Micellar Organocatalysis Using Smart Polymer Supports: Influence of Thermoresponsive Self-Assembly on Catalytic Activity



Abstract

1. Introduction
2. Materials and Methods
2.1. Characterization
2.2. Materials
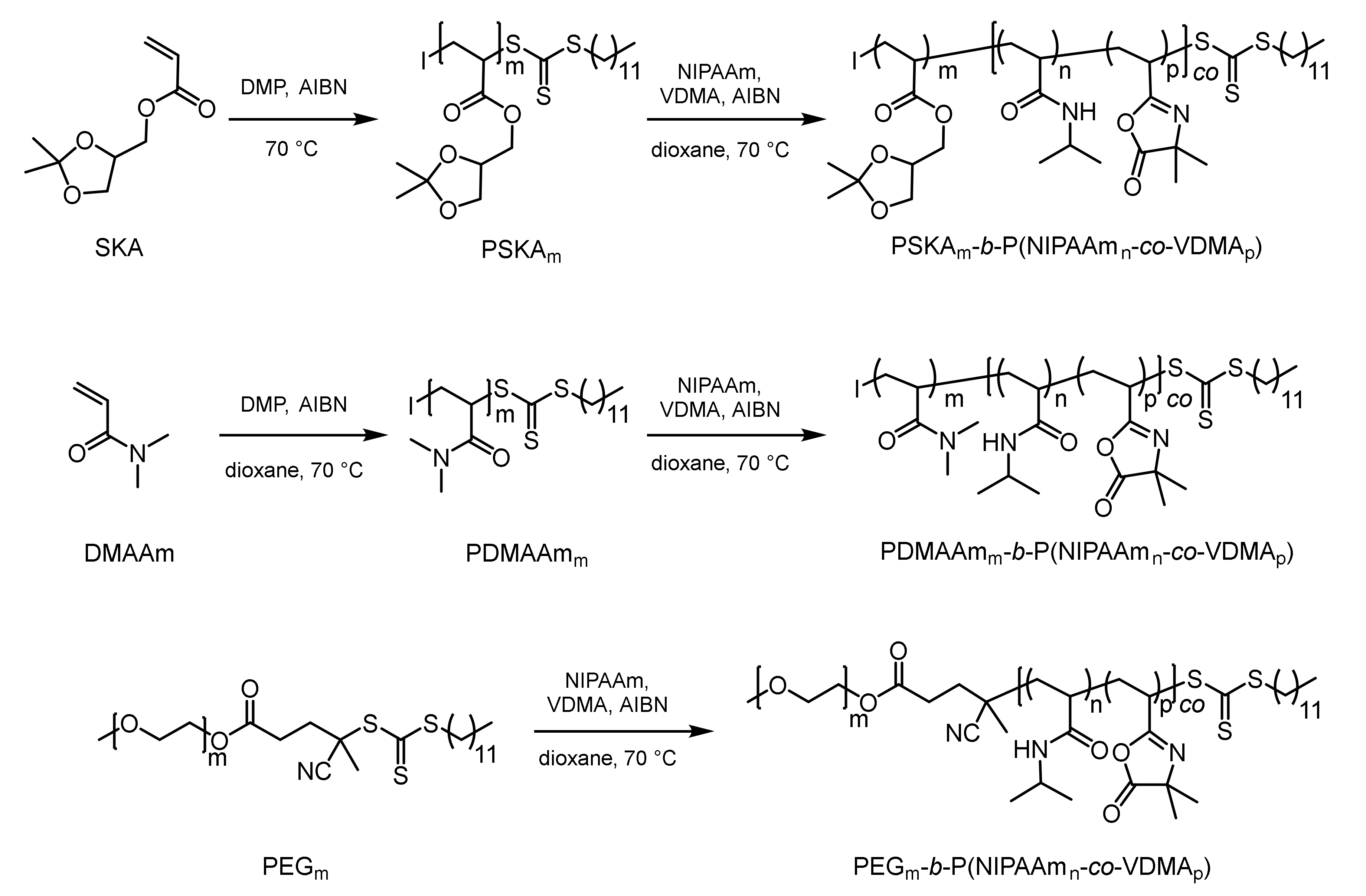
2.3. Synthesis of the Block Copolymer PSKA-b-P(NIPAAm-co-VDMA)
2.4. Synthesis of the Block Copolymer PDMAAm-b-P(NIPAAm-co-VDMA)
2.5. Synthesis of the Block Copolymer PEG-b-P(NIPAAm-co-VDMA)
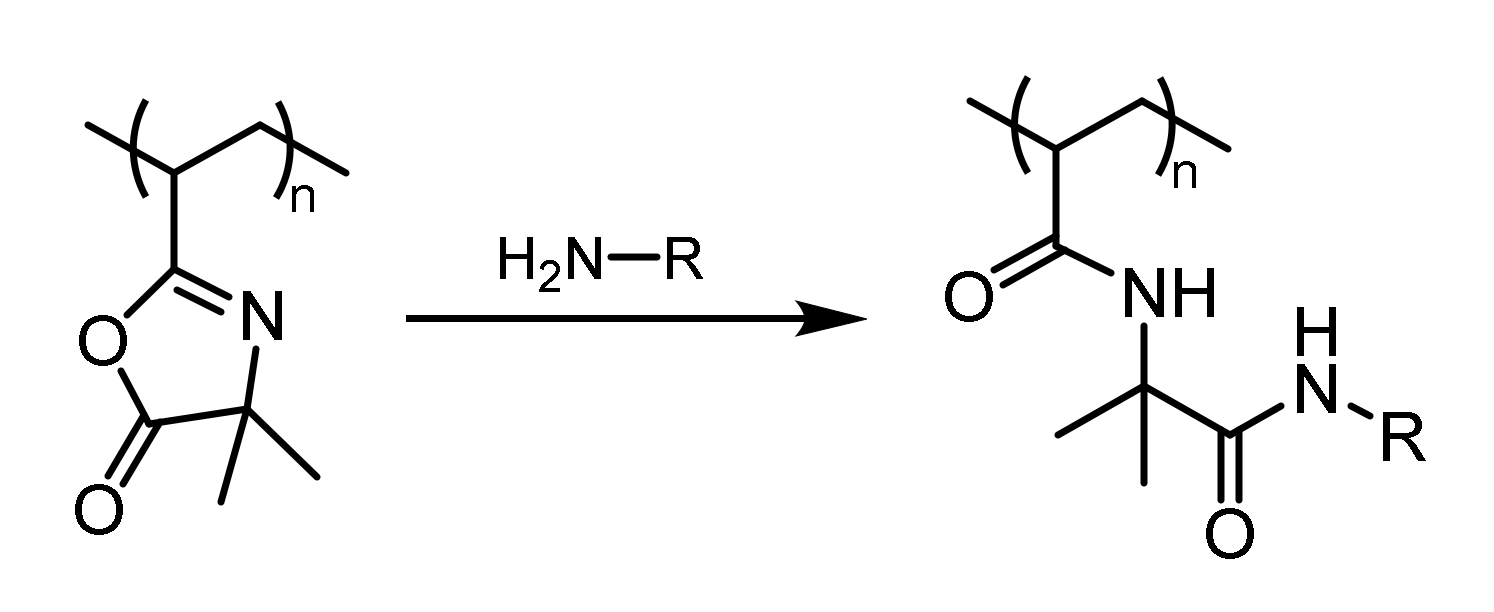
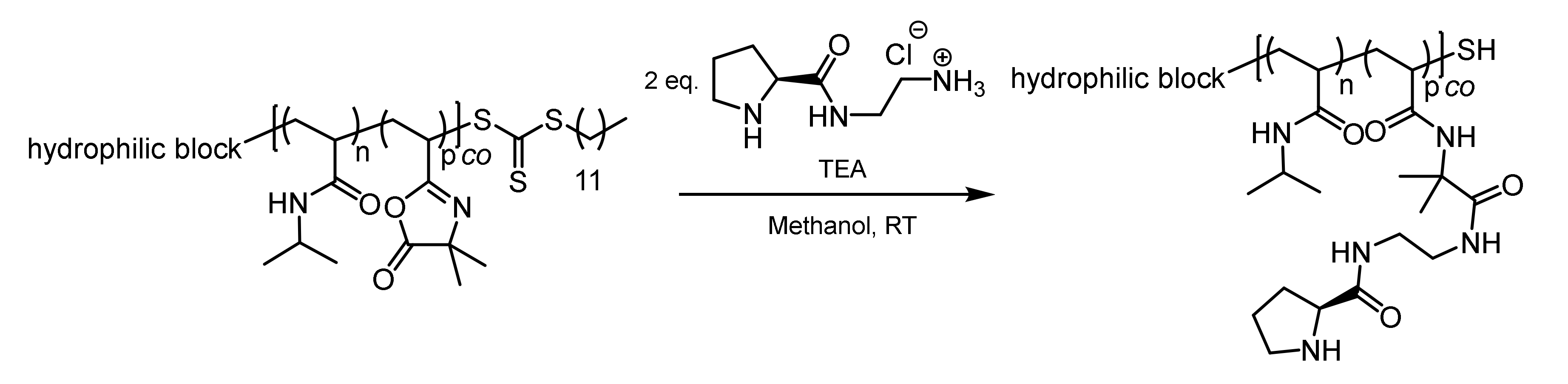
2.6. Post-Polymerization Attachment of the Organocatalysts
2.7. Hydrolysis of the Solketal Moieties
2.8. Temperature-Induced Aggregation of the Block Copolymers
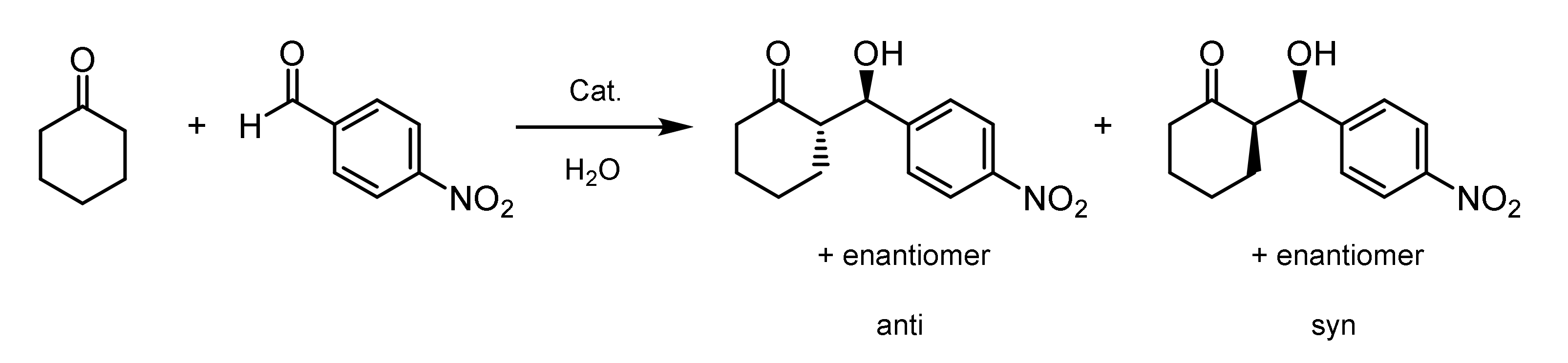
2.9. Asymmetric Aldol Reaction in Water
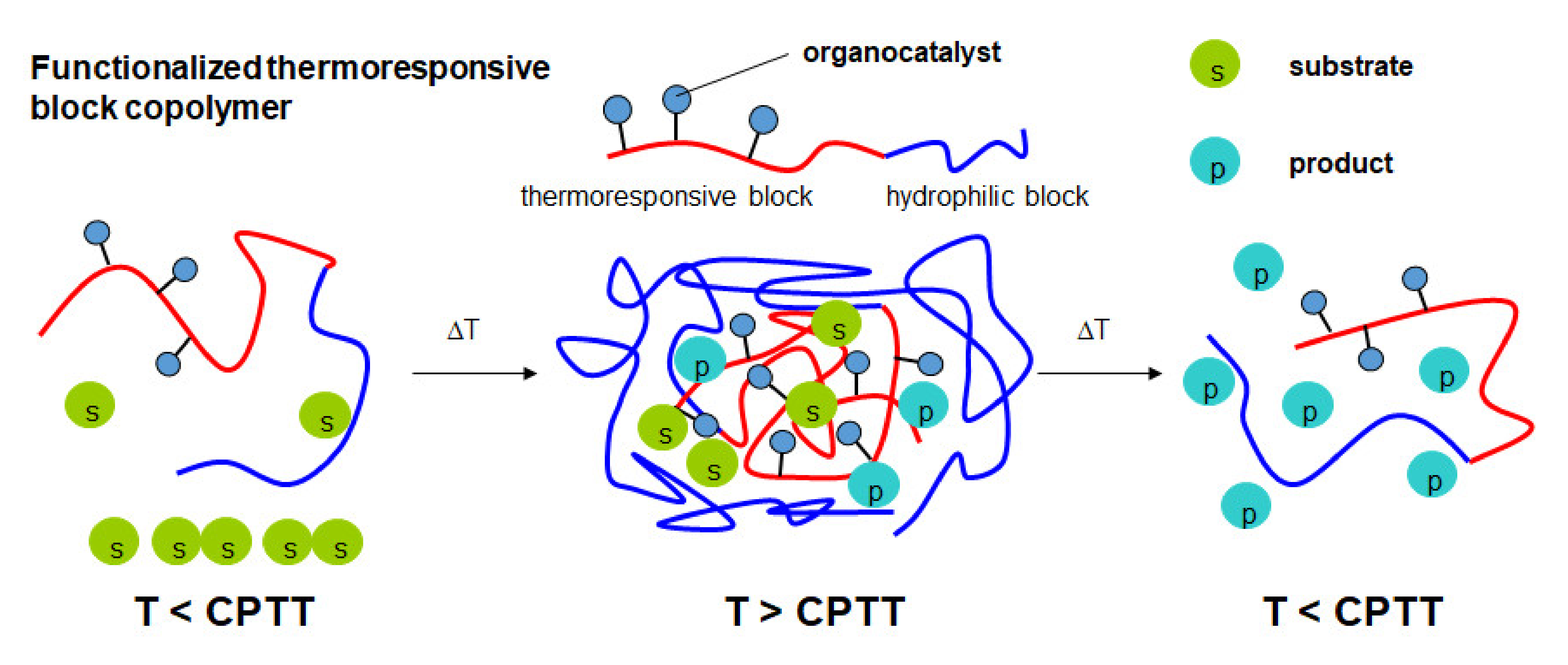
3. Results and Discussion
3.1. Synthesis of the Functionalized Block Copolymers
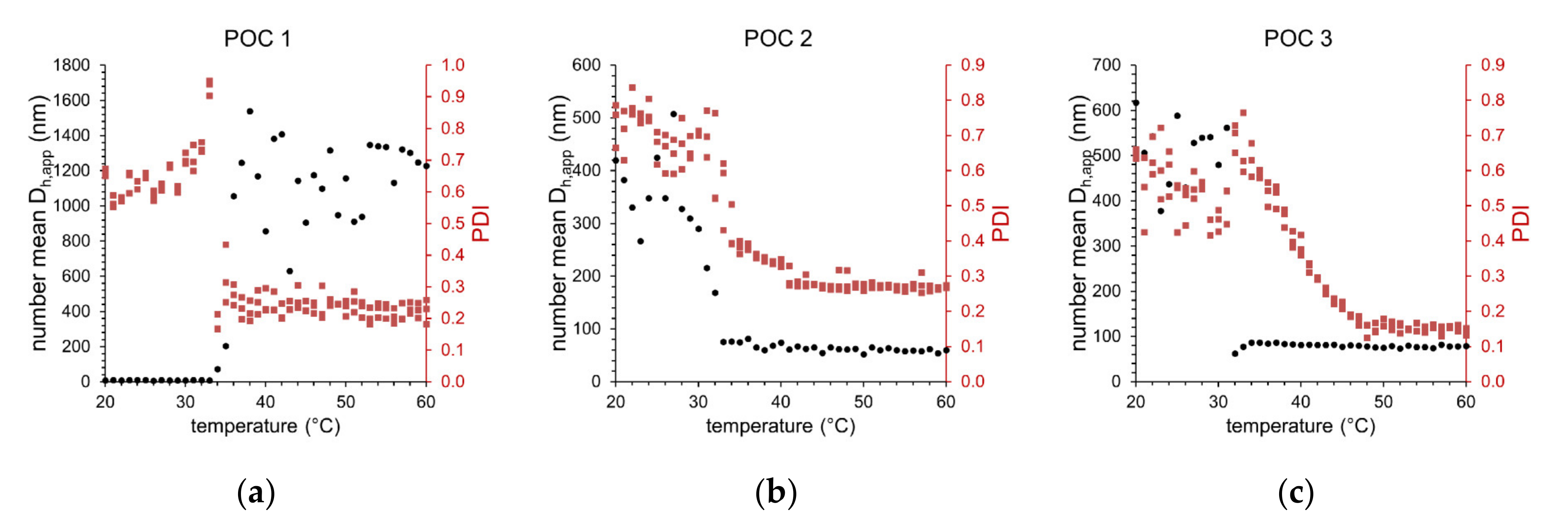
3.2. Temperature-Induced Self-Assembly of the Functionalized Thermoresponsive Block Copolymers
3.3. Micellar Organocatalysis in Water
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Anastas, P.; Eghbali, N. Green chemistry: Principles and practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Bergbreiter, D.E.; Kobayashi, S. Introduction to facilitated synthesis. Chem. Rev. 2009, 109, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Toy, P.H. Organic polymer supports for synthesis and for reagent and catalyst immobilization. Chem. Rev. 2009, 109, 815–838. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhao, S.; Zhang, Y.; Tang, Z. Metal-Organic Frameworks Encapsulating Active Nanoparticles as Emerging Composites for Catalysis: Recent Progress and Perspectives. Adv. Mater. 2018, 30, e1800702. [Google Scholar] [CrossRef]
- Bezerra, C.S.; Farias Lemos, C.M.G.d.; Sousa, M.d.; Gonçalves, L.R.B. Enzyme immobilization onto renewable polymeric matrixes: Past, present, and future trends. J. Appl. Polym. Sci. 2015, 132, 42125. [Google Scholar] [CrossRef]
