Polymerization-Induced Microphase Separation with Long-Range Order in Melts of Gradient Copolymers


Abstract
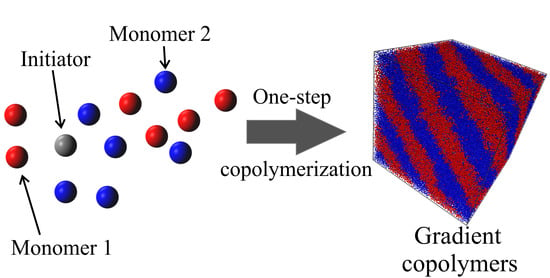
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Method and Model
3. Results and Discussion
4. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Zaremski, M.Y.; Kalugin, D.I.; Golubev, V.B. Gradient copolymers: Synthesis, structure, and properties. Polym. Sci. Ser. A 2009, 51, 103–122. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Ziegler, M.J.; Arehart, S.V.; Greszta, D.; Pakula, T. Gradient copolymers by atom transfer radical copolymerization. J. Phys. Org. Chem. 2000, 13, 775–786. [Google Scholar] [CrossRef]
- Lefebvre, M.D.; Dettmer, C.M.; McSwain, R.L.; Xu, C.; Davila, J.R.; Composto, R.J.; Nguyen, S.T.; Shull, K.R. Effect of Sequence Distribution on Copolymer Interfacial Activity. Macromolecules 2005, 38, 10494–10502. [Google Scholar] [CrossRef]
- Shull, K.R. Interfacial Activity of Gradient Copolymers. Macromolecules 2002, 35, 8631–8639. [Google Scholar] [CrossRef]
- Yuan, W.; Mok, M.M.; Kim, J.; Wong, C.L.H.; Dettmer, C.M.; Nguyen, S.T.; Torkelson, J.M.; Shull, K.R. Behavior of Gradient Copolymers at Liquid/Liquid Interfaces. Langmuir 2010, 26, 3261–3267. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Mok, M.M.; Sandoval, R.W.; Woo, D.J.; Torkelson, J.M. Uniquely Broad Glass Transition Temperatures of Gradient Copolymers Relative to Random and Block Copolymers Containing Repulsive Comonomers. Macromolecules 2006, 39, 6152–6160. [Google Scholar] [CrossRef]
- Wong, C.L.H.; Kim, J.; Torkelson, J.M. Breadth of glass transition temperature in styrene/acrylic acid block, random, and gradient copolymers: Unusual sequence distribution effects. J. Polym. Sci. Part B Polym. Phys. 2007, 45, 2842–2849. [Google Scholar] [CrossRef]
- Mok, M.M.; Pujari, S.; Burghardt, W.R.; Dettmer, C.M.; Nguyen, S.T.; Ellison, C.J.; Torkelson, J.M. Microphase Separation and Shear Alignment of Gradient Copolymers: Melt Rheology and Small-Angle X-Ray Scattering Analysis. Macromolecules 2008, 41, 5818–5829. [Google Scholar] [CrossRef]
- Ganesan, V.; Kumar, N.A.; Pryamitsyn, V. Blockiness and Sequence Polydispersity Effects on the Phase Behavior and Interfacial Properties of Gradient Copolymers. Macromolecules 2012, 45, 6281–6297. [Google Scholar] [CrossRef]
- Filippov, S.K.; Verbraeken, B.; Konarev, P.V.; Svergun, D.I.; Angelov, B.; Vishnevetskaya, N.S.; Papadakis, C.M.; Rogers, S.; Radulescu, A.; Courtin, T.; et al. Block and Gradient Copoly(2-oxazoline) Micelles: Strikingly Different on the Inside. J. Phys. Chem. Lett. 2017, 8, 3800–3804. [Google Scholar] [CrossRef]
- Kravchenko, V.S.; Potemkin, I.I. Micelles of Gradient vs Diblock copolymers: Difference in the internal structure and properties. J. Phys. Chem. B 2016, 120, 12211–12217. [Google Scholar] [CrossRef] [PubMed]
- Okabe, S.; Seno, K.; Kanaoka, S.; Aoshima, S.; Shibayama, M. Micellization Study on Block and Gradient Copolymer Aqueous Solutions by DLS and SANS. Macromolecules 2006, 39, 1592–1597. [Google Scholar] [CrossRef]
- Elsen, A.M.; Li, Y.; Li, Q.; Sheiko, S.S.; Matyjaszewski, K. Exploring quality in gradient copolymers. Macromol. Rapid Commun. 2014, 35, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Min, K.E.; Mei, L.I.; Matyjaszewski, K. Preparation of gradient copolymers via ATRP using a Simultaneous Reverse and Normal Initiation process. I. Spontaneous gradient. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 3616–3622. [Google Scholar] [CrossRef]
- Beginn, U. Gradient copolymers. Colloid Polym. Sci. 2008, 286, 1465–1474. [Google Scholar] [CrossRef]
- Ogura, Y.; Takenaka, M.; Sawamoto, M.; Terashima, T. Fluorous Gradient Copolymers via in-Situ Transesterification of a Perfluoromethacrylate in Tandem Living Radical Polymerization: Precision Synthesis and Physical Properties. Macromolecules 2018, 51, 864–871. [Google Scholar] [CrossRef]
