Biodegradable Starch/Chitosan Foam via Microwave Assisted Preparation: Morphology and Performance Properties


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
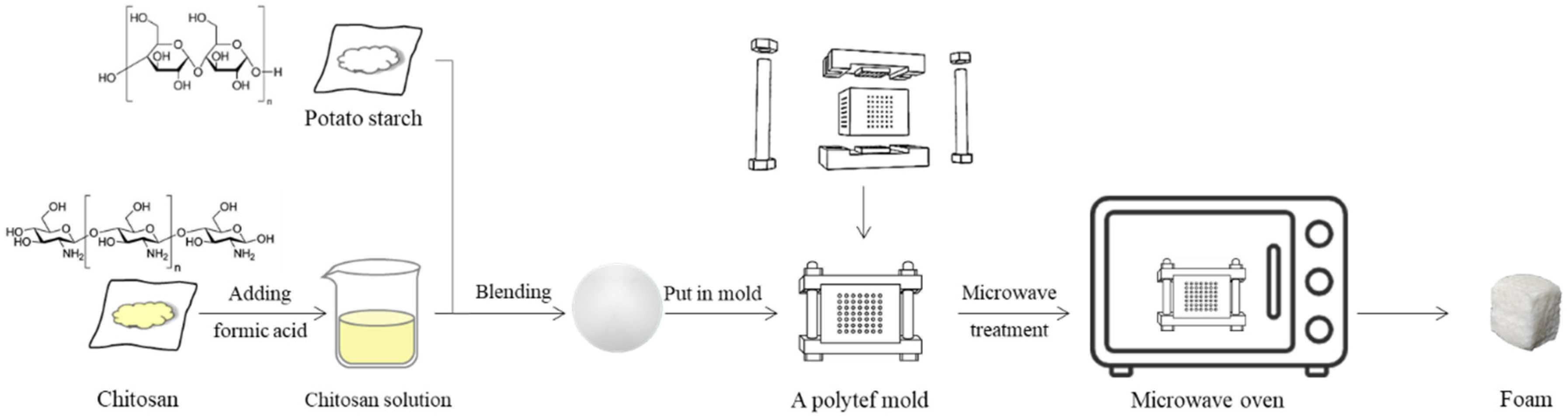
2.2. Sample Preparation
2.3. Characterization
3. Results and Discussion
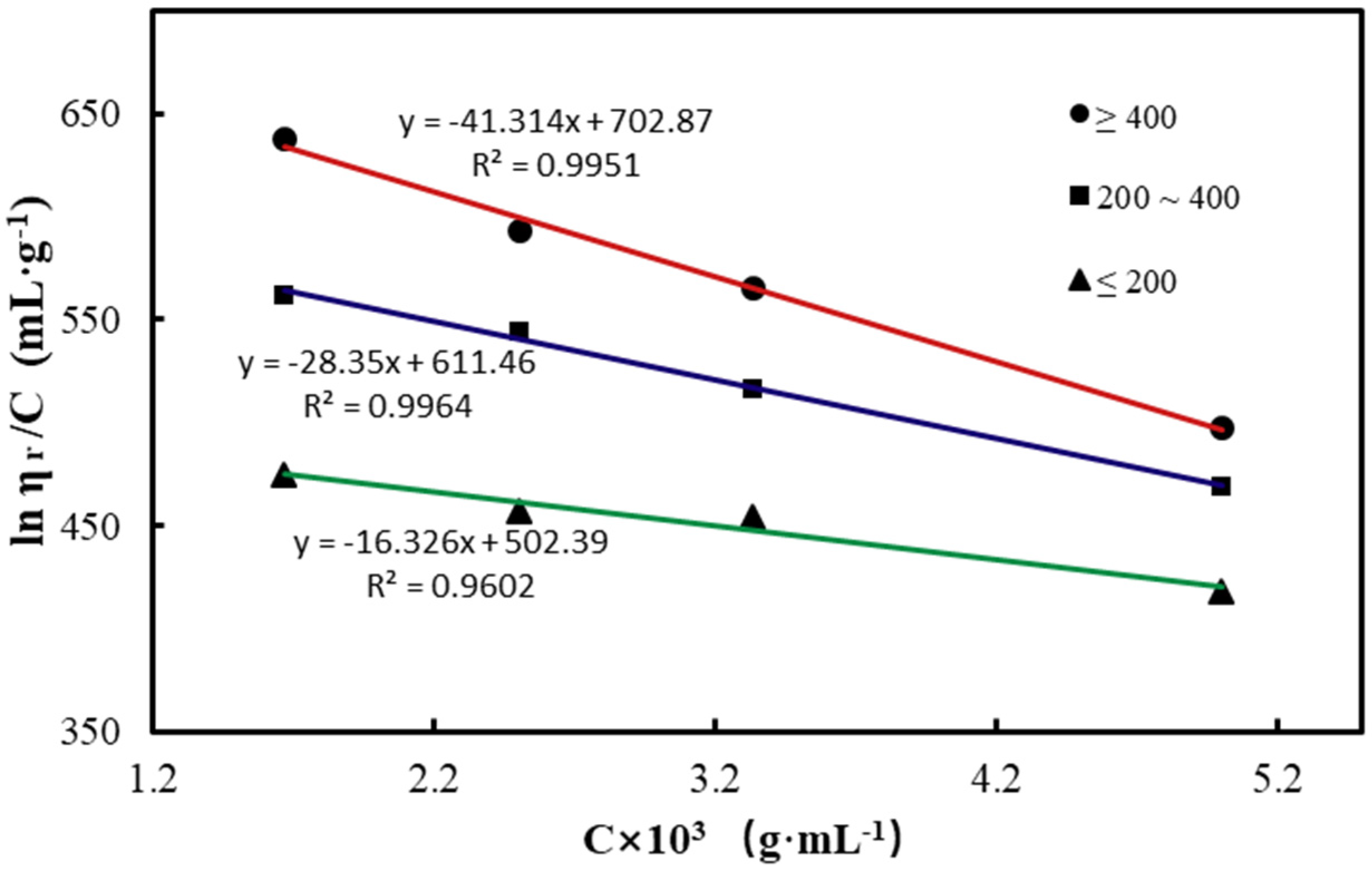
3.1. Molecular Weight of CTS
3.2. Morphology of Starch-Based Foams
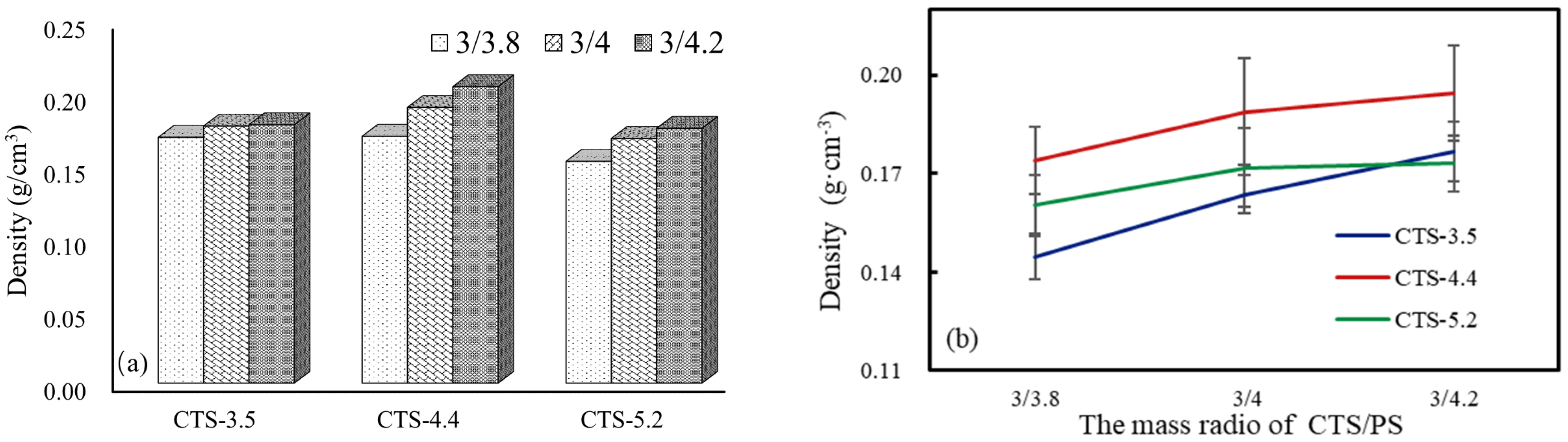
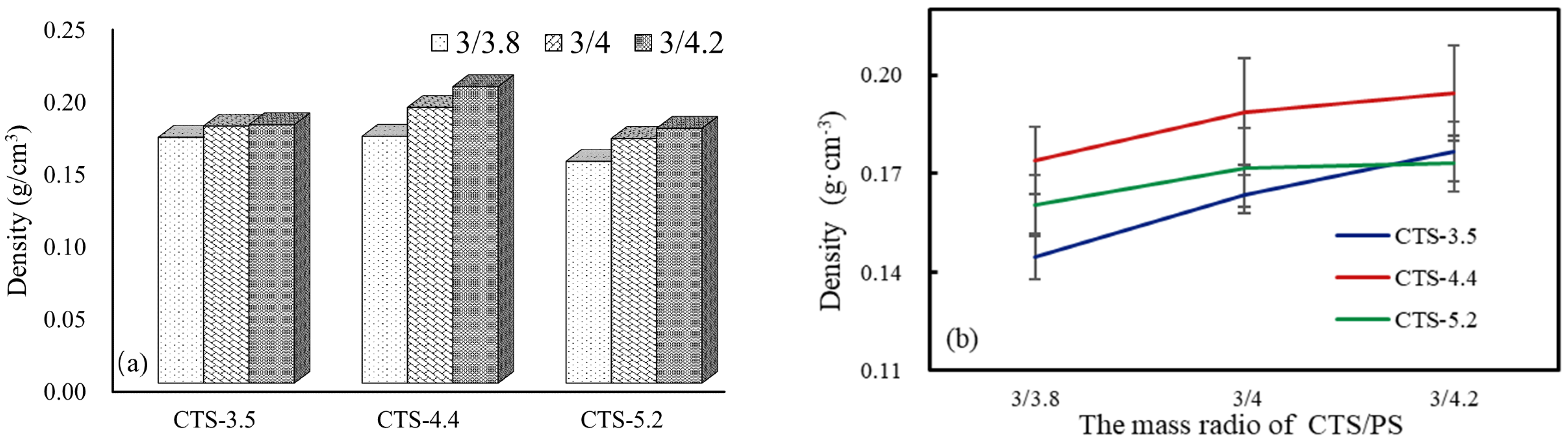
3.3. Pore Size, Porosity, and Density of CTS–PS Foams
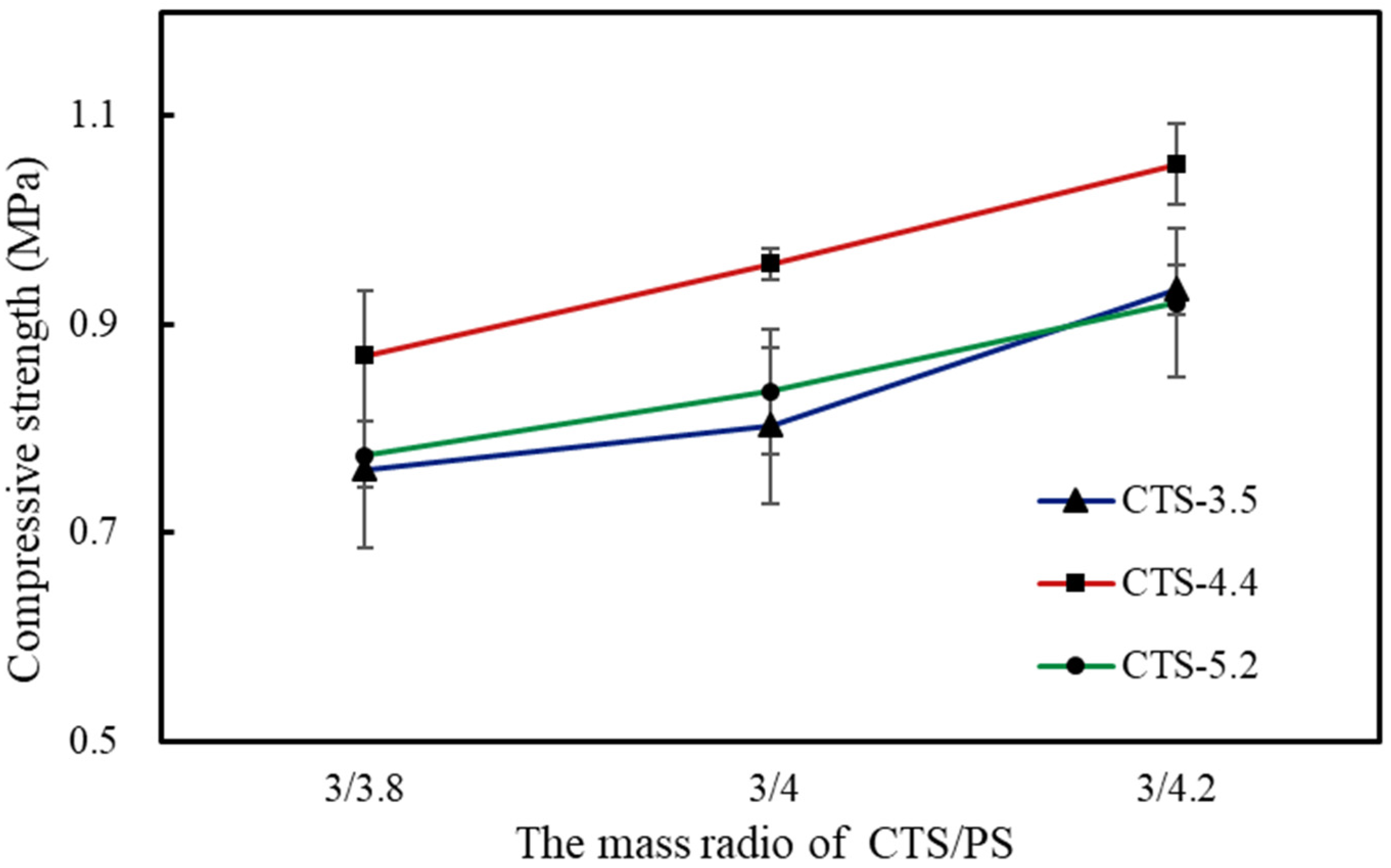
3.4. The Compressive Property of Foams
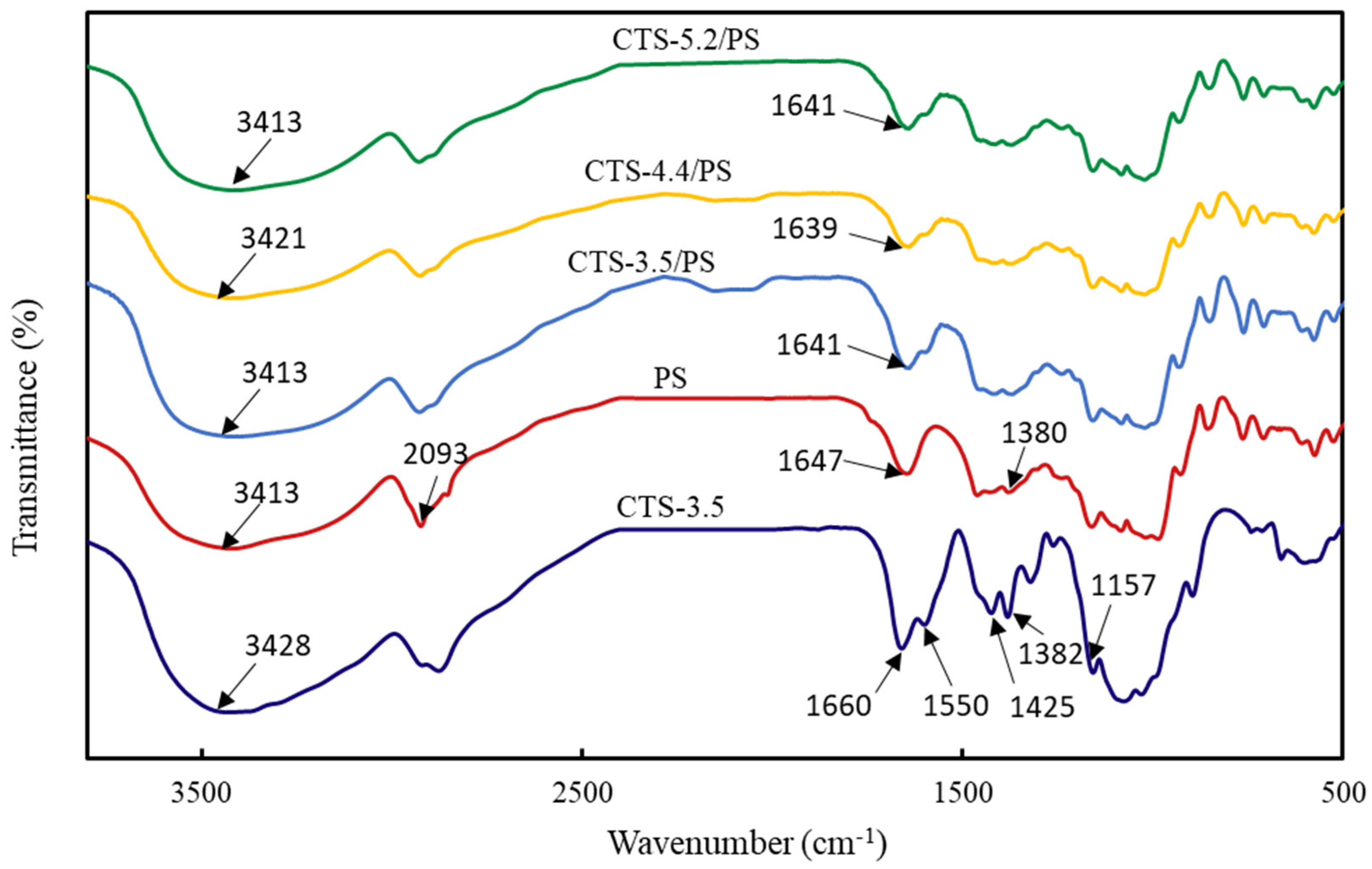
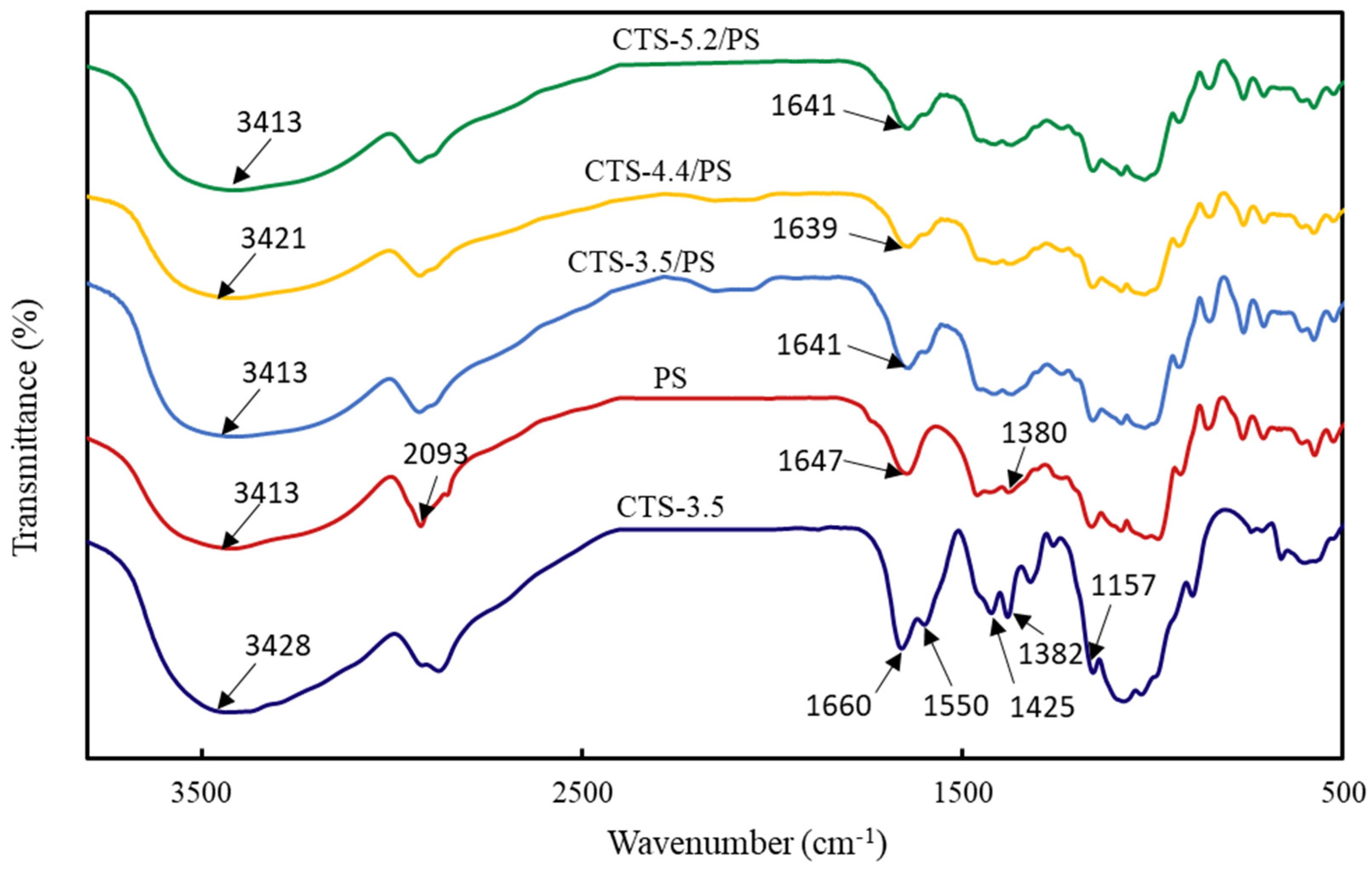
3.5. FTIR Analysis
3.6. Thermogravimetric Analysis (TGA)
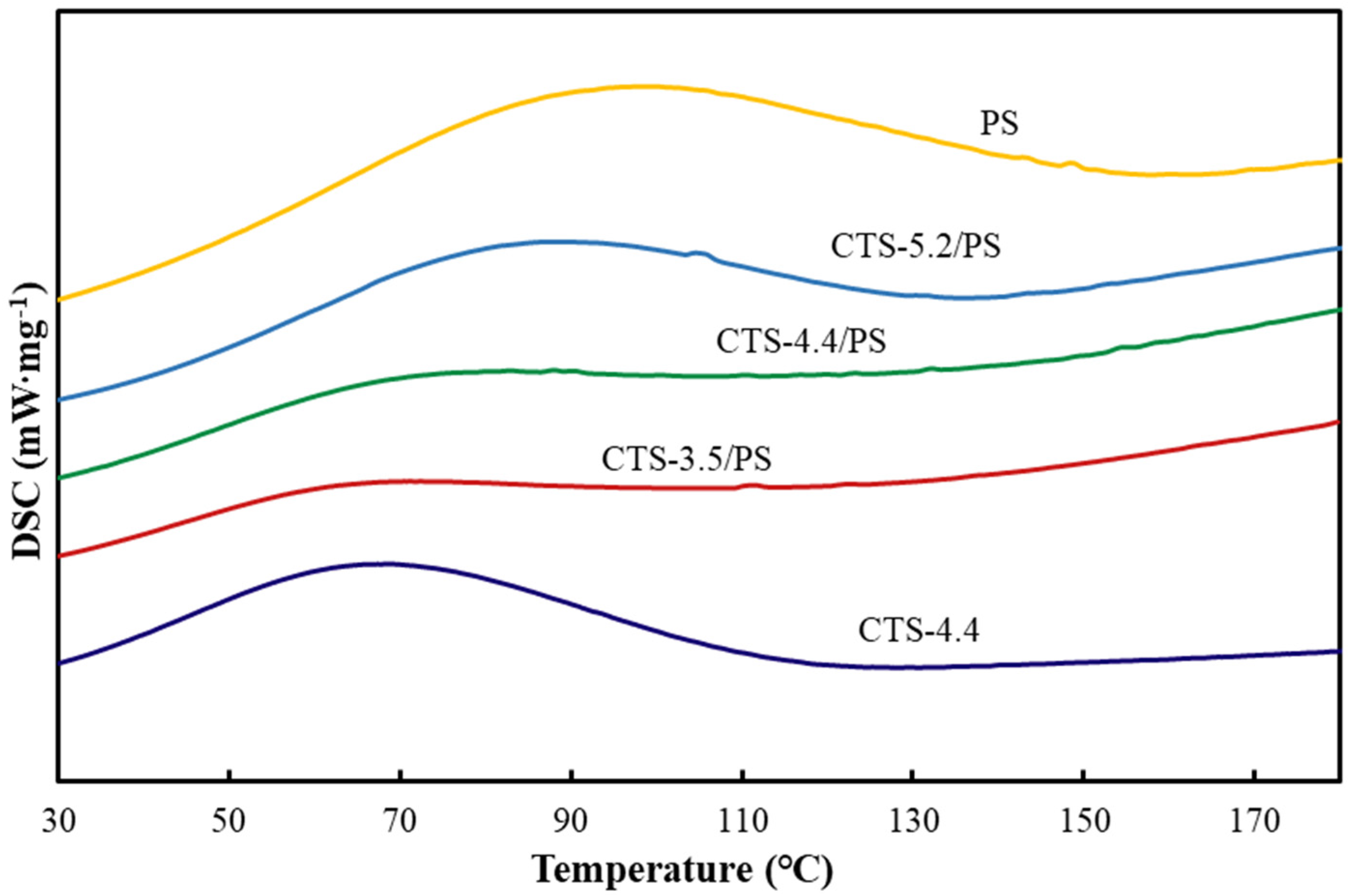
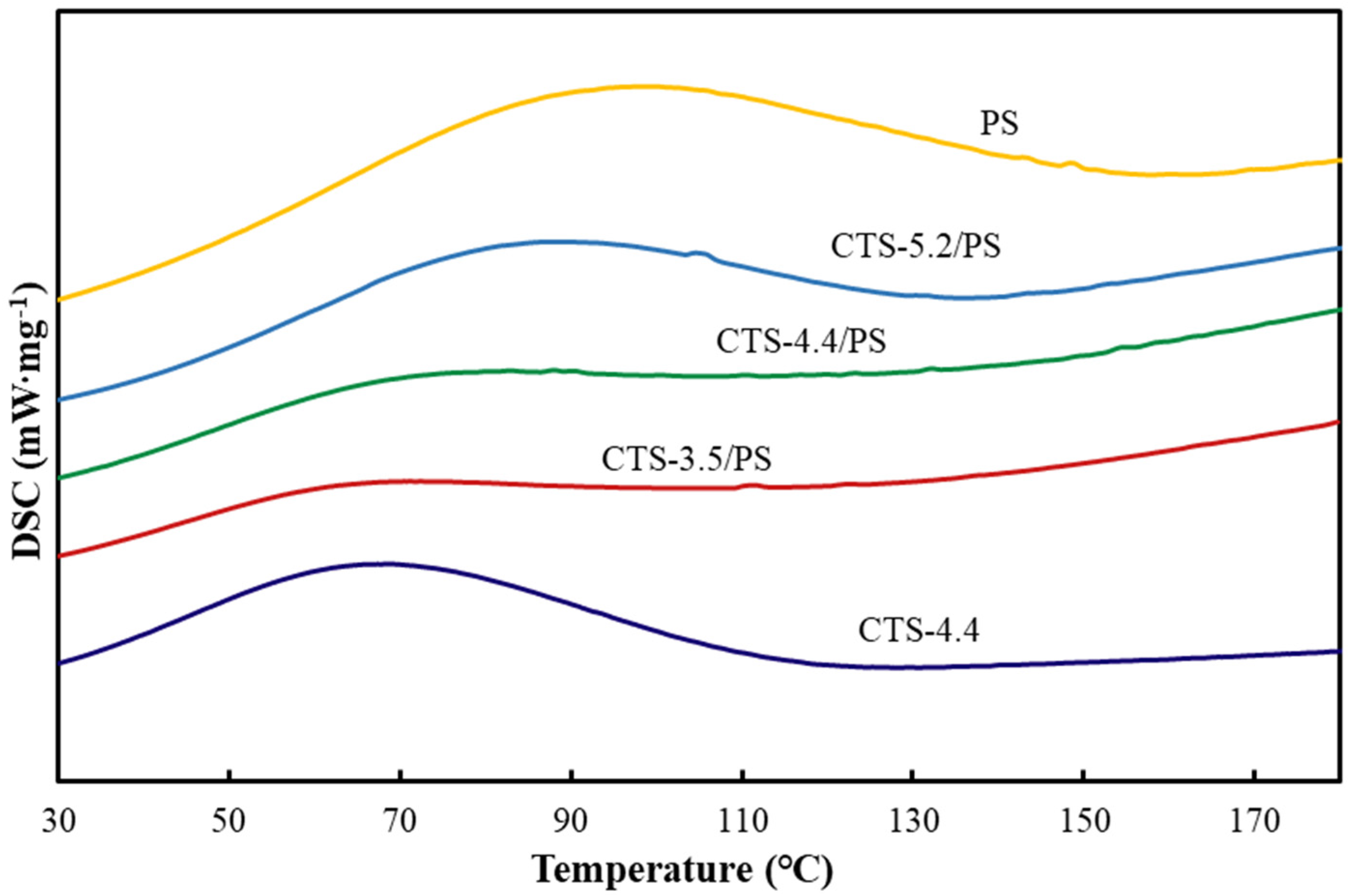
3.7. DSC Analysis
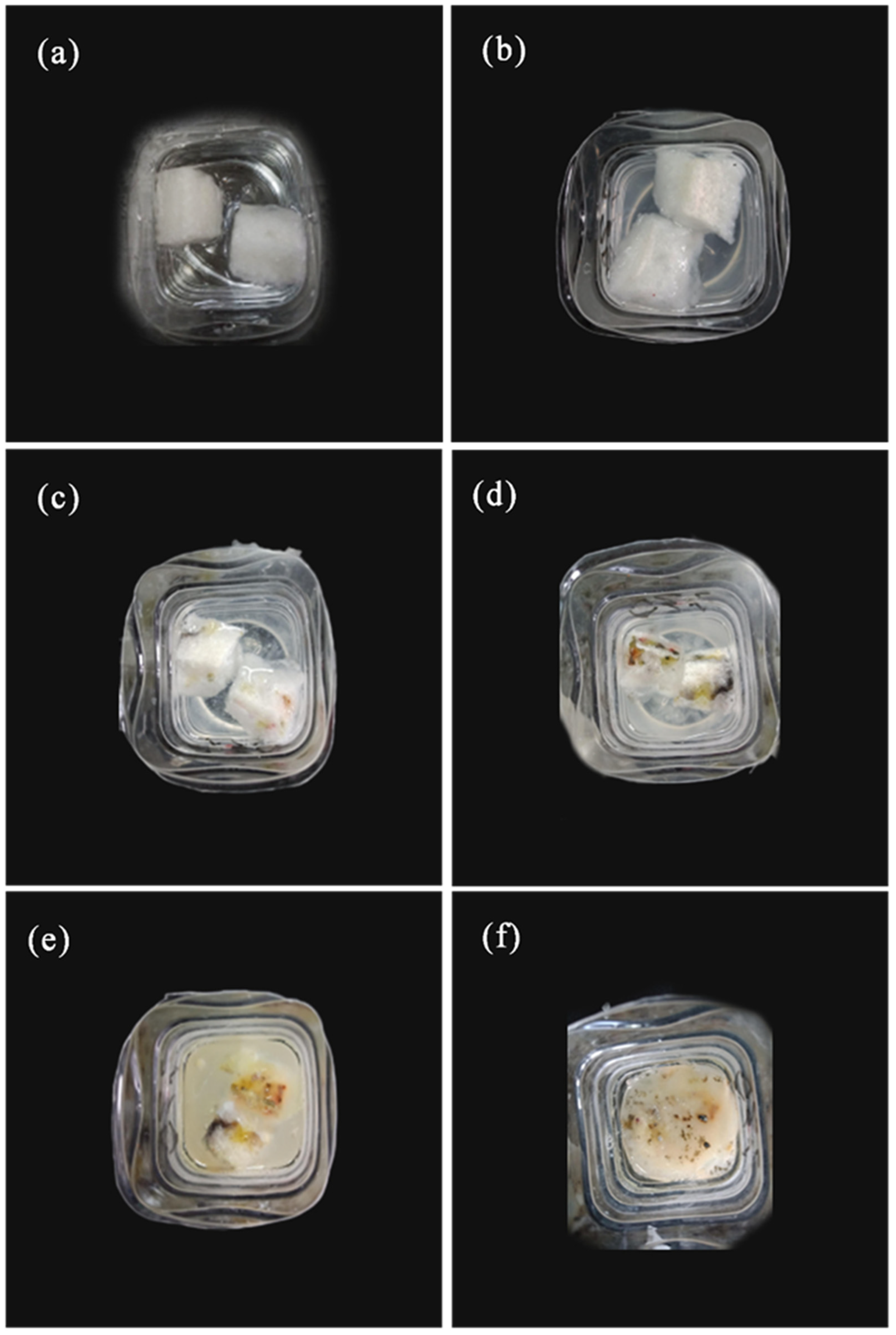
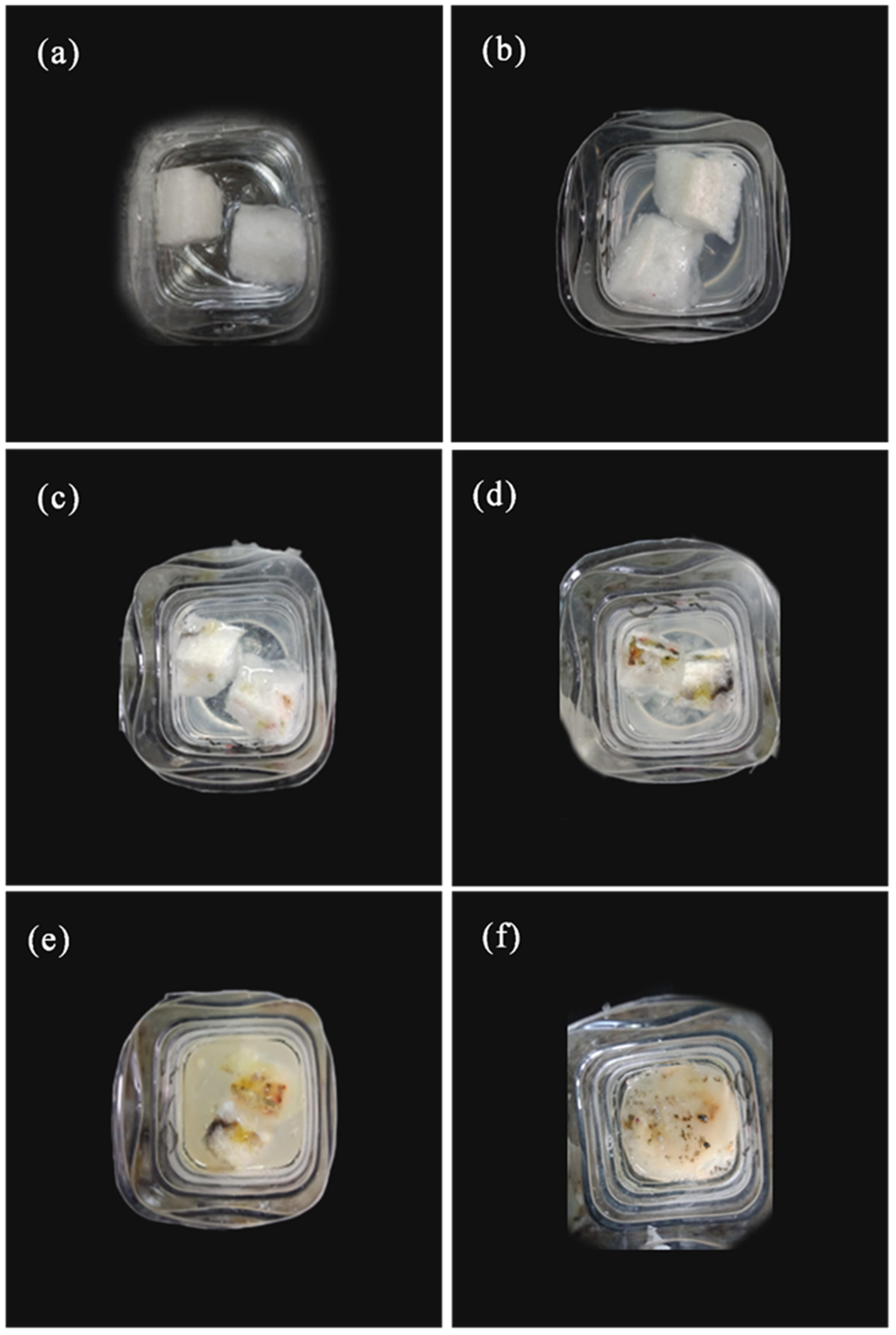
3.8. Water Solubility Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nofar, M.; Park, C.B. Poly (lactic acid) foaming. Prog. Polym. Sci. 2014, 39, 1721–1741. [Google Scholar] [CrossRef]
- Ventura, H.; Laguna-Gutiérrez, E.; Rodriguez-Perez, M.A.; Ardanuy, M. Effect of chain extender and water-quenching on the properties of poly(3-hydroxybutyrate-co-4-hydroxybutyrate) foams for its production by extrusion foaming. Eur. Polym. J. 2016, 85, 14–25. [Google Scholar] [CrossRef]
- Feng, Z.; Yu, L.; Hong, Y.; Wu, J.; Jian, Z.; Li, H.; Qi, R.; Jiang, P. Preparation of enhanced poly (butylene succinate) foams. Polym. Eng. Sci. 2016, 56, 1275–1282. [Google Scholar] [CrossRef]
- Gao, Z.; Zhan, W.; Wang, Y.; Yun, G.; Li, W.; Guo, Y.; Lu, G. Aldehyde-functionalized mesostructured cellular foams prepared by copolymerization method for immobilization of penicillin G acylase. Microporous Mesoporous Mater. 2015, 202, 90–96. [Google Scholar] [CrossRef]