- Döring, A.; Birnbaum, W.; Kuckling, D. Responsive hydrogels—Structurally and dimensionally optimized smart frameworks for applications in catalysis, micro-system technology and material science. Chem. Soc. Rev. 2013, 42, 7391–7420. [Google Scholar] [CrossRef]
- Li, S.; Lieberzeit, P.A.; Piletsky, S.; Turner, A.P.F. Smart Polymer Catalysts and Tunable Catalysis; Elsevier: Amsterdam, The Netherlands, 2019; ISBN 9780128118405. [Google Scholar]
- Dörr, M.; Zentel, R.; Dietrich, R.; Meerholz, K.; Bräuchle, C.; Wichern, J.; Zippel, S.; Boldt, P. Reactions on Vinyl Isocyanate/Maleimide Copolymers: NLO-functionalized Polymers with High Glass Transitions for Nonlinear Optical Applications. Macromolecules 1998, 31, 1454–1465. [Google Scholar] [CrossRef]
- Xu, F.J.; Cai, Q.J.; Li, Y.L.; Kang, E.T.; Neoh, K.G. Covalent immobilization of glucose oxidase on well-defined poly(glycidyl methacrylate)-Si(111) hybrids from surface-initiated atom-transfer radical polymerization. Biomacromolecules 2005, 6, 1012–1020. [Google Scholar] [CrossRef]
- Theato, P.; Kim, J.-U.; Lee, J.-C. Controlled Radical Polymerization of Active Ester Monomers: Precursor Polymers for Highly Functionalized Materials. Macromolecules 2004, 37, 5475–5478. [Google Scholar] [CrossRef]
- Gibson, M.I.; Fröhlich, E.; Klok, H.-A. Postpolymerization modification of poly(pentafluorophenyl methacrylate): Synthesis of a diverse water-soluble polymer library. J. Polym. Sci. A Polym. Chem. 2009, 47, 4332–4345. [Google Scholar] [CrossRef]
- Beyer, D.; Paulus, W.; Seitz, M.; Maxein, G.; Ringsdorf, H.; Eich, M. Second harmonic generation in self-assembled alternating multilayers of hemicyanine containing polymers and polyvinylamine. Thin Solid Films 1995, 271, 73–83. [Google Scholar] [CrossRef]
- Zhu, Y.; Batchelor, R.; Lowe, A.B.; Roth, P.J. Design of Thermoresponsive Polymers with Aqueous LCST, UCST, or Both: Modification of a Reactive Poly(2-vinyl-4,4-dimethylazlactone) Scaffold. Macromolecules 2016, 49, 672–680. [Google Scholar] [CrossRef]
- Rasmussen, J.K.; Heilmann, S.M.; Krepski, L.R.; Jensen, K.M.; Mickelson, J.; Johnson, K.; Coleman, P.L.; Milbrath, D.S.; Walker, M.M. Crosslinked, hydrophilic, azlactone-functional polymeric beads: A two-step approach. React. Polym. 1992, 16, 199–212. [Google Scholar] [CrossRef]
