Paclitaxel. Polymers 2020, 12, 380. https://doi.org/10.3390/polym12020380
Han Y, Liang N, Yan P, Kawashima Y, Cui F, Sun S. A Chitosan-Based Micellar System as Nanocarrier For the Delivery of Paclitaxel. Polymers. 2020; 12(2):380. https://doi.org/10.3390/polym12020380
Chicago/Turabian StyleHan, Yang, Na Liang, Pengfei Yan, Yoshiaki Kawashima, Fude Cui, and Shaoping Sun. 2020. "A Chitosan-Based Micellar System as Nanocarrier For the Delivery of Paclitaxel" Polymers 12, no. 2: 380. https://doi.org/10.3390/polym12020380
APA StyleHan, Y., Liang, N., Yan, P., Kawashima, Y., Cui, F., & Sun, S. (2020). A Chitosan-Based Micellar System as Nanocarrier For the Delivery of Paclitaxel. Polymers, 12(2), 380. https://doi.org/10.3390/polym12020380