- Jordi, G.; Espinach, F.X.; Neus, P.; Josep, T.; Manel, A.; Pere, M. High-Performance-tensile-strength alpha-grass reinforced starch-based fully biodegradable composites. Bioresources 2013, 8, 6121–6135. [Google Scholar]
- Guimares, J.L.; Wypych, F.; Saul, C.K.; Ramos, L.P.; Satyanarayana, K.G. Studies of the processing and characterization of corn starch and its composites with banana and sugarcane fibers from Brazil. Carbohydr. Polym. 2010, 80, 130–138. [Google Scholar] [CrossRef]
- Moraes, A.R.F.E.; Pola, C.C.; Bilck, A.P.; Yamashita, F.; Tronto, J.; Medeiros, E.A.A.; Soares, N.D.F.F. Starch, cellulose acetate and polyester biodegradable sheets: Effect of composition and processing conditions. Mater. Sci. Eng. C 2017, 78, 932–941. [Google Scholar] [CrossRef]
- Shariatinia, Z.; Fazli, M. Mechanical properties and antibacterial activities of novel nanobiocomposite films of chitosan and starch. Food Hydrocoll. 2015, 46, 112–124. [Google Scholar] [CrossRef]
- Akrami, M.; Ghasemi, I.; Azizi, H.; Karrabi, M.; Seyedabadi, M. A new approach in compatibilization of the poly(lactic acid)/thermoplastic starch (PLA/TPS) blends. Carbohydr. Polym. 2016, 144, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Mahieu, A.; Terrié, C.; Agoulon, A.; Leblanc, N.; Youssef, B. Thermoplastic starch and poly(ε-caprolactone) blends: Morphology and mechanical properties as a function of relative humidity. J. Polym. Res. 2013, 20, 229. [Google Scholar] [CrossRef]
- Yun, I.S.; Hwang, S.W.; Shim, J.K.; Seo, K.H. A study on the thermal and mechanical properties of poly (butylene succinate)/ thermoplastic starch binary blends. Int. J. Precis. Eng. Manuf. Green Technol. 2016, 3, 289–296. [Google Scholar] [CrossRef]