- Heilmann, S.M.; Rasmussen, J.K.; Krepski, L.R. Chemistry and technology of 2-alkenyl azlactones. J. Polym. Sci. A Polym. Chem. 2001, 39, 3655–3677. [Google Scholar] [CrossRef]
- Braunecker, W.A.; Matyjaszewski, K. Controlled/living radical polymerization: Features, developments, and perspectives. Prog. Polym. Sci. 2007, 32, 93–146. [Google Scholar] [CrossRef]
- Shipp, D.A. Reversible-Deactivation Radical Polymerizations. Polym. Rev. 2011, 51, 99–103. [Google Scholar] [CrossRef]
- Destarac, M. Industrial development of reversible-deactivation radical polymerization: Is the induction period over? Polym. Chem. 2018, 9, 4947–4967. [Google Scholar] [CrossRef]
- Guo, X.; Choi, B.; Feng, A.; Thang, S.H. Polymer Synthesis with More Than One Form of Living Polymerization Method. Macromol. Rapid Commun. 2018, 39, e1800479. [Google Scholar] [CrossRef]
- Gurnani, P.; Perrier, S. Controlled radical polymerization in dispersed systems for biological applications. Progr. Polym. Sci. 2020, 102, 101209. [Google Scholar] [CrossRef]
- Kurochkin, S.A.; Grachev, V.P. Reversible deactivation radical polymerization of polyfunctional monomers. Polym. Sci. Ser. C 2015, 57, 20–31. [Google Scholar] [CrossRef]
- Neve, J.d.; Haven, J.J.; Maes, L.; Junkers, T. Sequence-definition from controlled polymerization: The next generation of materials. Polym. Chem. 2018, 9, 4692–4705. [Google Scholar] [CrossRef]
- Xiong, Q.; Zhang, X.; Wei, W.; Wei, G.; Su, Z. Enzyme-mediated reversible deactivation radical polymerization for functional materials: Principles, synthesis, and applications. Polym. Chem. 2020, 11, 1673–1690. [Google Scholar] [CrossRef]
- Ni, Y.; Zhang, L.; Cheng, Z.; Zhu, X. Iodine-mediated reversible-deactivation radical polymerization: A powerful strategy for polymer synthesis. Polym. Chem. 2019, 10, 2504–2515. [Google Scholar] [CrossRef]
- Grubbs, R.B. Nitroxide-Mediated Radical Polymerization: Limitations and Versatility. Polym. Rev. 2011, 51, 104–137. [Google Scholar] [CrossRef]
