Cellulose Structural Changes during Mild Torrefaction of Eucalyptus Wood





Abstract
:1. Introduction
2. Materials and Methods
2.1. Raw Materials and Thermal Treatment
2.2. Analysis of Wood
2.3. Cellulose Isolation
2.4. Cellulose Characterization
3. Results and Discussion
3.1. Chemical Analysis of the Thermally Treated Woods
3.2. Changes in Cellulose Structure during the Thermal Treatment
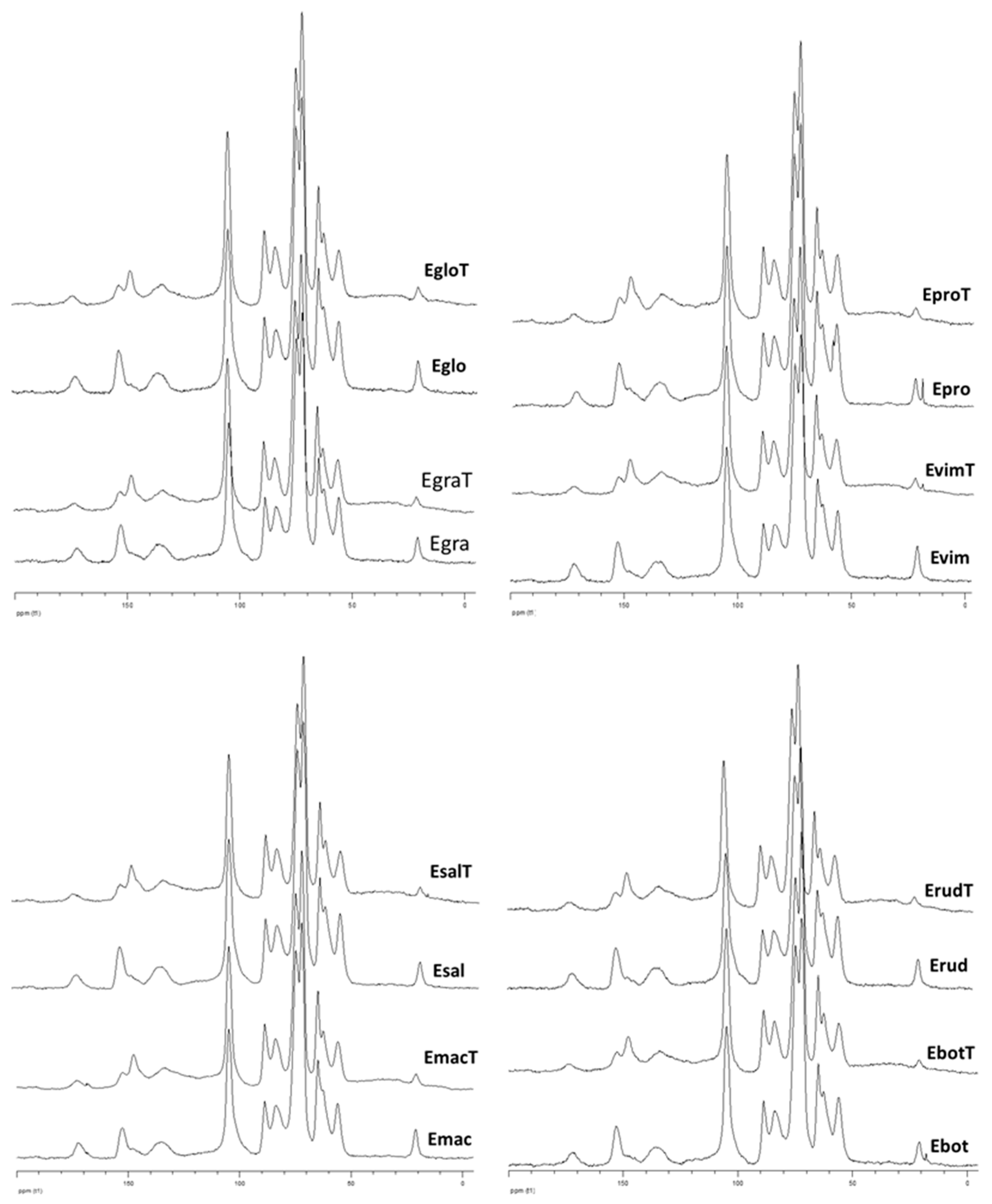
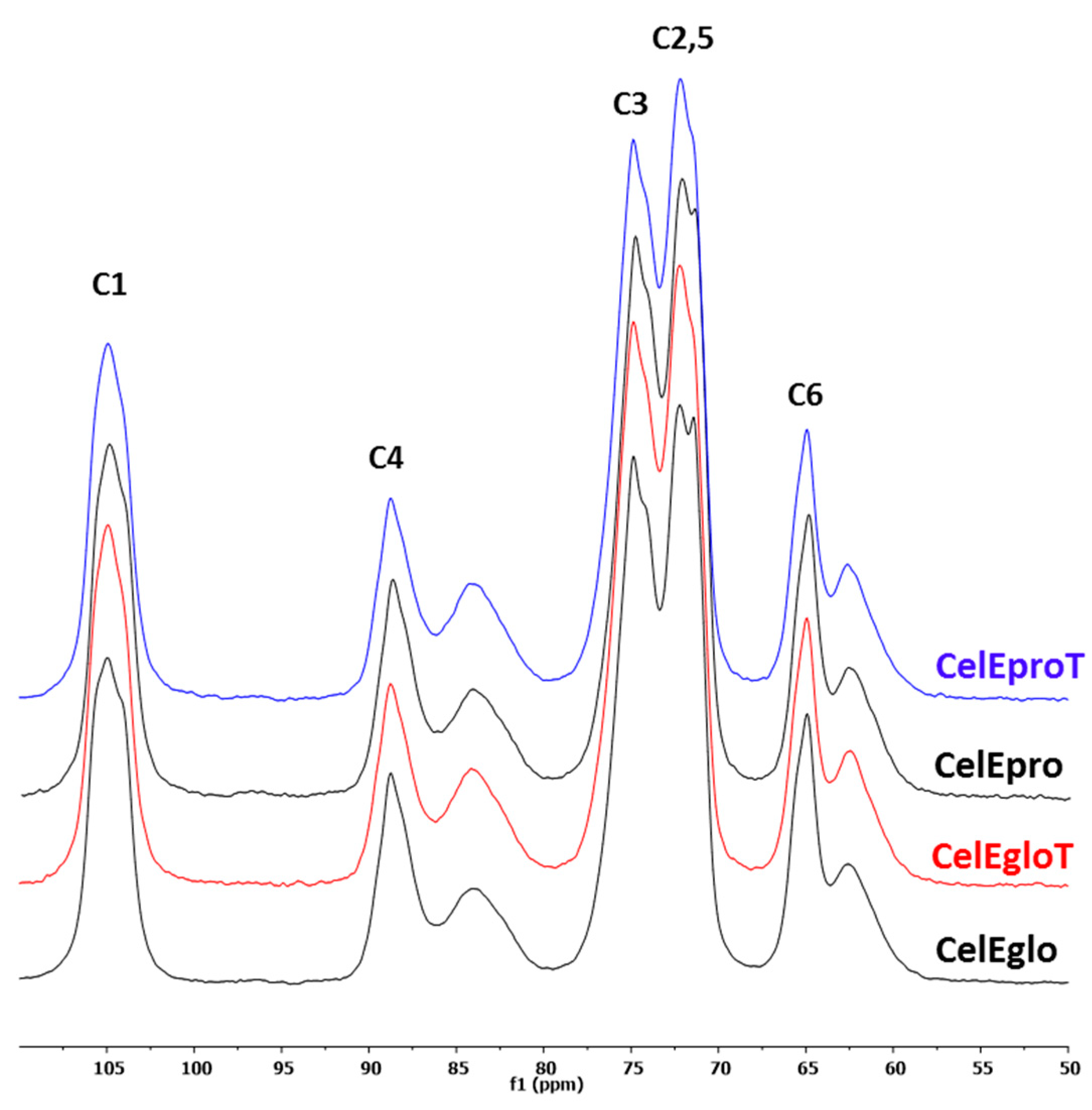
3.2.1. CP-MAS 13C NMR Analysis
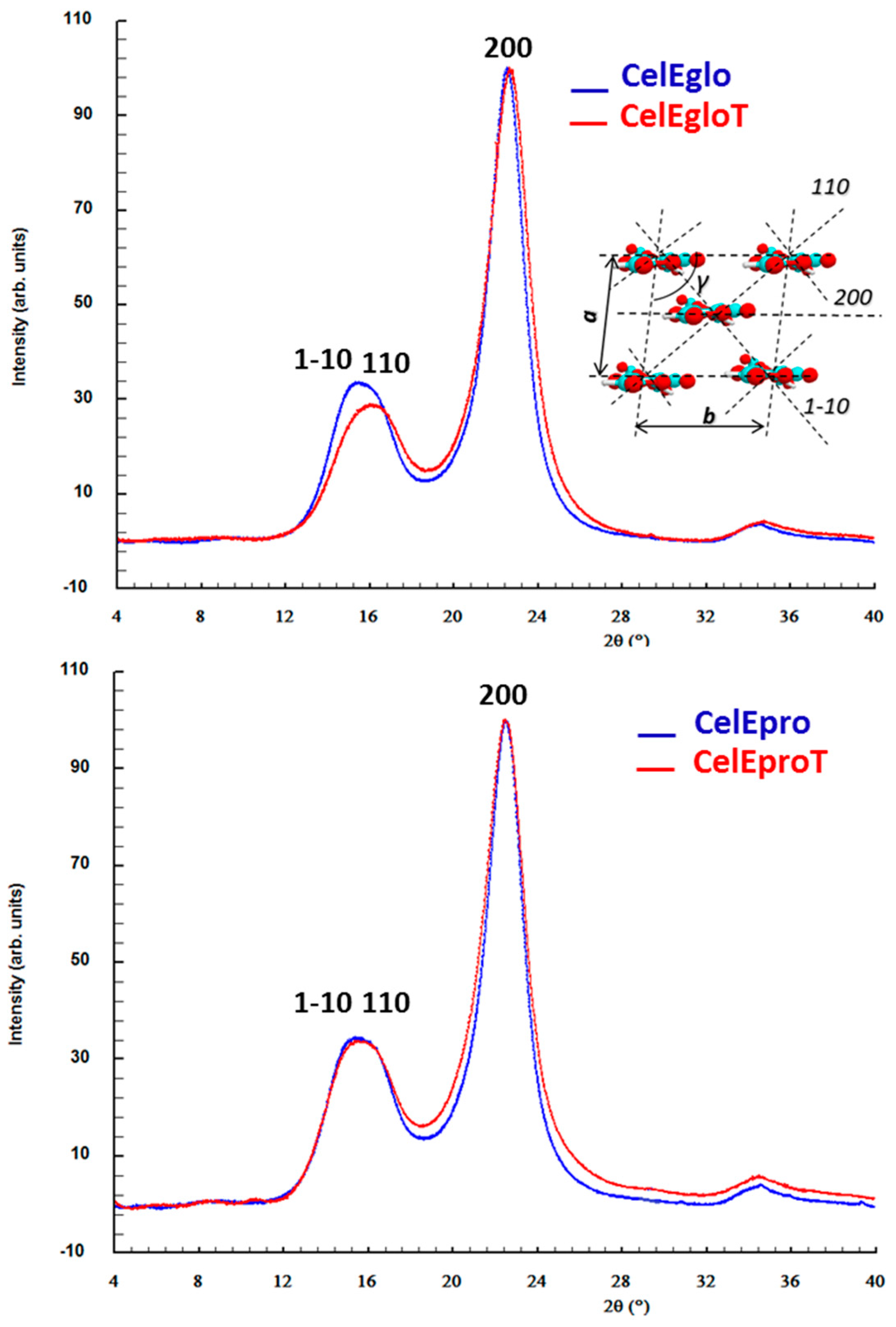
3.2.2. X-ray Scattering Analysis (WAXS)
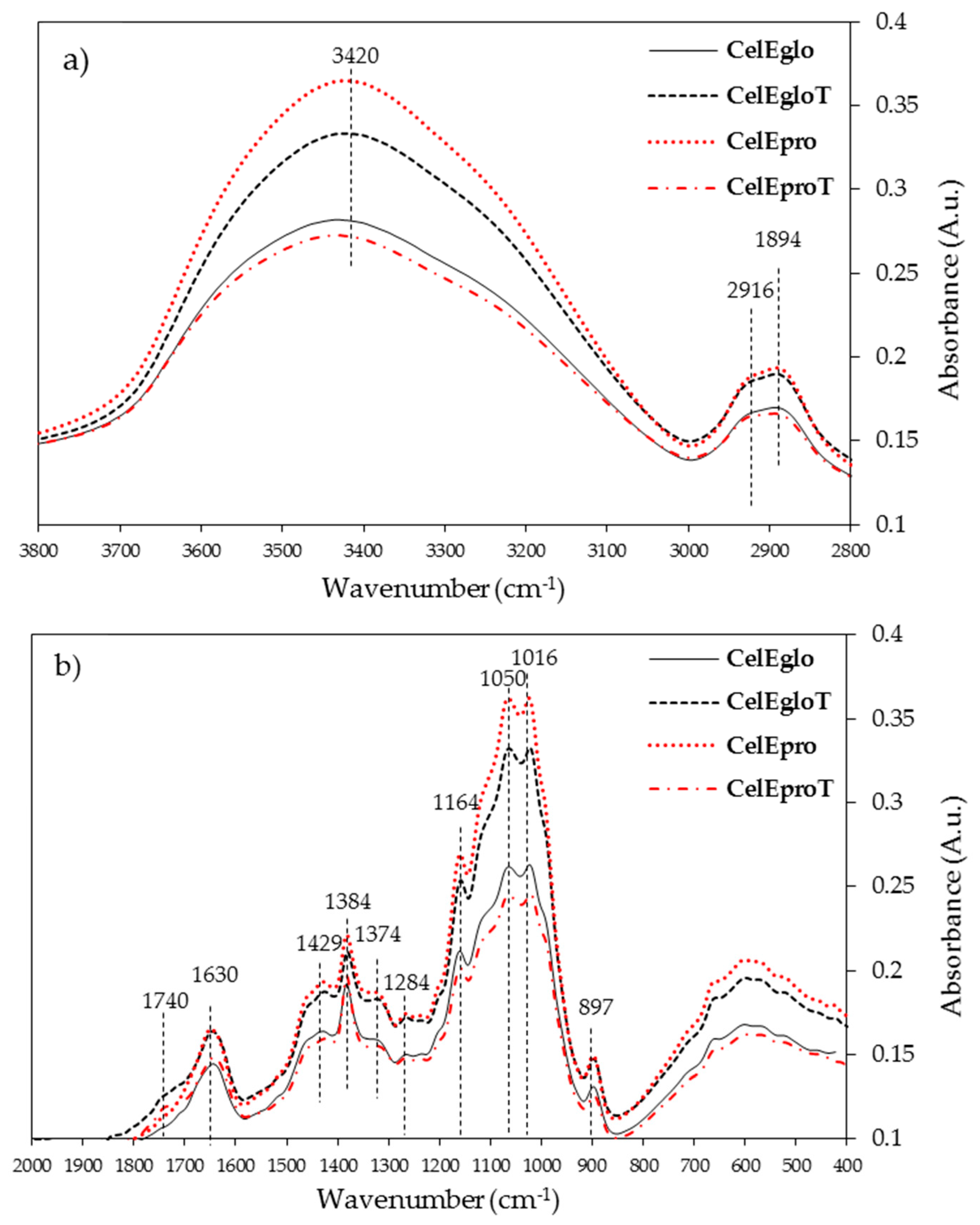
3.2.3. FTIR Analysis
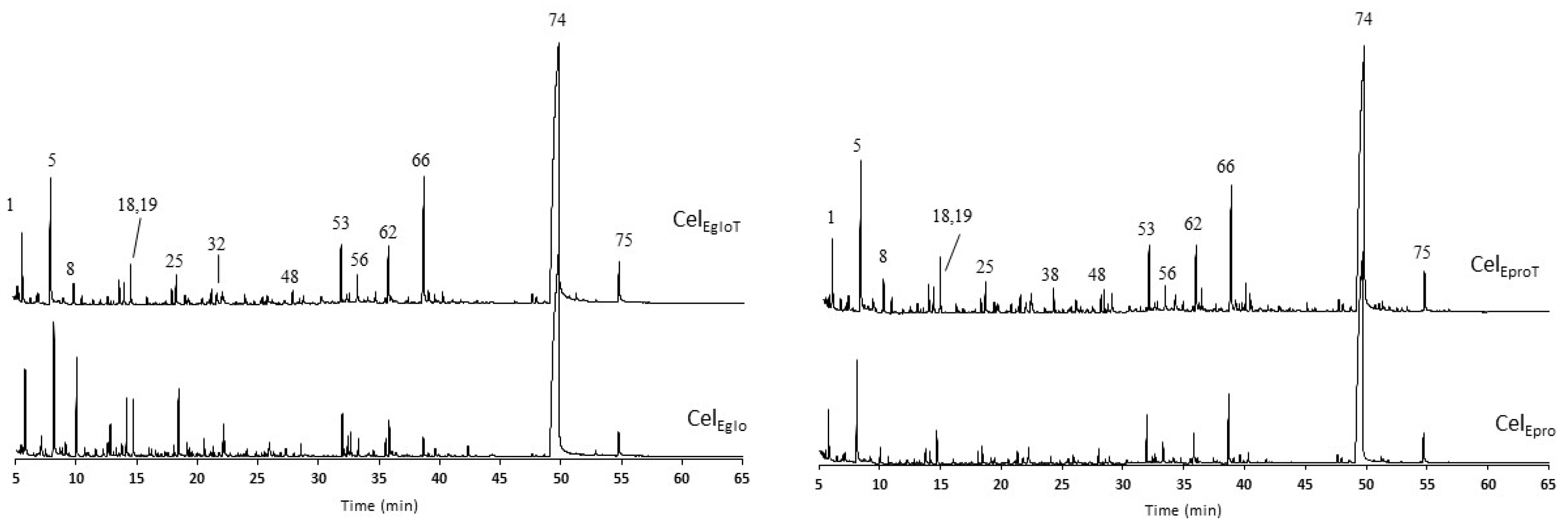
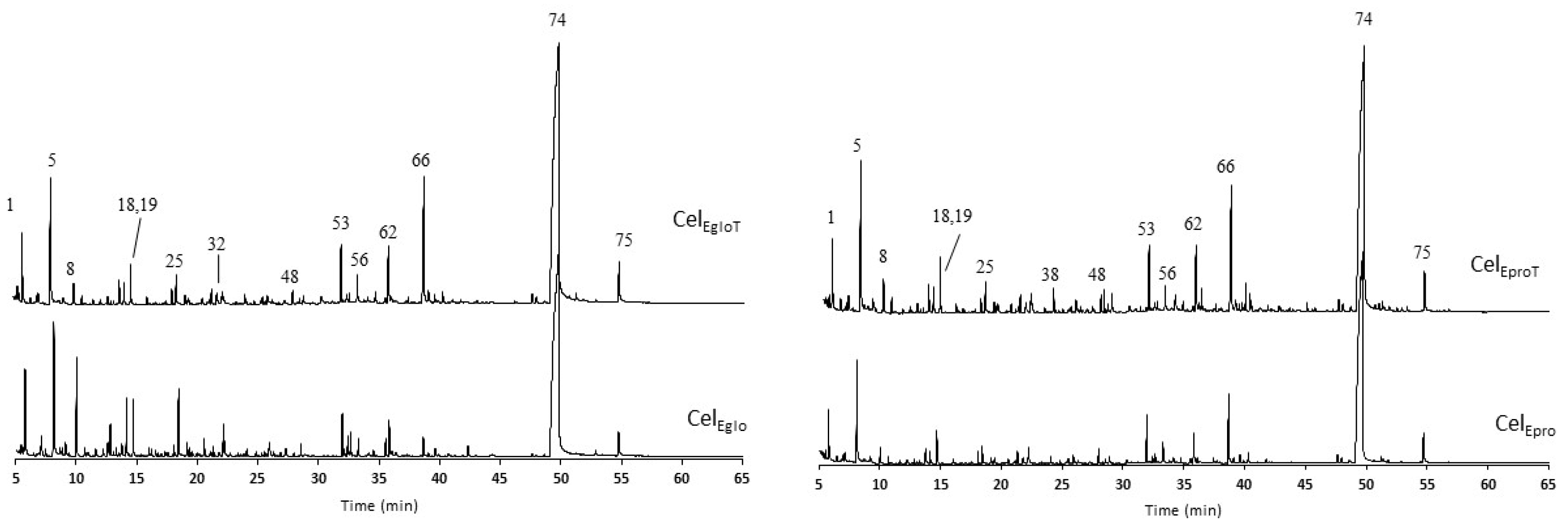
3.2.4. Pyrolysis Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Priadi, T.; Hiziroglu, S. Characterization of heat treated wood species. Mater. Des. 2013, 49, 575–582. [Google Scholar] [CrossRef]
- Rowell, R.M. Chemical Modification of Wood. Forest Prod. Abstr. 1983, 6, 363–382. [Google Scholar]
- Ibach, RE. Biological properties of wood. In Handbook of Chemistry and Wood Composites, 2nd ed.; Rowell, R.M., Ed.; Taylor Francis: Boca Raton, FL, USA, 2013; pp. 99–126. [Google Scholar]
- Evans, P.D. Weathering of wood and wood composites. In Handbook of Chemistry and Wood Composites, 2nd ed.; Rowell, R.M., Ed.; Taylor Francis: Boca Raton, FL, USA, 2013; pp. 152–200. [Google Scholar]
- Jones, D.; Brischke, C. (Eds.) Performance of Bio-Based Building Materials; Woodhead Publishing, Elsevier Ltd.: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Tiemann, H.D. The effect of different methods of drying on the strength of wood. Lumber World Rev. 1915, 28, 19–20. [Google Scholar]
- Stamm, A.J.; Hansen, L.A. Minimizing wood shrinkage and swelling effect of heating in various gasses. Ind. Eng. Chem. 1937, 29, 831–833. [Google Scholar] [CrossRef]