- Aydın, A.A.; Ilberg, V. Effect of different polyol-based plasticizers on thermal properties of polyvinyl alcohol (PVA): Starch blends films. Carbohydr. Polym. 2015, 136, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Guan, K.; Liu, P.; Li, G.; Li, J. Ingredient of biomass packaging material and compare study on cushion properties. Int. J. Polym. Sci. 2014, 2014, 146509. [Google Scholar] [CrossRef]
- Loercks, J.; Pommeranz, W.; Schmidt, H.; Timmermann, R.; Grigat, E.; Schulz-Schlitte, W. Thermoplastic Starch Utilizing a Biodegradable Polymer as Melting Aid. U.S. Patent 6472497, 29 October 2002. [Google Scholar]
- Ferri, J.M.; Garcia-Garcia, D.; Sánchez-Nacher, L.; Fenollar, O.; Balart, R. The effect of maleinized linseed oil (MLO) on mechanical performance of poly(lactic acid)-thermoplastic starch (PLA-TPS) blends. Carbohydr. Polym. 2016, 147, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Bénézet, J.-C.; Stanojlovic-Davidovic, A.; Bergeret, A.; Ferry, L.; Crespy, A. Mechanical and physical properties of expanded starch, reinforced by natural fibres. Ind. Crop. Prod. 2012, 37, 435–440. [Google Scholar] [CrossRef]
- Garcia, M.A.; Pinotti, A.; Zaritzky, N.E. Physicochemical, water vapor barrier and mechanical properties of corn starch and chitosan composite films. Starch Stärke 2006, 58, 453–463. [Google Scholar] [CrossRef]
- Vicentini, D.S.; Smania, A.; Laranjeira, M.C.M. Chitosan/poly (vinyl alcohol) films containing ZnO nanoparticles and plasticizers. Mater. Sci. Eng. C Mater. 2010, 30, 503–508. [Google Scholar] [CrossRef]
- Rinaudo, M. Chitin and chitosan: Properties and applications. Cheminform 2007, 31, 603–632. [Google Scholar] [CrossRef]
- Dang, K.M.; Yoksan, R. Morphological characteristics and barrier properties of thermoplastic starch/chitosan blown film. Carbohydr. Polym. 2016, 150, 40–47. [Google Scholar] [CrossRef]