- Ribelli, T.G.; Lorandi, F.; Fantin, M.; Matyjaszewski, K. Atom Transfer Radical Polymerization: Billion Times More Active Catalysts and New Initiation Systems. Macromol. Rapid Commun. 2019, 40, e1800616. [Google Scholar] [CrossRef]
- Theriot, J.C.; McCarthy, B.G.; Lim, C.-H.; Miyake, G.M. Organocatalyzed Atom Transfer Radical Polymerization: Perspectives on Catalyst Design and Performance. Macromol. Rapid Commun. 2017, 38. [Google Scholar] [CrossRef] [PubMed]
- Chmielarz, P.; Fantin, M.; Park, S.; Isse, A.A.; Gennaro, A.; Magenau, A.J.D.; Sobkowiak, A.; Matyjaszewski, K. Electrochemically mediated atom transfer radical polymerization (eATRP). Progr. Polym. Sci. 2017, 69, 47–78. [Google Scholar] [CrossRef]
- Poli, R.; Allan, L.E.N.; Shaver, M.P. Iron-mediated reversible deactivation controlled radical polymerization. Progr. Polym. Sci. 2014, 39, 1827–1845. [Google Scholar] [CrossRef]
- Moad, G. RAFT polymerization to form stimuli-responsive polymers. Polym. Chem. 2017, 8, 177–219. [Google Scholar] [CrossRef]
- Tully, D.C.; Roberts, M.J.; Geierstanger, B.H.; Grubbs, R.B. Synthesis of Reactive Poly(vinyl oxazolones) via Nitroxide-Mediated “Living” Free Radical Polymerization. Macromolecules 2003, 36, 4302–4308. [Google Scholar] [CrossRef]
- Fournier, D.; Pascual, S.; Fontaine, L. Copper-Mediated Living Radical Polymerization of 2-Vinyl-4,4-dimethyl-5-oxazolone. Macromolecules 2004, 37, 330–335. [Google Scholar] [CrossRef]
- Schilli, C.M.; Müller, A.H.E.; Rizzardo, E.; Thang, S.H.; Chong, Y.K. RAFT Polymers: Novel Precursors for Polymer—Protein Conjugates. In Advances in Controlled/Living Radical Polymerization; Matyjaszewski, K., Ed.; American Chemical Society: Washington, DC, USA, 2000; pp. 603–618. ISBN 0-8412-3854-5. [Google Scholar]
- Levere, M.E.; Ho, H.T.; Pascual, S.; Fontaine, L. Stable azlactone-functionalized nanoparticles prepared from thermoresponsive copolymers synthesized by RAFT polymerization. Polym. Chem. 2011, 2, 2878. [Google Scholar] [CrossRef]
- Ho, H.T.; Levere, M.E.; Pascual, S.; Montembault, V.; Casse, N.; Caruso, A.; Fontaine, L. Thermoresponsive block copolymers containing reactive azlactone groups and their bioconjugation with lysozyme. Polym. Chem. 2013, 4, 675–685. [Google Scholar] [CrossRef]