- Lefay, C.; Charleux, B.; Save, M.; Chassenieux, C.; Guerret, O.; Magnet, S. Amphiphilic gradient poly(styrene-co-acrylic acid) copolymer prepared via nitroxide-mediated solution polymerization. Synthesis, characterization in aqueous solution and evaluation as emulsion polymerization stabilizer. Polymer 2006, 47, 1935–1945. [Google Scholar] [CrossRef]
- D’hooge, D.; Van Steenberge, P.; Reyniers, M.-F.; Marin, G. Fed-Batch Control and Visualization of Monomer Sequences of Individual ICAR ATRP Gradient Copolymer Chains. Polymers 2014, 6, 1074–1095. [Google Scholar] [CrossRef]
- Kim, J.; Zhou, H.; Nguyen, S.T.; Torkelson, J.M. Synthesis and application of styrene/4-hydroxystyrene gradient copolymers made by controlled radical polymerization: Compatibilization of immiscible polymer blends via hydrogen-bonding effects. Polymer 2006, 47, 5799–5809. [Google Scholar] [CrossRef]
- Karaky, K.; Billon, L.; Pouchan, C.; Desbrières, J. Amphiphilic Gradient Copolymers Shape Composition Influence on the Surface/Bulk Properties. Macromolecules 2007, 40, 458–464. [Google Scholar] [CrossRef]
- Hutchings, L.R.; Brooks, P.P.; Shaw, P.; Ross-Gardner, P. Fire and Forget! One-Shot Synthesis and Characterization of Block-Like Statistical Terpolymers via Living Anionic Polymerization. J. Polym. Sci. Part A Polym. Chem. 2019, 57, 382–394. [Google Scholar] [CrossRef]
- Von Tiedemann, P.; Blankenburg, J.; Maciol, K.; Johann, T.; Müller, A.H.E.; Frey, H. Copolymerization of Isoprene with p-Alkylstyrene Monomers: Disparate Reactivity Ratios and the Shape of the Gradient. Macromolecules 2019, 52, 796–806. [Google Scholar] [CrossRef]
- Grune, E.; Johann, T.; Appold, M.; Wahlen, C.; Blankenburg, J.; Leibig, D.; Müller, A.H.E.; Gallei, M.; Frey, H. One-Step Block Copolymer Synthesis versus Sequential Monomer Addition: A Fundamental Study Reveals That One Methyl Group Makes a Difference. Macromolecules 2018, 51, 3527–3537. [Google Scholar] [CrossRef]
- Grune, E.; Bareuther, J.; Blankenburg, J.; Appold, M.; Shaw, L.; Müller, A.H.E.; Floudas, G.; Hutchings, L.R.; Gallei, M.; Frey, H. Towards bio-based tapered block copolymers: The behaviour of myrcene in the statistical anionic copolymerisation. Polym. Chem. 2019, 10, 1213–1220. [Google Scholar] [CrossRef]
- Canning, S.L.; Smith, G.N.; Armes, S.P. A Critical Appraisal of RAFT-Mediated Polymerization-Induced Self-Assembly. Macromolecules 2016, 49, 1985–2001. [Google Scholar] [CrossRef] [PubMed]
- Penfold, N.J.W.; Yeow, J.; Boyer, C.; Armes, S.P. Emerging Trends in Polymerization-Induced Self-Assembly. ACS Macro Lett. 2019, 8, 1029–1054. [Google Scholar] [CrossRef]
- Tritschler, U.; Pearce, S.; Gwyther, J.; Whittell, G.R.; Manners, I. 50th Anniversary Perspective: Functional Nanoparticles from the Solution Self-Assembly of Block Copolymers. Macromolecules 2017, 50, 3439–3463. [Google Scholar] [CrossRef]
- Xu, S.; Zhang, T.; Kuchel, R.P.; Yeow, J.; Boyer, C. Gradient Polymerization–Induced Self-Assembly: A One-Step Approach. Macromol. Rapid Commun. 2020, 41, 1900493. [Google Scholar] [CrossRef]
- Xu, S.; Corrigan, N.; Boyer, C. Forced gradient copolymerisation: A simplified approach for polymerisation-induced self-assembly. Polym. Chem. 2020, 64–69. [Google Scholar] [CrossRef]
- Seo, M.; Hillmyer, M.A. Reticulated Nanoporous Polymers by Controlled Polymerization-Induced Microphase Separation. Science 2012, 336, 1422–1425. [Google Scholar] [CrossRef]
- Lequieu, J.; Magenau, A.J.D. Reaction-induced phase transitions with block copolymers in solution and bulk. Polym. Chem. 2020. [Google Scholar] [CrossRef]