- Zare, E.N.; Makvandi, P.; Tay, F.R. Recent progress in the industrial and biomedical applications of tragacanth gum: A review. Carbohydr. Polym. 2019, 212, 450–467. [Google Scholar] [CrossRef]
- Mei, J.; Yuan, Y.; Wu, Y.; Li, Y. Characterization of edible starch–chitosan film and its application in the storage of Mongolian cheese. Int. J. Biol. Macromol. 2013, 57, 17–21. [Google Scholar] [CrossRef]
- Bonilla, J.; Talón Argente, E.; Atarés, L.; Vargas, M.; Chiralt, A. Effect of the incorporation of antioxidants on physicochemical and antioxidant properties of wheat starch–chitosan films. J. Food Eng. 2013, 118, 271–278. [Google Scholar] [CrossRef]
- Mathew, S.; Abraham, T.E. Characterisation of ferulic acid incorporated starch-chitosan blend films. Food Hydrocoll. 2008, 22, 826–835. [Google Scholar] [CrossRef]
- Zhong, Y.; Li, Y.; Zhao, Y. Physicochemical, microstructural, and antibacterial properties of β-chitosan and kudzu starch composite films. J. Food Sci. 2012, 77, E280–E286. [Google Scholar] [CrossRef] [PubMed]
- Makvandi, P.; Ghomi, M.; Padil, V.V.T.; Shalchy, F.; Ashrafizadeh, M.; Askarinejad, S.; Pourreza, N.; Zarrabi, A.; Zare, E.N.; Kooti, M.; et al. Biofabricated Nanostructures and Their Composites in Regenerative Medicine. ACS Appl. Nano Mater. 2020, 3, 6210–6238. [Google Scholar] [CrossRef]
- Flory, P.J. In Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA, 1953. [Google Scholar]
- Sabu, T. Physical Chemistry of Macromolecules; Wiley: Ithaca, NY, USA, 1961. [Google Scholar]
- Kasaai, M.R.; Arul, J.; Charlet, G. Intrinsic viscosity–molecular weight relationship for chitosan. J. Polym. Ence Part B Polym. Phys. 2015, 38, 2591–2598. [Google Scholar] [CrossRef]