- Pedersen, K.J. The Decomposition of α-Nitrocarboxylic Acids. With Some Remarks on the Decomposition of β-Ketocarboxylic Acids. J. Phys. Chem. 1934, 38, 559–571. [Google Scholar] [CrossRef]
- Westheimer, F.H.; Jones, W.A. The Effect of Solvent on Some Reaction Rates. J. Am. Chem. Soc. 1941, 63, 3283–3286. [Google Scholar] [CrossRef]
- Hajos, Z.G.; Parrish, D.R. Asymmetric synthesis of bicyclic intermediates of natural product chemistry. J. Org. Chem. 1974, 39, 1615–1621. [Google Scholar] [CrossRef]
- Barbas, C.F. Organocatalysis lost: Modern chemistry, ancient chemistry, and an unseen biosynthetic apparatus. Angew. Chem. Int. Ed. Engl. 2008, 47, 42–47. [Google Scholar] [CrossRef]
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef]
- Chauhan, P.; Mahajan, S.; Enders, D. Achieving Molecular Complexity via Stereoselective Multiple Domino Reactions Promoted by a Secondary Amine Organocatalyst. Acc. Chem. Res. 2017, 50, 2809–2821. [Google Scholar] [CrossRef]
- Oliveira, V.; Cardoso, M.; Forezi, L. Organocatalysis: A Brief Overview on Its Evolution and Applications. Catalysts 2018, 8, 605. [Google Scholar] [CrossRef]
- Heravi, M.M.; Zadsirjan, V.; Dehghani, M.; Hosseintash, N. Current applications of organocatalysts in asymmetric aldol reactions: An update. Tetrahedron Asymmetry 2017, 28, 587–707. [Google Scholar] [CrossRef]
- Kondo, K.; Yamano, T.; Takemoto, K. Asymmetric Robinson cyclization reaction catalyzed by polymer-bound L-proline. Makromol. Chem. 1985, 186, 1781–1785. [Google Scholar] [CrossRef]
- Kristensen, T.E.; Hansen, T. Polymer-Supported Chiral Organocatalysts: Synthetic Strategies for the Road Towards Affordable Polymeric Immobilization. Eur. J. Org. Chem. 2010, 2010, 3179–3204. [Google Scholar] [CrossRef]
- Lu, A.; Smart, T.P.; Epps, T.H.; Longbottom, D.A.; O’Reilly, R.K. L-Proline Functionalized Polymers Prepared by RAFT Polymerization and Their Assemblies as Supported Organocatalysts. Macromolecules 2011, 44, 7233–7241. [Google Scholar] [CrossRef]