- Pelaez-Samaniego, M.R.; Yadama, V.; Lowell, E.; Espinoza-Herrera, R. A review of wood thermal pretreatments to improve wood composite properties. Wood Sci. Technol. 2013, 47, 1285–1319. [Google Scholar] [CrossRef]
- Esteves, B.M.; Pereira, H. Wood modification by heat treatment: A Review. Bioresources 2009, 4, 370–404. [Google Scholar] [CrossRef]
- Sandberg, D.; Kutnar, A. Thermal modified timber: Recent developments in Europe and North America. Wood Fiber. Sci. 2016, 48, 28–39. Available online: https://wfs.swst.org/index.php/wfs/article/view/2296 (accessed on 1 October 2020).
- Tjeerdsma, B.F.; Boonstra, M.; Pizzi, A.; Tekely, P.; Militz, H. Characterisation of thermally modified wood: Molecular reasons for wood performance improvement. Holz als Roh-und Werkst. 1998, 56, 149–153. [Google Scholar] [CrossRef]
- Ru, B.; Wang, S.; Dai, G.; Zhang, L. Effect of torrefaction on biomass physicochemical characteristics and the resulting pyrolysis behavior. Energy Fuels 2015, 29, 5865–5874. [Google Scholar] [CrossRef]
- Silva, C.M.S.; Carneiro, A.C.O.; Vital, B.R.; Figueiró, C.G.; Fialho, L.F.; Magalhães, M.A.; Carvalho, A.G.; Cândido, W.L. Biomass Torrefaction for Energy Purposes—Definitions and an Overview of Challenges and Opportunities in Brazil. Renew. Sustain. Energy Rev. 2018, 82, 2426–2432. [Google Scholar] [CrossRef]
- Manninen, A.M.; Pasanen, P.; Holopainen, J.K. Comparing the VOC emissions between air-dried and heat-treated Scots pine wood. Atmos. Environ. 2002, 36, 1763–1768. [Google Scholar] [CrossRef]
- Kim, J.Y.; Hwang, H.; Oh, S.; Kim, Y.S.; Kim, U.J.; Choi, J.W. Investigation of structural modification and thermal characteristics of lignin after heat treatment. Int. J. Biol. Macromol. 2014, 66, 57–65. [Google Scholar] [CrossRef]
- Lourenço, A.; Araújo, S.; Gominho, J.; Pereira, H.; Evtuguin, D. Structural changes in lignin of thermally treated eucalyptus wood. J. Wood Chem. Technol. 2020, 40, 258–268. [Google Scholar] [CrossRef]
- Hon, D.N.C. Cellulose: A random walk along its historical path. Cellulose 1994, 1, 1–25. [Google Scholar] [CrossRef]