- Swinkels, I.J.J.M. Composition and properties of commercial native starches. Starch Stärke 1985, 37, 1–5. [Google Scholar] [CrossRef]
- Charles, W.P. Biochemistry for the Pharmaceutical Sciences; Jones & Bartlett Learning: Burlington, MA, USA, 2019. [Google Scholar]
- Zamudio-Flores, P.B.; Torres, A.V.; Salgado-Delgado, R.; Bello-Pérez, L.A. Influence of the oxidation and acetylation of banana starch on the mechanical and water barrier properties of modified starch and modified starch/chitosan blend films. J. Appl. Polym. Sci. 2010, 115, 991–998. [Google Scholar] [CrossRef]
- Park, P.J.; Je, J.Y.; Kim, S.K. Free radical scavenging activities of differently deacetylated chitosans using an ESR spectrometer. Carbohydr. Polym. 2004, 55, 17–22. [Google Scholar] [CrossRef]
- Shukur, M.F.; Yusof, Y.M.; Zawawi, S.M.M.; Illias, H.A.; Kadir, M.F.Z. Conductivity and transport studies of plasticized chitosan-based proton conducting biopolymer electrolytes. Phys. Scr. 2013, 2013, 014050. [Google Scholar] [CrossRef]
- Xu, Y.X.; Kim, K.M.; Hanna, M.A.; Nag, D. Chitosan-starch composite film: Preparation and characterization. Ind. Crop. Prod. 2005, 21, 185–192. [Google Scholar] [CrossRef]
- Mathew, S.; Brahmakumar, M.; Abraham, T.E. Microstructural imaging and characterization of the mechanical, chemical, thermal, and swelling properties of starch-chitosan blend films. Biopolymers 2010, 82, 176–187. [Google Scholar] [CrossRef]
- Yusof, Y.M.; Shukur, M.F.; Illias, H.A.; Kadir, M.F.Z. Conductivity and electrical properties of corn starch–chitosan blend biopolymer electrolyte incorporated with ammonium iodide. Phys. Scr. 2014, 89, 35701. [Google Scholar] [CrossRef]
- Kittur, F.S.; Harish Prashanth, K.V.; Udaya Sankar, K.; Tharanathan, R.N. Characterization of chitin, chitosan and their carboxymethyl derivatives by differential scanning calorimetry. Carbohydr. Polym. 2002, 49, 185–193. [Google Scholar] [CrossRef]