- Pedrosa, R.; Andrés, J.M.; Gamarra, A.; Manzano, R.; Pérez-López, C. Novel sulfonylpolystyrene-supported prolinamides as catalysts for enantioselective aldol reaction in water. Tetrahedron 2013, 69, 10811–10819. [Google Scholar] [CrossRef]
- Zhang, C.; Qiu, Y.; Bo, S.; Wang, F.; Wang, Y.; Liu, L.; Zhou, Y.; Niu, H.; Dong, H.; Satoh, T. Recyclable helical poly(phenylacetylene)-supported catalyst for asymmetric aldol reaction in aqueous media. J. Polym. Sci. A Polym. Chem. 2019, 57, 1024–1031. [Google Scholar] [CrossRef]
- Lu, A.; Moatsou, D.; Hands-Portman, I.; Longbottom, D.A.; O’Reilly, R.K. Recyclable l -Proline Functional Nanoreactors with Temperature-Tuned Activity Based on Core–Shell Nanogels. ACS Macro Lett. 2014, 3, 1235–1239. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, Q.; Wu, L.; Liu, K.; Wang, W.; Shen, Y.; Xue, Y.; Dai, S. L-proline functionalized pH-responsive copolymers as supported organocatalysts for asymmetric aldol reaction in water. React. Funct. Polym. 2020, 150, 104544. [Google Scholar] [CrossRef]
- Zayas, H.A.; Lu, A.; Valade, D.; Amir, F.; Jia, Z.; O’Reilly, R.K.; Monteiro, M.J. Thermoresponsive Polymer-Supported l -Proline Micelle Catalysts for the Direct Asymmetric Aldol Reaction in Water. ACS Macro Lett. 2013, 2, 327–331. [Google Scholar] [CrossRef]
- Kristensen, T.E.; Hansen, F.K.; Hansen, T. The Selective O -Acylation of Hydroxyproline as a Convenient Method for the Large-Scale Preparation of Novel Proline Polymers and Amphiphiles. Eur. J. Org. Chem. 2009, 2009, 387–395. [Google Scholar] [CrossRef]
- Yu, X.; Herberg, A.; Kuckling, D. Azlactone-functionalized smart block copolymers for organocatalyst immobilization. Eur. Polym. J. 2019, 120, 109207. [Google Scholar] [CrossRef]
- Lai, J.T.; Filla, D.; Shea, R. Functional Polymers from Novel Carboxyl-Terminated Trithiocarbonates as Highly Efficient RAFT Agents. Macromolecules 2002, 35, 6754–6756. [Google Scholar] [CrossRef]
- Yu, C.C.; Lee, Y.S.; Cheon, B.S.; Lee, S.H. Synthesis of Glycerol Monostearate with High Purity. Bull. Korean Chem. Soc. 2003, 24, 1229–1231. [Google Scholar]
