General-Purpose Coarse-Grained Toughened Thermoset Model for 44DDS/DGEBA/PES

 , , ,
, , ,
Abstract
Abstract
Dataset
Dataset License
1. Introduction
- Predicting how deviations from process specifications impact performance.
- Composite formulations optimized for manufacturing requirements.
- Temperature schedules (termed cure profiles) optimized for speed and reproducibility.
2. Model
3. Methods
4. Results
- Setup
- Validation
- Exploration
4.1. Setup Simulations
4.1.1. Forcefield Parameterization
4.1.2. Reaction Kinetics Calibration
4.1.3. Finite Size Effects
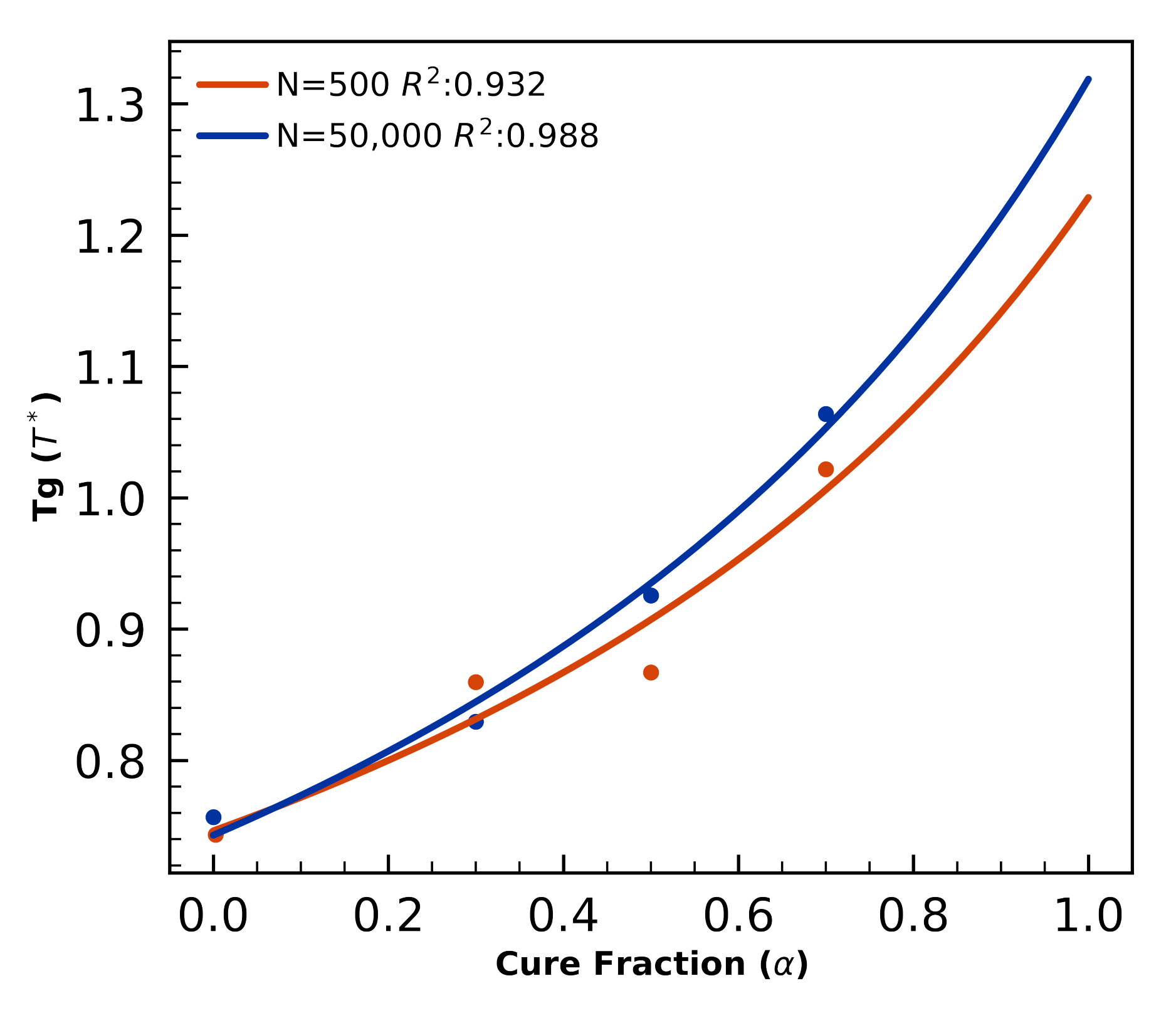
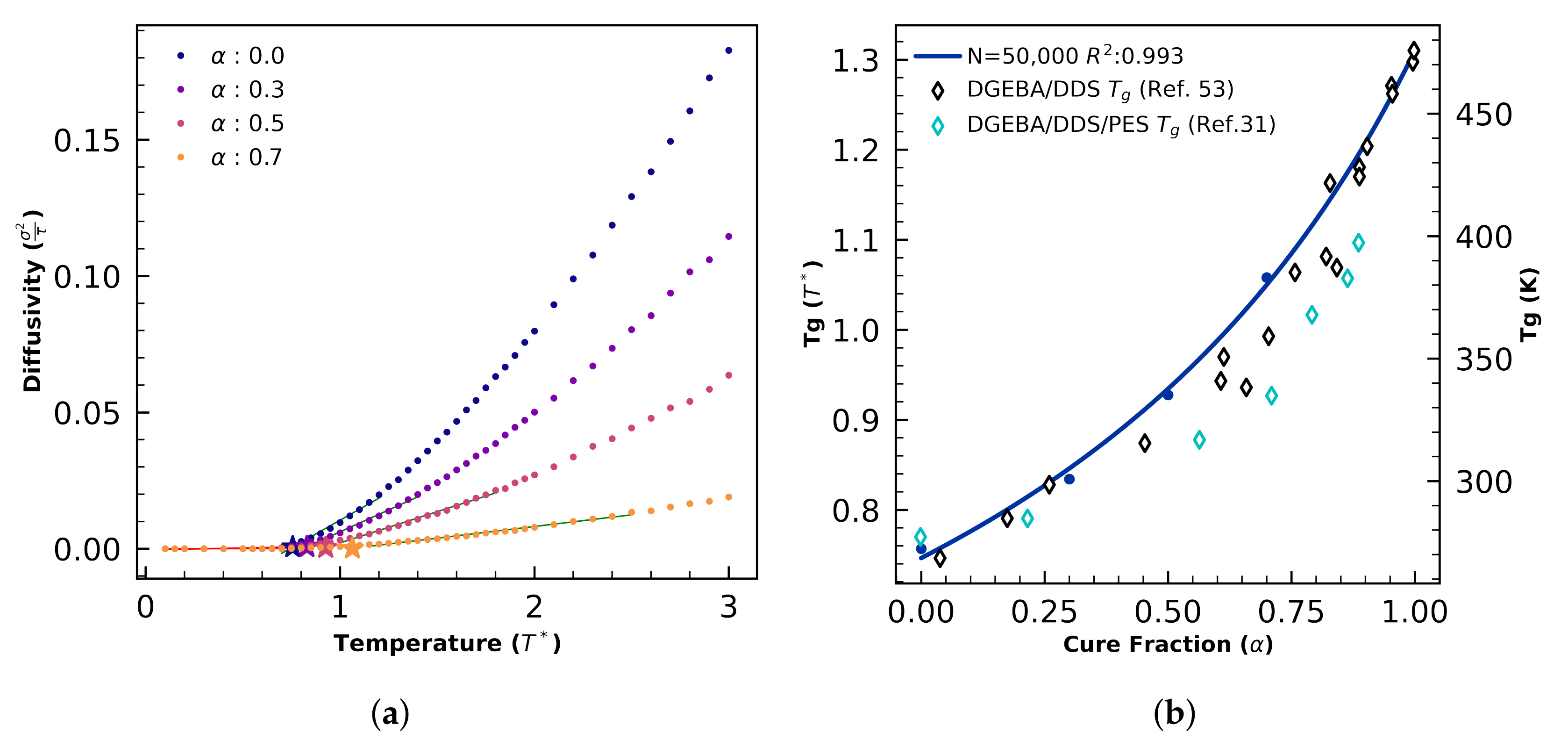
4.1.4. Glass Transition—Small Systems