- Bhuiyan, M.T.R.; Hirai, N.; Sobue, N. Changes of crystallinity in wood cellulose by heat treatment under drier and moist conditions. J. Wood Sci. 2000, 46, 431–436. [Google Scholar] [CrossRef]
- Bhuiyan, R.; Hirai, T.; Sobue, N.N. Effect of intermittent heat treatment on crystallinity in wood cellulose. J. Wood Sci. 2001, 47, 336–341. [Google Scholar] [CrossRef]
- Tarmian, A.; Mastouri, A. Changes in moisture exclusion efficiency and crystallinity of thermally modified wood with aging. iForest-Biogeosciences For. 2019, 12, 92–97. [Google Scholar] [CrossRef] [Green Version]
- Kubovský, I.; Kačíková, D.; Kačík, F. Structural changes of oak wood main components caused by thermal modification. Polymers 2020, 12, 485. [Google Scholar] [CrossRef] [Green Version]
- Melkior, T.; Jacob, S.; Gerbaud, G.; Hediger, S.; Le Pape, L.; Bonnefois, L.; Bardet, M. NMR analysis of the transformation of wood constituents by torrefaction. Fuel 2012, 92, 271–280. [Google Scholar] [CrossRef]
- Hill, S.J.; Grigsby, W.J.; Hall, P.W. Chemical and cellulose crystallite changes in Pinus radiata during torrefaction. Biomass Bioenergy 2013, 56, 92–98. [Google Scholar] [CrossRef]
- Wikberg, H.; Maunu, S.L. Characterization of thermally modified hard and softwoods by 13C CPMAS NMR. Carbohydr. Polym. 2004, 58, 461–466. [Google Scholar] [CrossRef]
- Yildiz, S.; Gümüskaya, E. The effects of thermal modification on crystalline structure of cellulose in soft and hardwood. Build. Environ. 2007, 42, 62–67. [Google Scholar] [CrossRef]
- Wentzel, M.; Fleckenstein, M.; Hofmann, T.; Militz, H. Relation of chemical and mechanical properties of Eucalyptus nitens wood thermally modified in open and closed systems. Wood Mater. Sci. Eng. 2019, 14, 165–173. [Google Scholar] [CrossRef]
- Baccile, N.; Falco, C.; Titirici, M.M. Characterization of biomass and its derived char using 13C solid state nuclear magnetic resonance. Green Chem. 2014, 16, 4839–4869. [Google Scholar] [CrossRef] [Green Version]