- Perez, J.J.; Francois, N.J. Chitosan-starch beads prepared by ionotropic gelation as potential matrices for controlled release of fertilizers. Carbohydr. Polym. 2016, 148, 134–142. [Google Scholar] [CrossRef]
- Zawadzki, J.; Kaczmarek, H. Thermal treatment of chitosan in various conditions. Carbohydr. Polym. 2010, 80, 394–400. [Google Scholar] [CrossRef]
- Tester, R.F.; Morrison, W.R. Swelling and gelatinization of cereal starches. II, Waxy rice starches. Cereal Chem. 1990, 67, 558–563. [Google Scholar]
- Liu, H.; Lv, M.; Wang, L.; Li, Y.; Fan, H.; Wang, M. Comparative study: How annealing and heat-moisture treatment affect the digestibility, textural, and physicochemical properties of maize starch. Starch Stärke 2016, 68, 268. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor | Viscosity of CTS (A) (mPa.s) | Mass Fraction of CTS (B) (wt%) | Weight Ratio of CTS/Starch (C) (g) | |
|---|---|---|---|---|
| Level | ||||
| 1 | ≤200 | 3.8 | 3/3.8 | |
| 2 | 200–400 | 4 | 3/4 | |
| 3 | ≥400 | 4.2 | 3/4.2 | |
| Viscosity (mPa.s) | [η] (mL·g−1) | R2 | Molecular Weight (Da) |
|---|---|---|---|
| ≤200 | 502.39 | 0.995 | 3.5 × 105 |
| 200–400 | 611.46 | 0.996 | 4.4 × 105 |
| ≥400 | 702.87 | 0.960 | 5.2 × 105 |
| CTS/PS | CTS-3.5 | CTS-4.4 | CTS-5.2 | |||
|---|---|---|---|---|---|---|
| Pore Size (μm) | Porosity (%) | Pore Size (μm) | Porosity (%) | Pore Size (μm) | Porosity (%) | |
| 3/3.8 | 413.4 (14.0) | 69.12 | 298.8 (24.2) | 53.24 | 407.8 (38.5) | 72.29 |
| 3/4 | 392.5 (23.9) | 66.98 | 281.7 (27.0) | 55.39 | 394.2 (54.2) | 71.72 |
| 3/4.2 | 391.7 (30.2) | 66.12 | 275.8 (36.3) | 51.62 | 389.8 (28.4) | 68.28 |
| Mass Fraction of CTS (wt%) | CTS/PS (g) | CTS-3.5 | CTS-4.4 | CTS-5.2 |