4.1.5. Morphology—Small Systems
4.2. Validation Simulations
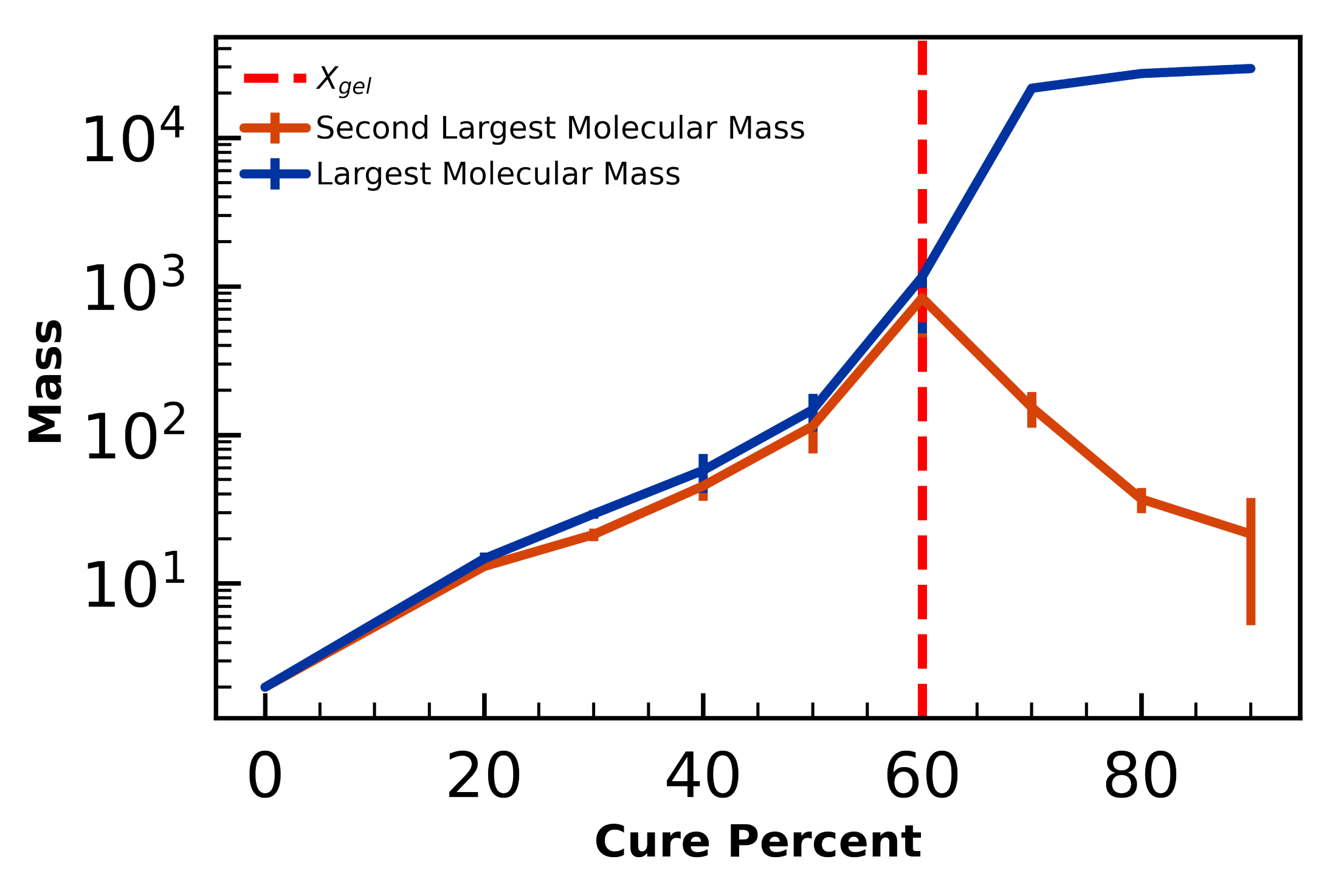
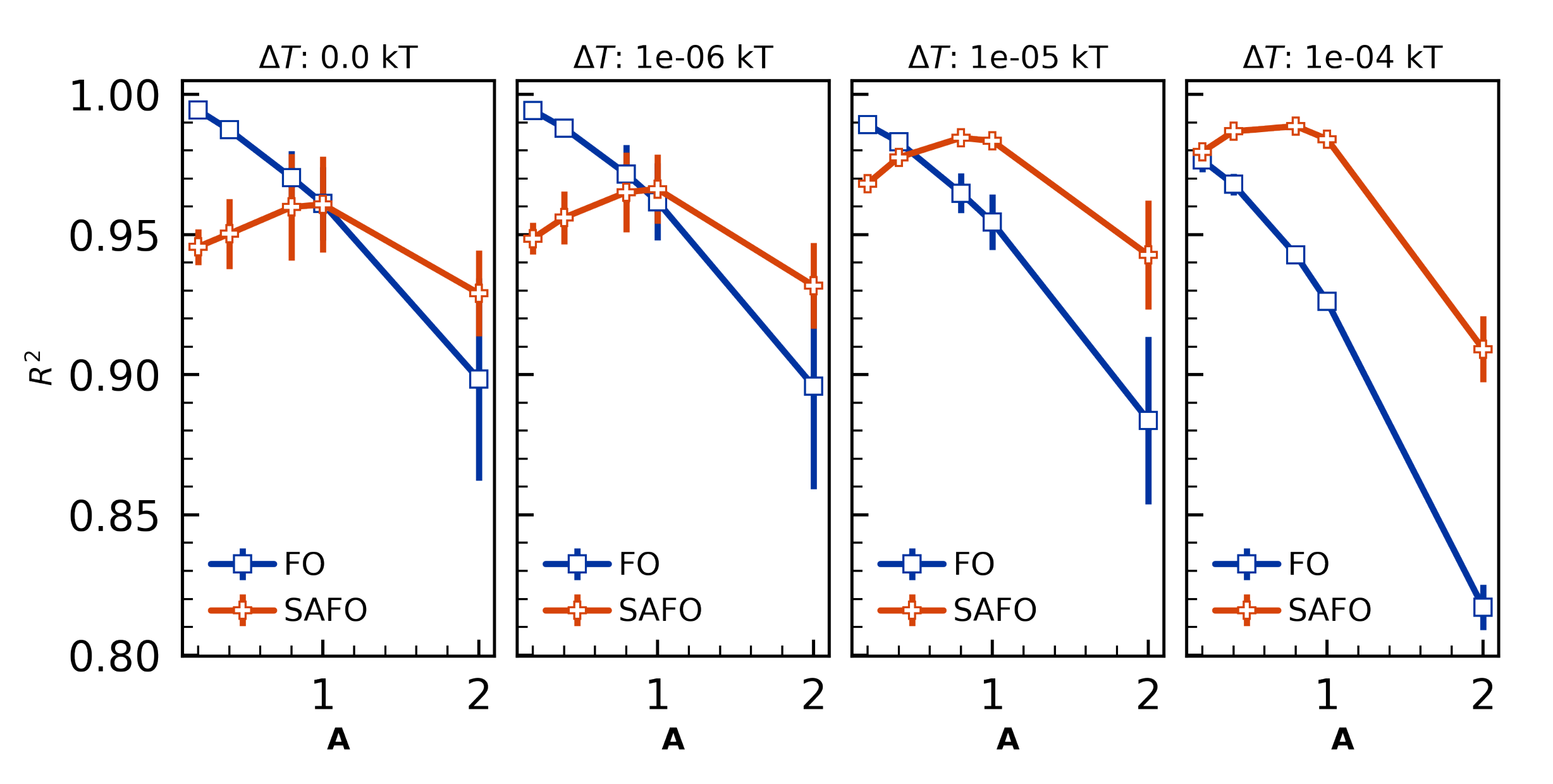
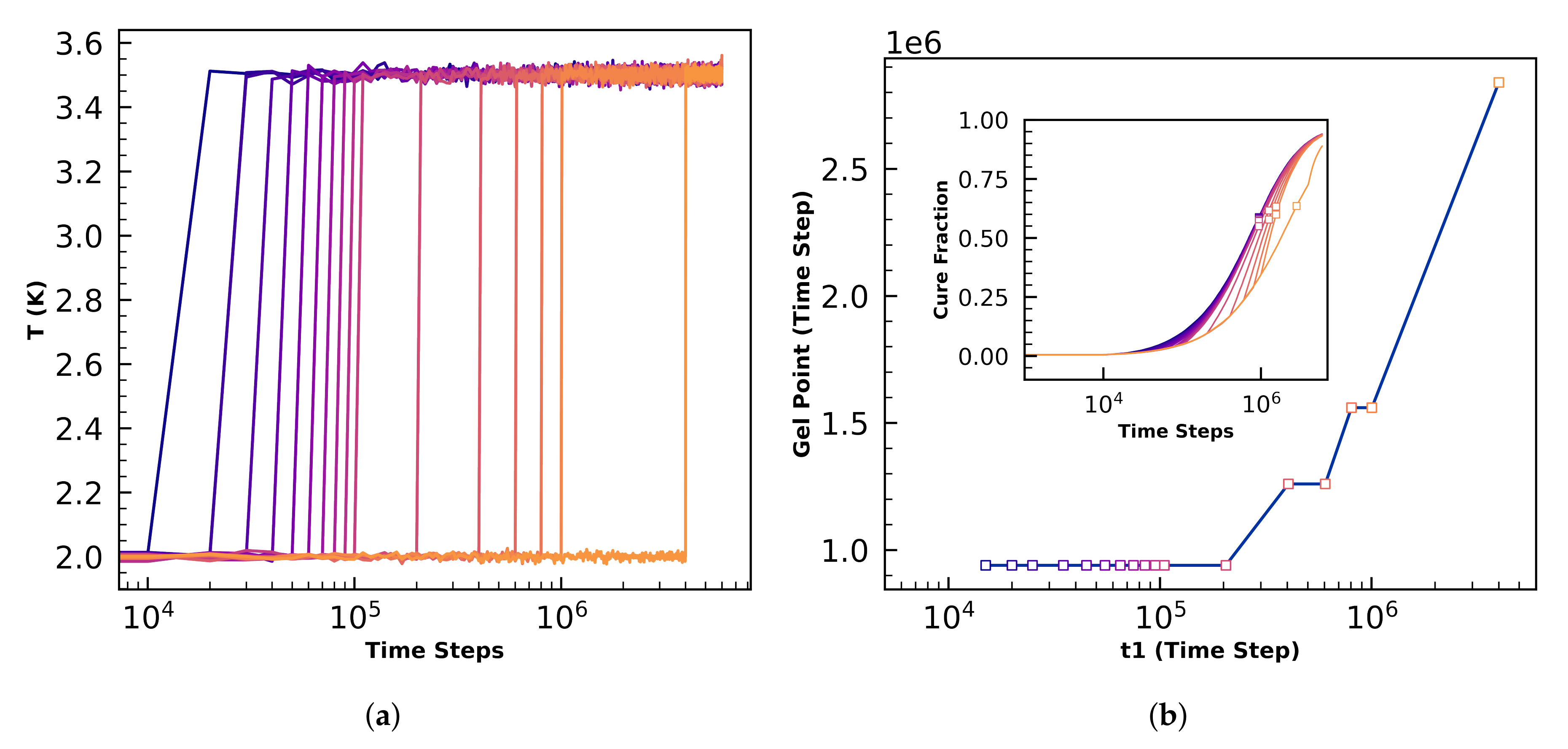
4.2.1. Gel-Point Validation
4.2.2. Glass Transition Validation
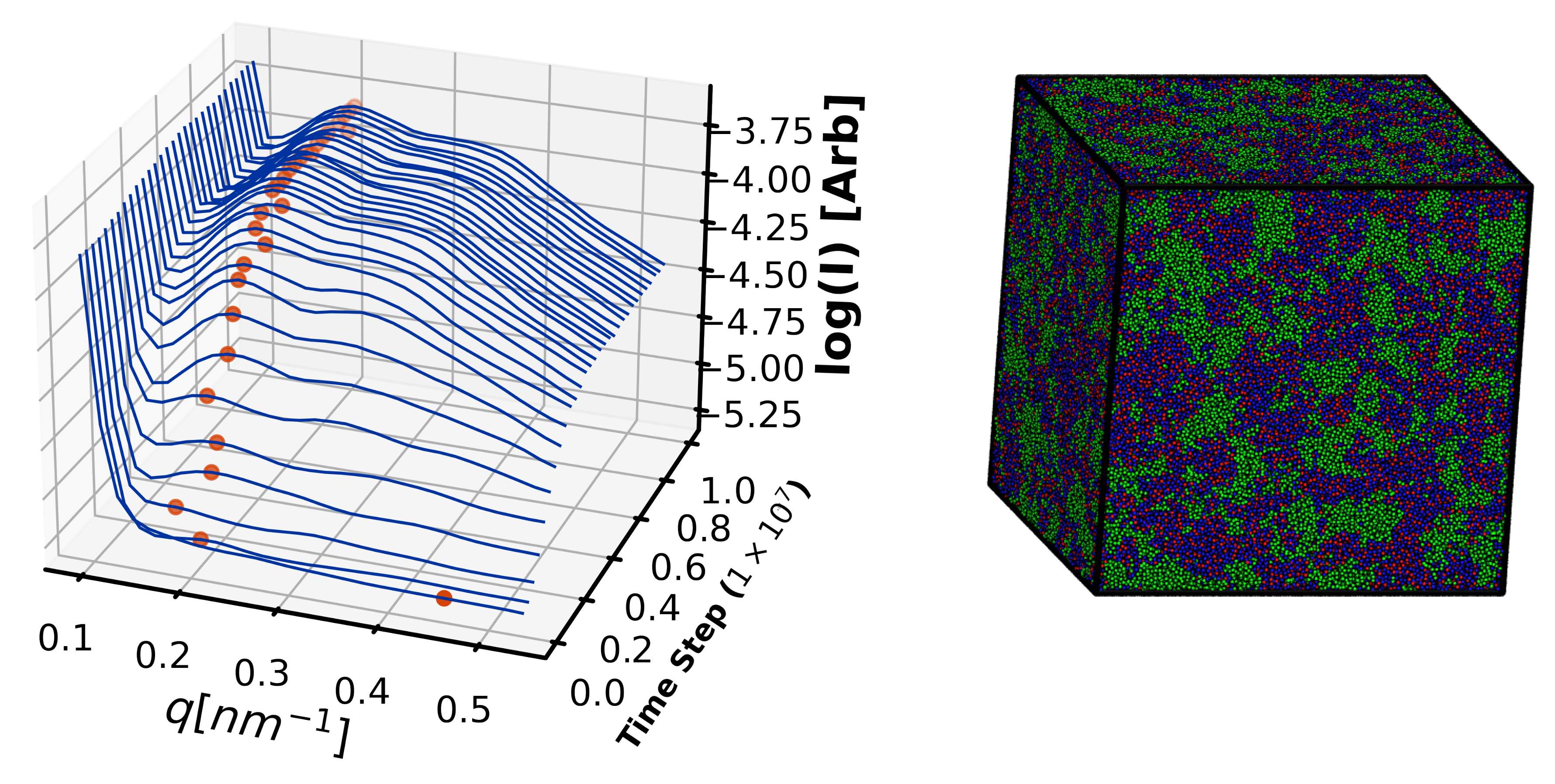
4.2.3. Morphology Validation