- Araújo, S.O.; Neiva, D.M.; Gominho, J.; Esteves, B.; Pereira, H. Chemical effects of a mild torrefaction on the wood of eight Eucalyptus species. Holzforschung 2017, 71, 291–298. [Google Scholar] [CrossRef]
- Browning, B.L. Methods in Wood Chemistry; John Wiley& Sons: New York, NY, USA, 1967; Volume II, pp. 406–727. [Google Scholar]
- Evtuguin, D.V.; Tomás, J.L.; Silva, A.M.S.; Neto, C. Characterization of acetylated heteroxylan from Eucalyptus globulus Labill. Carbohydr. Res. 2003, 338, 597–607. [Google Scholar] [CrossRef]
- Tribulová, T.; Kačík, F.; Evtuguin, D.V.; Čabalová, I.; Ďurkovičd, J. The effects of transition metal sulfates on cellulose crystallinity during accelerated ageing of silver fir wood. Cellulose 2019, 26, 2625–2638. [Google Scholar] [CrossRef]
- Ioelovitch, M.Y.; Tupureine, A.D.; Veveris, G.P. Study on the cellulose crystallinity in plant materials. Khimiya Drev. 1989, 5, 3–9. [Google Scholar]
- Tripp, V.W. Measurement of crystallinity. In Cellulose and Cellulose Derivatives; Bikales, N.M., Segal, L., Eds.; Part IV; Wiley-Interscience: New York, NY, USA, 1971; pp. 305–323. [Google Scholar]
- Auxenfans, T.; Crônie, D.; Chabbert, B.; Paёs, G. Understanding the structural and chemical changes of plant biomass following steam explosion pretreatment. Biotechnol. Biofuels 2017, 10, 36. [Google Scholar] [CrossRef] [Green Version]
- Liitiä, T.; Maunu, S.; Hortling, B. Solid-state NMR studies on cellulose crystallinity in fines and bulk fibres separated from refined kraft pulp. Holzforschung 2000, 54, 618–624. [Google Scholar] [CrossRef]
- Wickholm, K.; Larsson, PT.; Iversen, T. Assignment of non-crystalline forms in cellulose I by CP/MAS 13C NMR spectroscopy. Carbohydr. Res. 1998, 312, 123–129. [Google Scholar] [CrossRef]
- Ralph, J.; Hatfield, R.D. Pyrolysis-GC-MS characterization of forage materials. J. Agric. Food Chem. 1991, 39, 1426–1437. [Google Scholar] [CrossRef]