- Cai-Yuan, P.; Lei, T.; De-Cheng, W. Synthesis and characterizations of the four-armed amphiphilic block copolymer S[poly(2,3-dihydroxypropyl acrylate)-block-poly(methyl acrylate)]4. J. Polym. Sci. A Polym. Chem. 2001, 39, 3062–3072. [Google Scholar] [CrossRef]
- Kipping, M.; Krahl, F.; Döring, A.; Adler, H.-J.P.; Kuckling, D. Synthesis and characterization of particles consisting of a biodegradable poly(l-lactide) core and a functional hydrophilic shell. Eur. Polym. J. 2010, 46, 313–323. [Google Scholar] [CrossRef]
- Discher, D.E.; Eisenberg, A. Polymer vesicles. Science 2002, 297, 967–973. [Google Scholar] [CrossRef]
- Liu, K.; Xu, W.; Wang, Q.; Tang, Y.; Sheng, W.; Shen, Y.; Shi, L. Self-assembly of L-proline functional thermoresponsive double hydrophlic block copolymers for aldol reaction in water: The influence of POEGA block content. Colloid Polym. Sci. 2018, 296, 1109–1117. [Google Scholar] [CrossRef]
- Kuckling, D.; Doering, A.; Krahl, F.; Arndt, K.-F. Stimuli-Responsive Polymer Systems. In Polymer Science: A Comprehensive; Matyjaszewski, K., Möller, M., Eds.; Elseiver: Amsterdam, The Netherlands, 2012; pp. 377–413. ISBN 9780080878621. [Google Scholar]
- Schild, H.G. Poly(N-isopropylacrylamide): Experiment, theory and application. Progr. Polym. Sci. 1992, 17, 163–249. [Google Scholar] [CrossRef]
- Ge, Z.; Liu, S. Supramolecular self-assembly of nonlinear amphiphilic and double hydrophilic block copolymers in aqueous solutions. Macromol. Rapid Commun. 2009, 30, 1523–1532. [Google Scholar] [CrossRef]
- Gruttadauria, M.; Giacalone, F.; Noto, R. Water in Stereoselective Organocatalytic Reactions. Adv. Synth. Catal. 2009, 351, 33–57. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | m/na (NMR) | Weight Ratio m/n | Đb | Mn,b (g/mol) |
|---|---|---|---|---|
| PSKAm-b-P(NIPAAmn-co-VDMAp) | 1.0: 8.7 | 1.0: 6.8 | 1.49 | 46,800 |
| PDMAAmm-b-P(NIPAAmn-co-VDMAp) | 1.0: 6.8 | 1.0: 7.8 | 1.91 | 55,900 |
| PEGm-b-P(NIPAAmn-co-VDMAp) | 1.0: 3.1 | 1.0: 8.1 | 1.43 | 39,700 |
| Sample | Polymer Composition |
|---|---|
| POC 1 | PEGm-b-P(NIPAAmn-co-VDMA-prolinamidep) |
| POC 2 | PDHPAm-b-P(NIPAAmn-co-VDMA-prolinamidep) |
| POC 3 | PDMAAm-b-P(NIPAAmn-co-VDMA-prolinamidep) |