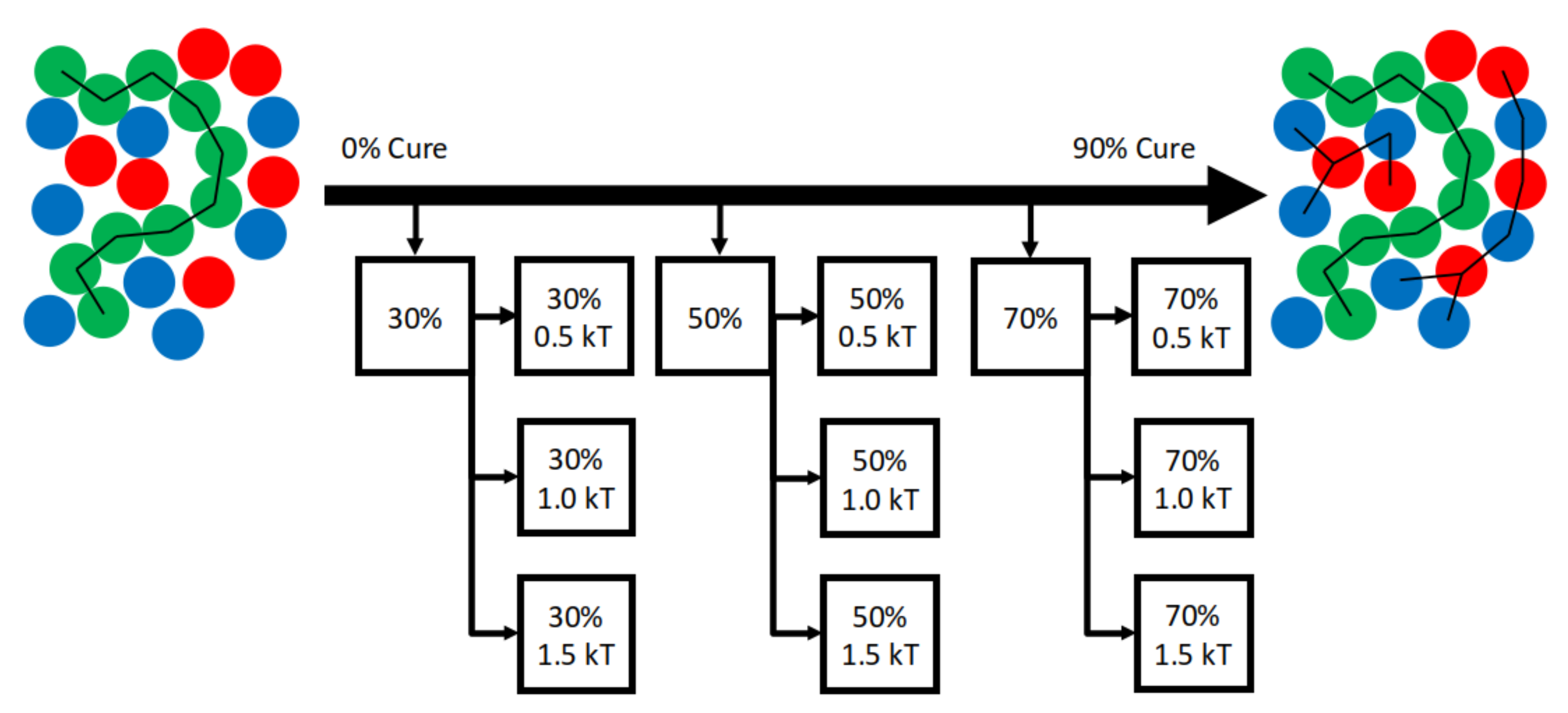
4.3. Exploration Simulations
4.3.1. Enthalpy Experiment
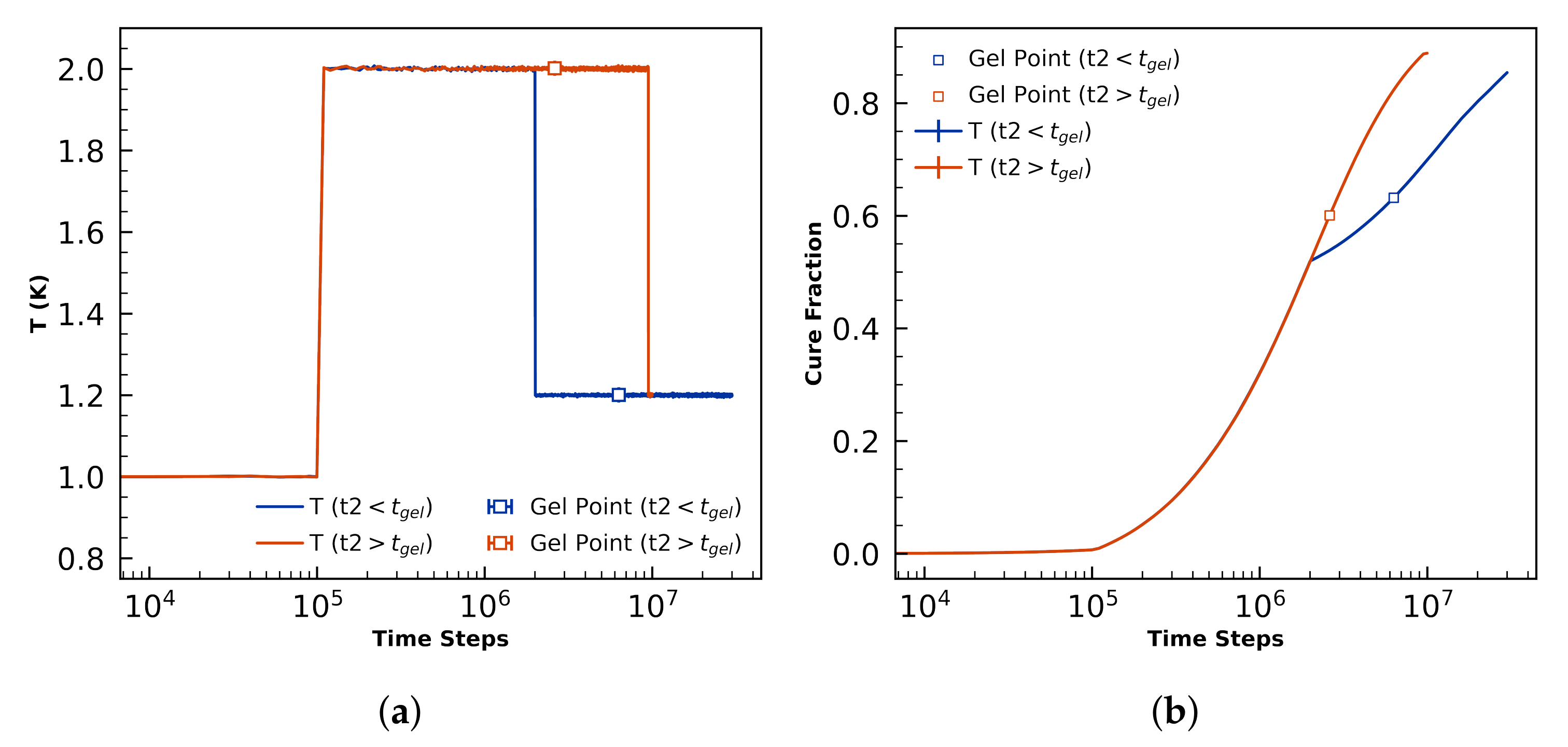
4.3.2. Sensitivity to Cure Profile
5. Conclusions and Outlook
- Offers straightforward forcefield parameterization.
- Can capture first-order and self-accelerated first order reaction dynamics.
- Is validated against experimental gel points, glass transition temperatures, and morphology for 44DDS/DGEBA/PES blends.
- Does not require backmapping for calculation.
- Can cure million-particle volumes (corresponding to 31-million atoms and (100 nm) periodic boxes) to in under one week.
- Demonstrates for the first time sensitivity of morphology to cure profile.