- Faix, O.; Fortman, I.; Bremer, J.; Meier, D. Thermal degradation products of wood. A collection of electron-impact (EI) mass spectra of polysaccharide derived products. Holz als Roh-und Werkst. 1991, 49, 299–304. [Google Scholar] [CrossRef]
- Evans, R.; Wallis, A.F.A. Cellulose molecular weights determined by viscometry. J. Appl. Polym. Sci. 1989, 37, 2331–2340. [Google Scholar] [CrossRef]
- Mafu, L.D.; Neomagus, H.W.J.P.; Everson, R.C.; Carrier, M.; Strydom, C.A.; Bunt, J.R. Structural and chemical modifications of typical South African biomasses during torrefaction. Biores. Technol. 2016, 202, 192–197. [Google Scholar] [CrossRef]
- Shoulaifar, T.K.; DeMartini, N.; Willför, S.; Pranovich, A.; Smeds, A.I.; Virtanen, T.A.P.; Maunu, S.-L.; Verhoeff, F.; Kiel, J.H.A.; Hupa, M. Impact of torrefaction on the chemical structure of birch wood. Energy Fuels 2014, 28, 3863–3872. [Google Scholar] [CrossRef]
- Zhu, C.; Krumm, C.; Facas, G.G.; Neurock, M.; Dauenhauer, P.J. Energetics of cellulose and cyclodextrin glycosidic bond cleavage. React. Chem. Eng. 2017, 2, 201–214. [Google Scholar] [CrossRef]
- Kim, S.; Chmely, S.C.; Nimlos, M.R.; Bomble, Y.J.; Foust, T.D.; Paton, R.S.; Beckham, G.T. Computational study of bond dissociation enthalpies for a large range of native and modified lignins. J. Phys. Chem. Lett. 2011, 2, 2846–2852. [Google Scholar] [CrossRef]
- Alonso, E.R.; Capucine Dupont, C.; Heux, L.; Da Silva Perez, D.; Commandre, J.-M.; Gourdon, C. Study of solid chemical evolution in torrefaction of different biomasses through solid-state 13C cross-polarization/magic angle spinning NMR (nuclear magnetic resonance) and TGA (thermogravimetric analysis). Energy 2016, 97, 381–390. [Google Scholar] [CrossRef]
- Cheng, X.Y.; Li, X.J.; Xu, K.; Huang, Q.T.; Sun, H.N.; Wu, Y.Q. Effect of thermal treatment on functional groups and degree of cellulose crystallinity of eucalyptus wood (Eucalyptus grandis x Eucalyptus urophylla). For. Prod. J. 2017, 67, 135–140. [Google Scholar] [CrossRef]