|---|---|---|---|---|
| 3/3.8 | 0.170 (0.018) | 0.170 (0.008) | 0.153 (0.013) | |
| 3.8 | 3/4 | 0.177 (0.013) | 0.190 (0.013) | 0.169 (0.008) |
| 3/4.2 | 0.178 (0.017) | 0.205 (0.037) | 0.176 (0.008) | |
| 3/3.8 | 0.154 (0.018) | 0.187 (0.011) | 0.151 (0.009) | |
| 4 | 3/4 | 0.174 (0.010) | 0.186 (0.017) | 0.169 (0.009) |
| 3/4.2 | 0.175 (0.015) | 0.197 (0.011) | 0.173 (0.008) | |
| 3/3.8 | 0.144 (0.007) | 0.174 (0.010) | 0.161 (0.009) | |
| 4.2 | 3/4 | 0.164 (0.006) | 0.189 (0.016) | 0.172 (0.012) |
| 3/4.2 | 0.177 (0.009) | 0.194 (0.015) | 0.173 (0.008) |
| Samples | CTS-3.8 wt% | CTS-4 wt% | CTS-4.2 wt% | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 3/3.8 | 3/4 | 3/4.2 | 3/3.8 | 3/4 | 3/4.2 | 3/3.8 | 3/4 | 3/4.2 | |
| CTS-3.5/PS | 0.768 | 0.811 | 0.983 | 0.860 | 0.916 | 0.958 | 0.761 | 0.803 | 0.934 |
| CTS-4.4/PS | 0.847 | 0.953 | 1.062 | 0.938 | 0.969 | 1.077 | 0.870 | 0.958 | 1.053 |
| CTS-5.2/PS | 0.745 | 0.792 | 0.939 | 0.822 | 0.892 | 0.918 | 0.775 | 0.836 | 0.920 |
| Molecular Weight of CTS (A) | Mass Fraction of CTS (B) | Weight Ratio of CTS/PS (C) | Compressive Strength/mPa | |
|---|---|---|---|---|
| 1 | 1 | 1 | 1 | 0.768 |
| 2 | 1 | 2 | 2 | 0.916 |
| 3 | 1 | 3 | 3 | 0.934 |
| 4 | 2 | 1 | 2 | 0.953 |
| 5 | 2 | 2 | 3 | 1.077 |
| 6 | 2 | 3 | 1 | 0.870 |
| 7 | 3 | 1 | 3 | 0.939 |
| 8 | 3 | 2 | 1 | 0.822 |
| 9 | 3 | 3 | 2 | 0.836 |
| K1 | 2.618 | 2.660 | 2.460 | |
| K2 | 2.900 | 2.815 | 2.705 | |
| K3 | 2.597 | 2.640 | 2.950 | |
| k1 | 0.873 | 0.887 | 0.820 | |
| k2 | 0.967 | 0.938 | 0.902 | |
| k3 | 0.866 | 0.880 | 0.983 | |
| R | 0.101 | 0.058 | 0.163 | |
| Optimal level | A2 | B2 | C3 | |
| Optimal combination | A2B2C3 |
| Factor | Square Sum (SS) | Degree of Freedom (DF) | Mean Square (MS) | F-Value | Significance Probability (P) |