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 44DDS | 4,4’-Diaminodiphenyl Sulfone |
| AAMD | All-Atom Molecular Dynamics |
| CG | coarse-grained |
| DETA | Diethylenetriamine |
| DGEBA | Bisphenol A diglycidyl ether |
| DPD | Dissipative Particle Dynamics |
| FO | first order |
| GPU | graphics processing unit |
| LB | Lorentz-Berthelot |
| LJ | Lennard-Jones |
| MD | molecular dynamics |
| PES | Poly(oxy-1,4-phenylsulfonyl-1,4-phenyl) |
| PRM | piecewise regression method |
| SAFO | self-accelerated first order |
References
- Marsh, G. Reclaiming value from post-use carbon composite. Reinf. Plast. 2008, 52, 36–39. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, Y.C.; Huang, P. Effect of cure cycle on curing process and hardness for epoxy resin. Express Polym. Lett. 2009, 3, 534–541. [Google Scholar] [CrossRef]
- Sinclair, J.W. Effects of Cure Temperature on Epoxy Resin Properties. J. Adhes. 1992, 38, 219–234. [Google Scholar] [CrossRef]
- Lapique, F.; Redford, K. Curing effects on viscosity and mechanical properties of a commercial epoxy resin adhesive. Int. J. Adhes. Adhes. 2002, 22, 337–346. [Google Scholar] [CrossRef]
- Jenninger, W.; Schawe, J.E.; Alig, I. Calorimetric studies of isothermal curing of phase separating epoxy networks. Polymer 2000, 41, 1577–1588. [Google Scholar] [CrossRef]
- Kim, B.S.; Chiba, T.; Inoue, T. Morphology development via reaction-induced phase separation in epoxy/poly(ether sulfone) blends: Morphology control using poly(ether sulfone) with functional end-groups. Polymer 1995, 36, 43–47. [Google Scholar] [CrossRef]
- Rosetti, Y.; Alcouffe, P.; Pascault, J.P.; Gérard, J.F.; Lortie, F. Polyether Sulfone-Based Epoxy Toughening: From Micro- to Nano-Phase Separation via PES End-Chain Modification and Process Engineering. Materials 2018, 11, 1960. [Google Scholar] [CrossRef]
- Yoon, T.H.; Priddy, D.B.; Lyle, G.D.; McGrath, J.E. Mechanical and morphological investigations of reactive polysulfone toughened epoxy networks. Macromol. Symp. 1995, 98, 673–686. [Google Scholar] [CrossRef]
- Williams, R.J.J.; Rozenberg, B.A.; Pascault, J.P. Reaction-induced phase separation in modified thermosetting polymers. In Polymer Analysis Polymer Physics; Springer: Berlin/Heidelberg, Germany, 1997; pp. 95–156. [Google Scholar] [CrossRef]
- Sultan, J.N.; McGarry, F.J. Effect of rubber particle size on deformation mechanisms in glassy epoxy. Polym. Eng. Sci. 1973, 13, 29–34. [Google Scholar] [CrossRef]
- MacKinnon, A.J.; Jenkins, S.D.; McGrail, P.T.; Pethrick, R.A. A dielectric, mechanical, rheological and electron microscopy study of cure and properties of a thermoplastic-modified epoxy resin. Macromolecules 1992, 25, 3492–3499. [Google Scholar] [CrossRef]
- MacKinnon, A.J.; Jenkins, S.D.; McGrail, P.T.; Pethrick, R.A. Dielectric, mechanical and rheological studies of phase separation and cure of a thermoplastic modified epoxy resin: Incorporation of reactively terminated polysulfones. Polymer 1993, 34, 3252–3263. [Google Scholar] [CrossRef]
- Kinloch, A.J.; Yuen, M.L.; Jenkins, S.D. Thermoplastic-toughened epoxy polymers. J. Mater. Sci. 1994, 29, 3781–3790. [Google Scholar] [CrossRef]
- Huang, P.; Zheng, S.; Huang, J.; Guo, Q.; Zhu, W. Miscibility and mechanical properties of epoxy resin/polysulfone blends. Polymer 1997, 38, 5565–5571. [Google Scholar] [CrossRef]
- Jong, S.R.; Yu, T.L. Physical aging of epoxy resin blended with a medium molecular weight poly(ether sulfone). Macromol. Chem. Phys. 1999, 200, 87–94. [Google Scholar] [CrossRef]
- Varley, R.J.; Hodgkin, J.H.; Hawthorne, D.G.; Simon, G.P.; McCulloch, D. Toughening of a trifunctional epoxy system Part {III}. Kinetic and morphological study of the thermoplastic modified cure process. Polymer 2000, 41, 3425–3436. [Google Scholar] [CrossRef]
- Hodgkin, J.H.; Simon, G.P.; Varley, R.J. Thermoplastic toughening of epoxy resins: A critical review. Polym. Adv. Technol. 1998, 9, 3–10. [Google Scholar] [CrossRef]
- Ruiz-Pérez, L.; Royston, G.J.; Fairclough, J.P.A.; Ryan, A.J. Toughening by nanostructure. Polymer 2008, 49, 4475–4488. [Google Scholar] [CrossRef]
- Liu, J.D.; Thompson, Z.J.; Sue, H.J.; Bates, F.S.; Hillmyer, M.A.; Dettloff, M.; Jacob, G.; Verghese, N.; Pham, H. Toughening of Epoxies with Block Copolymer Micelles of Wormlike Morphology. Macromolecules 2010, 43, 7238–7243. [Google Scholar] [CrossRef]
- Blanco, M.; López, M.; Kortaberria, G.; Mondragon, I. Nanostructured thermosets from self-assembled amphiphilic block copolymer/epoxy resin mixtures: Effect of copolymer content on nanostructures. Polym. Int. 2010, 59, 523–528. [Google Scholar] [CrossRef]
- Hillmyer, M.A.; Lipic, P.M.; Hajduk, D.A.; Almdal, K.; Bates, F.S. Self-Assembly and Polymerization of Epoxy Resin-Amphiphilic Block Copolymer Nanocomposites. J. Am. Chem. Soc. 1997, 119, 2749–2750. [Google Scholar] [CrossRef]
- George, S.M.; Puglia, D.; Kenny, J.M.; Parameswaranpillai, J.; Thomas, S. Reaction-induced phase separation and thermomechanical properties in epoxidized Styrene- block -butadiene- block -styrene triblock copolymer modified epoxy/DDM system. Ind. Eng. Chem. Res. 2014, 53, 6941–6950. [Google Scholar] [CrossRef]
- Li, C.; Strachan, A. Molecular scale simulations on thermoset polymers: A review. J. Polym. Sci. Part Polym. Phys. 2015, 53, 103–122. [Google Scholar] [CrossRef]
- Lee, A.; McKenna, G.B. Effect of crosslink density on physical ageing of epoxy networks. Polymer 1988, 29, 1812–1817. [Google Scholar] [CrossRef]
- Rajagopalan, G.; Gillespie, J.W.; McKnight, S.H. Diffusion of reacting epoxy and amine monomers in polysulfone: A diffusivity model. Polymer 2000, 41, 7723–7733. [Google Scholar] [CrossRef]