- Hult, E.L.; Larsson, P.T.; Iversen, T. Cellulose fibril aggregation—An inherent property of kraft pulps. Polymer 2001, 42, 3309–3314. [Google Scholar] [CrossRef]
- Newman, R.H. Carbon-13 NMR evidence for cocrystallization of cellulose as a mechanism for hornification of bleached kraft pulp. Cellulose 2004, 11, 45–52. [Google Scholar] [CrossRef]
- Atalla, R.H.; VanderHart, D.L. The role of solid state 13C NMR spectroscopy in studies of the nature of native celluloses. Solid State Nucl. Magn. Reson. 1999, 15, 1–19. [Google Scholar] [CrossRef]
- Pérez, S.; Samain, D. Structure and Engineering of Celluloses. Adv. Carbohydr. Chem. Biochem. 2010, 64, 25–116. [Google Scholar] [CrossRef]
- Wada, M.; Okano, T. Localization of Iα and Iβ phases in algal cellulose revealed by acid treatments. Cellulose 2001, 8, 183–188. [Google Scholar] [CrossRef]
- Polettoa, M.; Pistorc, V.; Santana, R.M.C.; Zattera, A.J. Materials Produced From Plant Biomass. Part II: Evaluation of Crystallinity and Degradation Kinetics of Cellulose. Mater. Res. 2012, 15, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Lourenço, A.; Gominho, J.; Pereira, H. Chemical characterization of lignocellulosic materials by analytical pyrolysis. In Analytical Pyrolysis; Chapter 2; Kusch, P., Ed.; InTechOpen: London, UK, 2019; pp. 9–30. ISBN 978-1-78984-958-5. Available online: https://www.intechopen.com/books/analytical-pyrolysis/chemical-characterization-of-lignocellulosic-materials-by-analytical-pyrolysis (accessed on 1 October 2020). [CrossRef] [Green Version]
- Li, S.; Lyons-Hart, J.; Banyasz, J.; Shafer, K. Real-time evolved gas analysis by FTIR method: An experimental study of cellulose pyrolysis. Fuel 2001, 80, 1809–1817. [Google Scholar] [CrossRef]
- Wang, S.; Guo, X.; Liang, T.; Zhou, Y.; Luo, Z. Mechanism research on cellulose pyrolysis by Py-GC/MS and subsequent density functional theory studies. Bioresour. Technol. 2012, 104, 722–728. [Google Scholar] [CrossRef]