|---|---|---|---|---|---|
| A | 0.030 | 2 | 0.015 | 40.411 | 0.024 |
| B | 0.005 | 2 | 0.002 | 6.112 | 0.141 |
| C | 0.043 | 2 | 0.021 | 56.613 | 0.017 |
| Error | 0.001 | 2 |
| Samples | T1 a (°C) | TP (°C) | Residue (%) |
|---|---|---|---|
| CTS-3.5 | 72.11 | 292.98 | 31.44 |
| CTS-4.4 | 74.70 | 295.40 | 32.80 |
| CTS-5.2 | 72.17 | 292.94 | 32.02 |
| PS | 92.77 | 293.64 | 18.41 |
| CTS-3.5-PS | 72.77 | 302.88 | 15.97 |
| CTS-4.4-PS | 74.22 | 305.31 | 18.83 |
| CTS-5.2-PS | 73.60 | 305.22 | 19.18 |
| Samples | To (°C) | Tp (°C) | Tc (°C) | ΔH (J/g) |
|---|---|---|---|---|
| CTS-4.4 | 34.65 | 69.57 | 104.72 | 133.53 |
| PS | 43.67 | 94.48 | 153.09 | 131.33 |
| CTS-3.5/PS | 27.93 | 62.38 | 104.67 | 86.32 |
| CTS-4.4/PS | 39.02 | 72.08 | 126.83 | 123.91 |
| CTS-5.2/PS | 50.96 | 86.32 | 116.99 | 167.86 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Teng, Z.; Huang, R.; Catchmark, J.M. Biodegradable Starch/Chitosan Foam via Microwave Assisted Preparation: Morphology and Performance Properties. Polymers 2020, 12, 2612. https://doi.org/10.3390/polym12112612
Zhang X, Teng Z, Huang R, Catchmark JM. Biodegradable Starch/Chitosan Foam via Microwave Assisted Preparation: Morphology and Performance Properties. Polymers. 2020; 12(11):2612. https://doi.org/10.3390/polym12112612
Chicago/Turabian StyleZhang, Xian, Zhuangzhuang Teng, Runzhou Huang, and Jeffrey M. Catchmark. 2020. "Biodegradable Starch/Chitosan Foam via Microwave Assisted Preparation: Morphology and Performance Properties" Polymers 12, no. 11: 2612. https://doi.org/10.3390/polym12112612
APA StyleZhang, X., Teng, Z., Huang, R., & Catchmark, J. M. (2020). Biodegradable Starch/Chitosan Foam via Microwave Assisted Preparation: Morphology and Performance Properties. Polymers, 12(11), 2612. https://doi.org/10.3390/polym12112612