- Pascault, J.P.; Williams, R.J. Glass transition temperature versus conversion relationships for thermosetting polymers. J. Polym. Sci. Part Polym. Phys. 1990, 28, 85–95. [Google Scholar] [CrossRef]
- DiBenedetto, A.T. Prediction of the glass transition temperature of polymers: A model based on the principle of corresponding states. J. Polym. Sci. Part Polym. Phys. 1987, 25, 1949–1969. [Google Scholar] [CrossRef]
- Enns, J.B.; Gillham, J.K. Time-temperature-transformation (TTT) cure diagram: Modeling the cure behavior of thermosets. J. Appl. Polym. Sci. 1983, 28, 2567–2591. [Google Scholar] [CrossRef]
- Bidstrup, S.A.; Sheppard, N.F.; Senturia, S.D. Dielectric analysis of the cure of thermosetting epoxy/amine systems. Polym. Eng. Sci. 1989, 29, 325–328. [Google Scholar] [CrossRef]
- Georjon, O.; Galy, J.; Pascault, J.P. Isothermal curing of an uncatalyzed dicyanate ester monomer: Kinetics and modeling. J. Appl. Polym. Sci. 1993, 49, 1441–1452. [Google Scholar] [CrossRef]
- Jordan, C.; Galy, J.; Pascault, J.P. Measurement of the extent of reaction of an epoxy–cycloaliphatic amine system and influence of the extent of reaction on its dynamic and static mechanical properties. J. Appl. Polym. Sci. 1992, 46, 859–871. [Google Scholar] [CrossRef]
- Li, C.; Medvedev, G.A.; Lee, E.W.; Kim, J.; Caruthers, J.M.; Strachan, A. Molecular dynamics simulations and experimental studies of the thermomechanical response of an epoxy thermoset polymer. Polymer 2012, 53, 4222–4230. [Google Scholar] [CrossRef]
- Li, C.; Coons, E.; Strachan, A. Material property prediction of thermoset polymers by molecular dynamics simulations. Acta Mech. 2014, 225, 1187–1196. [Google Scholar] [CrossRef]
- Abbott, L.J.; Hughes, J.E.; Colina, C.M. Virtual synthesis of thermally cross-linked copolymers from a novel implementation of polymatic. J. Phys. Chem. B 2014, 118, 1916–1924. [Google Scholar] [CrossRef] [PubMed]
- Shudo, Y.; Izumi, A.; Hagita, K.; Nakao, T.; Shibayama, M. Large-scale molecular dynamics simulation of crosslinked phenolic resins using pseudo-reaction model. Polymer 2016, 103, 261–276. [Google Scholar] [CrossRef]
- Khare, K.S.; Phelan, F.R. Quantitative Comparison of Atomistic Simulations with Experiment for a Cross-Linked Epoxy: A Specific Volume-Cooling Rate Analysis. Macromolecules 2018, 51, 564–575. [Google Scholar] [CrossRef]
- Prasad, A.; Grover, T.; Basu, S. Coarse–grained molecular dynamics simulation of cross—linking of DGEBA epoxy resin and estimation of the adhesive strength. Int. J. Eng. Sci. Technol. 2010, 2, 17–30. [Google Scholar] [CrossRef]
- Liu, H.; Li, M.; Lu, Z.Y.; Zhang, Z.G.; Sun, C.C.; Cui, T. Multiscale Simulation Study on the Curing Reaction and the Network Structure in a Typical Epoxy System. Macromolecules 2011, 44, 8650–8660. [Google Scholar] [CrossRef]
- Langeloth, M.; Sugii, T.; Böhm, M.C.; Müller-Plathe, F. The glass transition in cured epoxy thermosets: A comparative molecular dynamics study in coarse-grained and atomistic resolution. J. Chem. Phys. 2015, 143, 243158. [Google Scholar] [CrossRef]
- Kacar, G.; Peters, E.A.J.F.; De With, G. Multi-scale simulations for predicting material properties of a cross-linked polymer. Comput. Mater. Sci. 2015, 102, 68–77. [Google Scholar] [CrossRef]
- Pervaje, A.K.; Tilly, J.C.; Detwiler, A.T.; Spontak, R.J.; Khan, S.A.; Santiso, E.E. Molecular Simulations of Thermoset Polymers Implementing Theoretical Kinetics with Top-Down Coarse-Grained Models. Macromolecules 2020, 53, 2310–2322. [Google Scholar] [CrossRef]
- Thomas, S.; Alberts, M.; Henry, M.M.; Estridge, C.E.; Jankowski, E. Routine million-particle simulations of epoxy curing with dissipative particle dynamics. J. Theor. Comput. Chem. 2018, 17, S0219633618400059. [Google Scholar] [CrossRef]
- Gissinger, J.R.; Jensen, B.D.; Wise, K.E. Modeling chemical reactions in classical molecular dynamics simulations. Polymer 2017, 128, 211–217. [Google Scholar] [CrossRef]
- Komarov, P.V.; Yu-Tsung, C.; Shih-Ming, C.; Khalatur, P.G.; Reineker, P. Highly Cross-Linked Epoxy Resins: An Atomistic Molecular Dynamics Simulation Combined with a Mapping/Reverse Mapping Procedure. Macromolecules 2007, 40, 8104–8113. [Google Scholar] [CrossRef]
- Pérez-Maciá, M.A.; Curcó, D.; Bringué, R.; Iborra, M.; Alemán, C. Atomistic simulations of the structure of highly crosslinked sulfonated poly (styrene-co-divinylbenzene) ion exchange resins. Soft Matter 2015, 11, 2251–2267. [Google Scholar] [CrossRef]
- Liu, Z.; Li, J.; Zhou, C.; Zhu, W. A molecular dynamics study on thermal and rheological properties of BNNS-epoxy nanocomposites. Int. J. Heat Mass Transf. 2018, 126, 353–362. [Google Scholar] [CrossRef]
- Srikanth, A.; Kinaci, E.; Vergara, J.; Palmese, G.; Abrams, C.F. The effect of alkyl chain length on mechanical properties of fatty-acid-functionalized amidoamine-epoxy systems. Comput. Mater. Sci. 2018, 150, 70–76. [Google Scholar] [CrossRef]
- Aldridge, M.; Wineman, A.; Waas, A.; Kieffer, J. In situ analysis of the relationship between cure kinetics and the mechanical modulus of an epoxy resin. Macromolecules 2014, 47, 8368–8376. [Google Scholar] [CrossRef]
- Thomas, S.; Henry, M. Dybond Plugin; Zenodo; CERN: Geneva, Switzerland, 2017. [Google Scholar] [CrossRef]
- Anderson, J.A.; Glaser, J.; Glotzer, S.C. HOOMD-blue: A Python package for high-performance molecular dynamics and hard particle Monte Carlo simulations. Comput. Mater. Sci. 2020, 173, 109363. [Google Scholar] [CrossRef]
- Adorf, C.S.; Ramasubramani, V.; Dice, B.D.; Henry, M.M.; Dodd, P.M.; Glotzer, S.C. Glotzerlab/Signac; Zenodo; CERN: Geneva, Switzerland, 2019. [Google Scholar] [CrossRef]