- Cuba-Torres, C.M.; Had, S.; Pecha, B.; Garcia-Perez, M. Effect of cellulose crystallinity on the formation of a liquid intermediate and on product distribution during pyrolysis. J. Anal. Appl. Pyrolysis 2013, 100, 56–66. [Google Scholar] [CrossRef]
- Junior, I.I.; Nascimento, M.A.; Souza, R.O.M.A.; Dufour, A.; Wojcieszak, R. Levoglucosan: A promising platform molecule? Green Chem. 2020, 22, 5859. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Weight Loss (%) | Total Lignin (%) | Holocellulose (%) | Glucose (%) | Xylose (%) | Acetyl Groups (%) | |
|---|---|---|---|---|---|---|---|
| E. globulus | NT | - | 23.9 | 66.2 | 68.1 | 20.6 | 7.1 |
| T | 9.7 | 30.0 | 55.6 | 82.2 | 11.8 | 4.1 | |
| E. propinqua | NT | - | 31.5 | 63.6 | 69.2 | 19.6 | 7.0 |
| T | 12.7 | 39.8 | 51.1 | 82.7 | 10.6 | 4.2 | |
| E. botryoides | NT | - | 29.9 | 67.5 | 70.2 | 19.1 | 7.0 |
| T | 10.2 | 37.6 | 51.1 | 85.1 | 8.8 | 4.0 | |
| E. viminalis | NT | - | 28.4 | 61.4 | 59.2 | 27.0 | 9.4 |
| T | 12.5 | 34.9 | 51.9 | 81.0 | 11.2 | 4.5 | |
| E. grandis | NT | - | 26.0 | 60.4 | 69.1 | 19.5 | 7.5 |
| T | 9.7 | 34.8 | 54.0 | 85.1 | 8.8 | 4.0 | |
| E. rudis | NT | - | 27.6 | 57.6 | 64.9 | 22.5 | 8.5 |
| T | 11.4 | 38.1 | 51.5 | 85.4 | 8.8 | 3.7 | |
| E. maculata | NT | - | 22.5 | 65.6 | 61.7 | 26.4 | 8.0 |
| T | 9.6 | 36.1 | 52.5 | 82.0 | 10.5 | 4.6 | |
| E. saligna | NT | - | 28.3 | 63.5 | 70.6 | 18.3 | 7.2 |
| T | 12.4 | 36.8 | 52.8 | 84.8 | 9.1 | 4.2 |
| Species | CrI * | ICO2 ** | |
|---|---|---|---|
| E. globulus | NT | 0.37 | 0.08 |
| T | 0.44 | 0.09 | |
| E. propinqua | NT | 0.38 | 0.12 |
| T | 0.43 | 0.12 | |
| E. botryoides | NT | 0.38 | 0.10 |
| T | 0.44 | 0.09 | |
| E. viminalis | NT | 0.39 | 0.16 |
| T | 0.44 | 0.06 | |
| E. grandis | NT | 0.38 | 0.11 |
| T | 0.43 | 0.04 | |
| E. rudis | NT | 0.35 | 0.14 |
| T | 0.43 | 0.13 | |
| E. maculata | NT | 0.36 | 0.16 |
| T | 0.46 | 0.11 | |
| E. saligna | NT | 0.42 | 0.14 |
| T | 0.45 | 0.14 |
| CelEglo | CelEgloT | CelEpro | CelEproT | |
|---|---|---|---|---|