| Sample | m/n/p (molar) a | Prolineamide-Content b (mol%) | m*/n*/p* (mass) c | Prolinamide-Content d/ (% w/w) |
|---|---|---|---|---|
| POC 1 | 1.00/3.09/0.13 | 3 | 44/349/42 | 10 |
| POC 2 | 1.00/10.00/0.53 | 5 | 140/1130/157 | 12 |
| POC 3 | 1.00/6.40/0.26 | 3 | 99/712/80 | 9 |
| Sample | CPTT (°C) | 25 °C | 40 °C | ||
|---|---|---|---|---|---|
| PDI | Number Mean Dh,app (nm) | PDI | Number Mean Dh,app (nm) | ||
| POC 1 + substrates | 35 | 0.65 | 11 | 0.25 | 856 |
| POC 2 + substrates | 32 | 0.67 | 425 | 0.34 | 74 |
| POC 3 + substrates | 32 | 0.51 | 588 | 0.38 | 82 |
| Catal. | T °C | V(H2O) (mL) | n(Catalyst)/ n(p-NBA) (%) | Conv. (%) | dra,b (Anti/Syn) NMR; HPLC | eeb (%) (Anti; Syn) |
|---|---|---|---|---|---|---|
| POC 1 | 25 | 3 | 8.1 | 10 | 75/25; 75/25 | 14; 23 |
| 40 | 3 | 8.1 | 58 | 83/17; 77/23 | 20; 6 | |
| 40 | 1 | 8.1 | 74 | 77/23; 79/21 | 33; 1 | |
| POC 2 | 25 | 3 | 8.0 | 6 | 85/15; 80/20 | 43; 22 |
| 40 | 3 | 8.0 | 55 | 80/20; 75/25 | 60; 23 | |
| 40 | 1 | 8.0 | 83 | 81/19; 65/35 | 82; 40 | |
| POC 3 | 25 | 3 | 8.0 | 10 | 85/15; 80/20 | 23; 25 |
| 40 | 3 | 8.0 | 39 | 82/18; 77/23 | 45; 2 | |
| 40 | 1 | 8.0 | 83 | 80/20; 59/41 | 61; 64 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, X.; Herberg, A.; Kuckling, D. Micellar Organocatalysis Using Smart Polymer Supports: Influence of Thermoresponsive Self-Assembly on Catalytic Activity. Polymers 2020, 12, 2265. https://doi.org/10.3390/polym12102265
Yu X, Herberg A, Kuckling D. Micellar Organocatalysis Using Smart Polymer Supports: Influence of Thermoresponsive Self-Assembly on Catalytic Activity. Polymers. 2020; 12(10):2265. https://doi.org/10.3390/polym12102265
Chicago/Turabian StyleYu, Xiaoqian, Artjom Herberg, and Dirk Kuckling. 2020. "Micellar Organocatalysis Using Smart Polymer Supports: Influence of Thermoresponsive Self-Assembly on Catalytic Activity" Polymers 12, no. 10: 2265. https://doi.org/10.3390/polym12102265
APA StyleYu, X., Herberg, A., & Kuckling, D. (2020). Micellar Organocatalysis Using Smart Polymer Supports: Influence of Thermoresponsive Self-Assembly on Catalytic Activity. Polymers, 12(10), 2265. https://doi.org/10.3390/polym12102265