- Adorf, C.S.; Dodd, P.M.; Ramasubramani, V.; Glotzer, S.C. Simple data and workflow management with the signac framework. Comput. Mater. Sci. 2018, 146, 220–229. [Google Scholar] [CrossRef]
- Klein, C.; Sallai, J.; Jones, T.J.; Iacovella, C.R.; McCabe, C.; Cummings, P.T. A Hierarchical, Component Based Approach to Screening Properties of Soft Matter. In Foundations of Molecular Modeling and Simulation; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Thomas, S.; Henry, M. Cmelab/Notebooks-for-2020-Epoxy-Paper v0.1 (Version v0.1); Zenodo; CERN: Geneva, Switzerland, 2020. [Google Scholar] [CrossRef]
- Jones, M.L.; Jankowski, E. Computationally connecting organic photovoltaic performance to atomistic arrangements and bulk morphology. Mol. Simul. 2017, 43, 1–18. [Google Scholar] [CrossRef]
- Thomas, S. New Methods for Understanding and Controlling the Self-Assembly of Reacting Systems Using Coarse-Grained Molecular Dynamics. Ph.D. Thesis, Boise State University, Boise, ID, USA, 2018. [Google Scholar]
- Nielsen, L.E. Cross-Linking–Effect on Physical Properties of Polymers. J. Macromol. Sci. Rev. Macromol. Chem. C 1969, 3, 69–103. [Google Scholar] [CrossRef]
- Eloundou, J.P.; Ayina, O.; Nga, H.N.; Gerard, J.F.; Pascault, J.P.; Boiteux, G.; Seytre, G. Simultaneous kinetic and microdielectric studies of some epoxy-amine systems. J. Polym. Sci. Part Polym. Phys. 1998, 36, 2911–2921. [Google Scholar] [CrossRef]
- Pascault, J.P.; Williams, R.J. Relationships between glass transition temperature and conversion—Analyses of limiting cases. Polym. Bull. 1990, 24, 115–121. [Google Scholar] [CrossRef]
- Xu, W.S.; Douglas, J.F.; Freed, K.F. Influence of Cohesive Energy on the Thermodynamic Properties of a Model Glass-Forming Polymer Melt. Macromolecules 2016, 49, 8341–8354. [Google Scholar] [CrossRef]
- Li, C.; Strachan, A. Cohesive energy density and solubility parameter evolution during the curing of thermoset. Polymer 2018, 135, 162–170. [Google Scholar] [CrossRef]
- Hagberg, A.a.; Schult, D.a.; Swart, P.J. Exploring network structure, dynamics, and function using NetworkX. In Proceedings of the 7th Python in Science Conference (SciPy2008), Pasadena, CA, USA, 19–24 August 2008; Volume 836, pp. 11–15. [Google Scholar]
- Flory, P.J. Molecular Size Distribution in Three Dimensional Polymers. I. Gelation 1. J. Am. Chem. Soc. 1941, 63, 3083–3090. [Google Scholar] [CrossRef]
- Stockmayer, W.H. Theory of Molecular Size Distribution and Gel Formation in Branched Polymers II. General Cross Linking. J. Chem. Phys. 1944, 12, 125–131. [Google Scholar] [CrossRef]
- Pramanik, M.; Fowler, E.W.; Rawlins, J.W. Another look at epoxy thermosets correlating structure with mechanical properties. Polym. Eng. Sci. 2014, 54, 1990–2004. [Google Scholar] [CrossRef]
- Stauffer, D.; Coniglio, A.; Adam, M. Gelation and critical phenomena. In Polymer Networks; Springer: Berlin/Heidelberg, Germany, 1982; pp. 103–158. [Google Scholar] [CrossRef]
- White, S.R.; Mather, P.T.; Smith, M.J. Characterization of the cure-state of DGEBA-DDS epoxy using ultrasonic, dynamic mechanical, and thermal probes. Polym. Eng. Sci. 2002, 42, 51–67. [Google Scholar] [CrossRef]
- Bonnet, A.; Pascault, J.P.; Sautereau, H.; Taha, M.; Camberlin, Y. Epoxy-diamine thermoset/thermoplastic blends. 1. Rates of reactions before and after phase separation. Macromolecules 1999, 32, 8517–8523. [Google Scholar] [CrossRef]
- Min, B.G.; Stachurski, Z.H.; Hodgkin, J.H. Cure kinetics of elementary reactions of a DGEBA/DDS epoxy resin: 1. Glass transition temperature versus conversion. Polymer 1993, 34, 4908–4912. [Google Scholar] [CrossRef]
- Schawe, J.E.K. The Interplay between Molecular Dynamics and Reaction Kinetics during Curing Reactions. Aip Conf. Proc. 2017, 100002, 1–6. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) 44DDS | (B) DGEBA | (C) PES | |
|---|---|---|---|
| (A) 44DDS | 0.9216 | 0.9600 | 0.9026 |
| (B) DGEBA | 1.0000 | 0.9402 | |
| (C) PES | 0.8840 |
| Parameter | Value |
|---|---|
| Bond equilibrium (A-B,C-C) () | 1.0 |
| Bond force constant (A-B,C-C) (k) | 100 |
| Angle equilibrium (C-C-C) () | 109.5 |
| Angle force constant (C-C-C) () | 25 |
| Non-bonded interaction cutoff | 2.5 |
| Number density () | 1.0 |
| Activation Energy () | 3.0 |
| Bonding distance maximum | 1.0 |
| Secondary bond weight () | 1.2 |
| Enthalpy of Reaction () | 0.0 |
| Bond Period () | 1.0 |
| Maximum attempted bonds () | |
| Langevin drag () | 4.5 |
| %monomers 44DDS:DGEBA:PES | 20:40:40 |
| Cure temperature (T) | 3.0 |
| Step size () | 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Henry, M.M.; Thomas, S.; Alberts, M.; Estridge, C.E.; Farmer, B.; McNair, O.; Jankowski, E. General-Purpose Coarse-Grained Toughened Thermoset Model for 44DDS/DGEBA/PES. Polymers 2020, 12, 2547. https://doi.org/10.3390/polym12112547
Henry MM, Thomas S, Alberts M, Estridge CE, Farmer B, McNair O, Jankowski E. General-Purpose Coarse-Grained Toughened Thermoset Model for 44DDS/DGEBA/PES. Polymers. 2020; 12(11):2547. https://doi.org/10.3390/polym12112547
Chicago/Turabian StyleHenry, Michael M., Stephen Thomas, Mone’t Alberts, Carla E. Estridge, Brittan Farmer, Olivia McNair, and Eric Jankowski. 2020. "General-Purpose Coarse-Grained Toughened Thermoset Model for 44DDS/DGEBA/PES" Polymers 12, no. 11: 2547. https://doi.org/10.3390/polym12112547
APA StyleHenry, M. M., Thomas, S., Alberts, M., Estridge, C. E., Farmer, B., McNair, O., & Jankowski, E. (2020). General-Purpose Coarse-Grained Toughened Thermoset Model for 44DDS/DGEBA/PES. Polymers, 12(11), 2547. https://doi.org/10.3390/polym12112547