| A (±0.3 nm) | 12.5 | 9.9 | 11.5 | 9.6 |
| CrI (±1%) | 58 | 52 | 57 | 53 |
| DPv (±20) | 1300 | 530 | 1330 | 590 |
| CelEglo | CelEgloT | CelEpro | CelEproT | |
|---|---|---|---|---|
| DC (±0.5%) | 71.3 | 68.8 | 71.6 | 69.8 |
| D200 (±0.1 nm) | 5.4 | 4.6 | 5.5 | 4.9 |
| a (±0.002 nm) | 0.797 | 0.788 | 0.798 | 0.795 |
| b (±0.002 nm) | 0.825 | 0.835 | 0.828 | 0.832 |
| γ (±0.1 deg.) | 97.0 | 96.1 | 97.2 | 96.5 |
| CelEglo | CelEgloT | CelEpro | CelEproT | |
|---|---|---|---|---|
| CrP (T1372/T2892 × 100%) | 97.9 | 97.2 | 96.0 | 97.2 |
| TCI (A1375/A2900) | 1.10 | 1.10 | 1.13 | 1.12 |
| LOI (A1429/A897) | 1.25 | 1.27 | 1.29 | 1.27 |
| HBI (A3336/A1335) | 1.77 | 1.82 | 1.95 | 1.76 |
| CelEglo | CelEgloT | CelEpro | CelEproT | |
|---|---|---|---|---|
| Total carbohydrates (% of total area) | 87.1 | 84.2 | 84.5 | 71.1 |
| Furan | 6.7 | 5.1 | 5.2 | 6.0 |
| Pyran | 53.6 | 63.3 | 60.8 | 47.6 |
| Low molecular compounds (LMC) | 16.6 | 8.3 | 9.4 | 9.4 |
| Others | 10.2 | 7.5 | 9.1 | 8.0 |
| Total lignin (% of total area) | 0.1 | 0.1 | 0.5 | 3.3 |
| % of identified compounds | 87.2 | 84.3 | 85.0 | 74.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lourenço, A.; Araújo, S.; Gominho, J.; Evtuguin, D. Cellulose Structural Changes during Mild Torrefaction of Eucalyptus Wood. Polymers 2020, 12, 2831. https://doi.org/10.3390/polym12122831
Lourenço A, Araújo S, Gominho J, Evtuguin D. Cellulose Structural Changes during Mild Torrefaction of Eucalyptus Wood. Polymers. 2020; 12(12):2831. https://doi.org/10.3390/polym12122831
Chicago/Turabian StyleLourenço, Ana, Solange Araújo, Jorge Gominho, and Dmitry Evtuguin. 2020. "Cellulose Structural Changes during Mild Torrefaction of Eucalyptus Wood" Polymers 12, no. 12: 2831. https://doi.org/10.3390/polym12122831
APA StyleLourenço, A., Araújo, S., Gominho, J., & Evtuguin, D. (2020). Cellulose Structural Changes during Mild Torrefaction of Eucalyptus Wood. Polymers, 12(12), 2831. https://doi.org/10.3390/polym12122831