- Chopade, S.A.; So, S.; Hillmyer, M.A.; Lodge, T.P. Anhydrous Proton Conducting Polymer Electrolyte Membranes via Polymerization-Induced Microphase Separation. ACS Appl. Mater. Interfaces 2016, 8, 6200–6210. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, L.D.; Schulze, M.W.; Irwin, M.T.; Hillmyer, M.A.; Lodge, T.P. Evolution of morphology, modulus, and conductivity in polymer electrolytes prepared via polymerization-induced phase separation. Macromolecules 2015, 48, 1418–1428. [Google Scholar] [CrossRef]
- Zofchak, E.S.; LaNasa, J.A.; Torres, V.M.; Hickey, R.J. Deciphering the Complex Phase Behavior during Polymerization-Induced Nanostructural Transitions of a Block Polymer/Monomer Blend. Macromolecules 2020, 53, 835–843. [Google Scholar] [CrossRef]
- Zofchak, E.S.; LaNasa, J.A.; Mei, W.; Hickey, R.J. Polymerization-Induced Nanostructural Transitions Driven by In Situ Polymer Grafting. ACS Macro Lett. 2018, 7, 822–827. [Google Scholar] [CrossRef]
- Van Steenberge, P.H.M.; D’Hooge, D.R.; Wang, Y.; Zhong, M.; Reyniers, M.F.; Konkolewicz, D.; Matyjaszewski, K.; Marin, G.B. Linear gradient quality of ATRP copolymers. Macromolecules 2012, 45, 8519–8531. [Google Scholar] [CrossRef]
- Yañez-Macias, R.; Kulai, I.; Ulbrich, J.; Yildirim, T.; Sungur, P.; Hoeppener, S.; Guerrero-Santos, R.; Schubert, U.S.; Destarac, M.; Guerrero-Sanchez, C.; et al. Thermosensitive spontaneous gradient copolymers with block- and gradient-like features. Polym. Chem. 2017, 8, 5023–5032. [Google Scholar] [CrossRef]
- Groot, R.D.; Warren, P.B. Dissipative particle dynamics: Bridging the gap between atomistic and mesoscopic simulation. J. Chem. Phys. 1997, 107, 4423–4435. [Google Scholar] [CrossRef]
- Gavrilov, A.A.; Chertovich, A.V. Copolymerization of Partly Incompatible Monomers: An Insight from Computer Simulations. Macromolecules 2017, 50, 4677–4685. [Google Scholar] [CrossRef]
- Greenley, R.Z. Recalculation of Some Reactivity Ratios. J. Macromol. Sci. Part A Chem. 1980, 14, 445–515. [Google Scholar] [CrossRef]
- Brandrup, J.; Immergut, E.H.; Grulke, E.A.; Abe, A.; Bloch, D.R. Polymer Handbook, 4th ed.; Brandrup, J., Immergut, E.H., Grulke, E.A., Abe, A., Bloch, D.R., Eds.; John Wiley and Sons: New York, NY, USA, 1999. [Google Scholar]
- Li, L.; Jiang, Z.; Xu, J.; Fang, T. Predicting poly(vinyl pyrrolidone)’s solubility parameter and systematic investigation of the parameters of electrospinning with response surface methodology. J. Appl. Polym. Sci. 2014, 131, 1–9. [Google Scholar] [CrossRef]
- Harwood, H.J. Structures and compositions of copolymers. Makromol. Chemie. Macromol. Symp. 1987, 10–11, 331–354. [Google Scholar] [CrossRef]
- Bork, J.F.; Coleman, L.E. Nitrogen-containing monomers. II. Reactivity ratios of n-vinyloxazolidone and N-vinylpyrrolidone with vinyl monomers. J. Polym. Sci. 1960, 43, 413–421. [Google Scholar] [CrossRef]
- Gavrilov, A.A.; Kudryavtsev, Y.V.; Chertovich, A.V. Phase diagrams of block copolymer melts by dissipative particle dynamics simulations. J. Chem. Phys. 2013, 139, 224901. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gavrilov, A.A.; Chertovich, A.V. Polymerization-Induced Microphase Separation with Long-Range Order in Melts of Gradient Copolymers. Polymers 2020, 12, 2637. https://doi.org/10.3390/polym12112637
Gavrilov AA, Chertovich AV. Polymerization-Induced Microphase Separation with Long-Range Order in Melts of Gradient Copolymers. Polymers. 2020; 12(11):2637. https://doi.org/10.3390/polym12112637
Chicago/Turabian StyleGavrilov, Alexey A., and Alexander V. Chertovich. 2020. "Polymerization-Induced Microphase Separation with Long-Range Order in Melts of Gradient Copolymers" Polymers 12, no. 11: 2637. https://doi.org/10.3390/polym12112637
APA StyleGavrilov, A. A., & Chertovich, A. V. (2020). Polymerization-Induced Microphase Separation with Long-Range Order in Melts of Gradient Copolymers. Polymers, 12(11), 2637. https://doi.org/10.3390/polym12112637