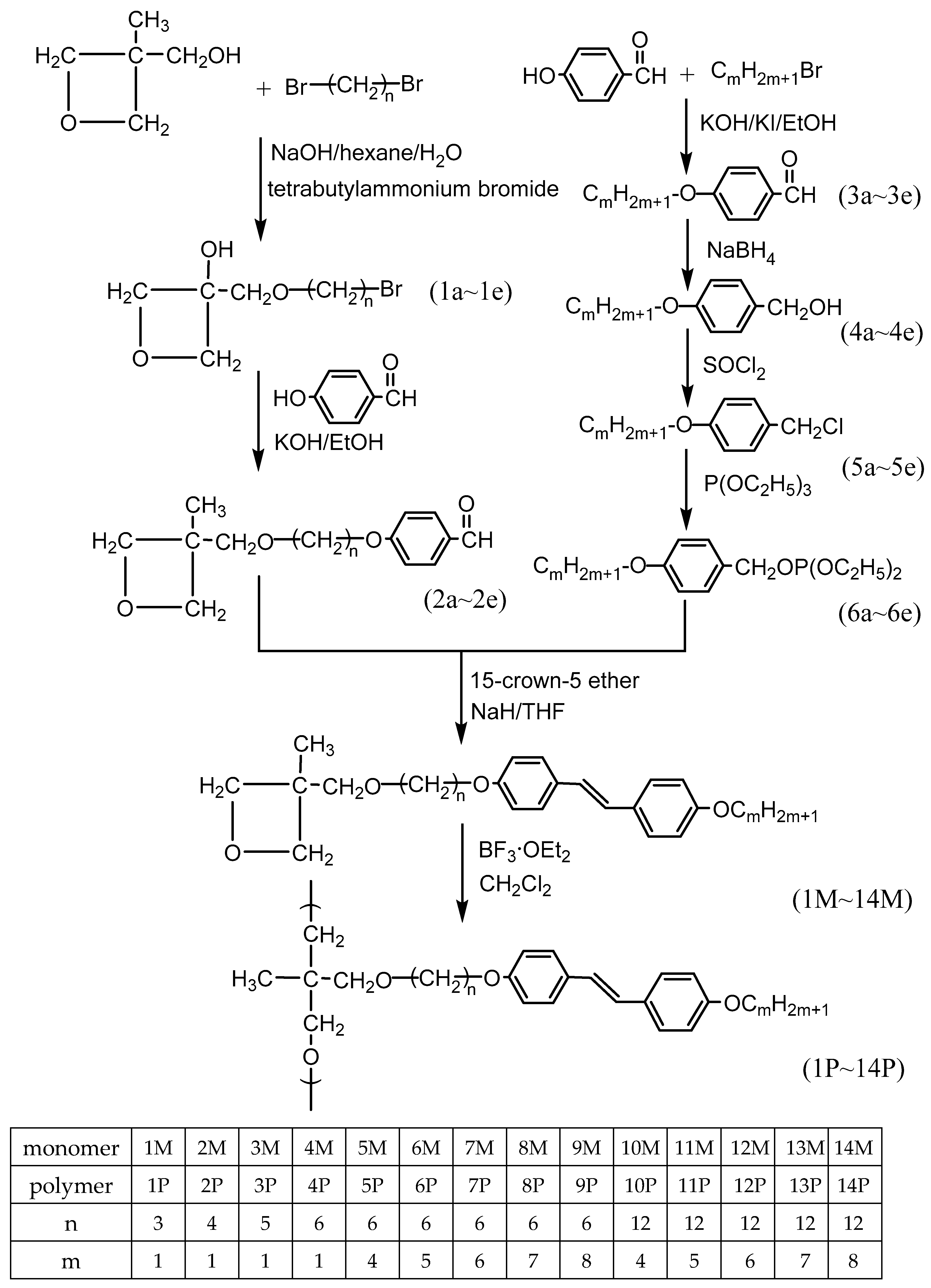
2.2. Synthesis
The intermediates and targets compound synthetic routes were represented in
Scheme 1. TLC and
1H-NMR spectroscopy were verified as the chemical structures and purity of the intermediates and target compounds. The synthesis methods and analysis of each product are described below.
3-[(3-Bromopropoxy)methyl]-3-methyloxetane (1a)
3-[(4-Bromobutoxy)methyl]-3-methyloxetane (1b)
3-[(5-Bromopentoxy)methyl]-3-methyloxetane (1c)
3-[(6-Bromohexoxy)methyl]-3-methyloxetane (1d)
3-[(12-Bromododecoxy)methyl]-3-methyloxetane (1e)
All five compounds were prepared by the same method. Taking compound 1d as an example, the synthesis is described below.
3-(Hydroxymethyl)-3-methyloxetane (10.0 g, 0.098 mol), dibromohexane (73.2 g, 0.299 mol), and hexane (120 mL) was added to a stirred solution of sodium hydroxide (64.7 g, 1.618 mol) in 150 mL of water. Then, tetrabutylammonium bromide (1.0 g) was added to the solution. The solution was stirred for 12 h at room temperature, then heated to reflux for 0.5 h. The reaction solution cooled to room temperature, 1000 mL of water was added, and the organic layer was extracted three times with hexane. The extraction solution was dried through anhydrous magnesium sulfate and after removal of the solvent under reduced pressure. The crude product was purified by distillation, to yield 20.85 g (80.3%) of a colorless transparent liquid. 1H-NMR (300 MHz, CDCl3, δ, ppm): 1.30 (s, 3H, –CH3 on the oxetane ring), 1.46–1.90 (m, 8H, –OCH2(CH2)4CH2–), 3.39–3.48 (m, 6H, –CH2OCH2(CH2)4CH2Br), 4.34, 4.51 (AB quartet, each 2H, –CH2–O on the oxetane ring).
3-[[3-(4-Hydroxybenzaldehyde)propoxy]methyl]-3-methyl oxetane (2a)
3-[[4-(4-Hydroxybenzaldehyde)butoxy]methyl]-3-methyl oxetane (2b)
3-[[5-(4-Hydroxybenzaldehyde)pentoxy]methyl]-3-methyl oxetane (2c)
3-[[6-(4-Hydroxybenzaldehyde)hexoxy]methyl]-3-methyl oxetane (2d)
3-[[12-(4-Hydroxybenzaldehyde)dodecanoxy]methyl]-3-methyl oxetane (2e)
4-Butoxy-benzaldehyde (3a)
4-Pentoxy-benzaldehyde (3b)
4-Hexoxy-benzaldehyde (3c)
4-Heptoxy-benzaldehyde (3d)
4-Octoxy-benzaldehyde (3e)
All ten compounds were prepared by the same method. Taking compound 2d as an example, the synthesis is described below.
4-Hydroxybenzaldehyde (4.45 g, 0.036 mol) was added to a stirred solution of potassium hydroxide (2.04 g, 0.036 mol) and potassium iodide (0.2 g) in 100 mL of 95% ethanol. Compound 1d (8.00 g, 0.030 mol) was added dropwise after the solution mentioned above was refluxed for 1 h. The solution refluxed for 12 h and then cooled to room temperature. The solution was extracted with water and ethyl acetate. The extraction solution was washed with 10% KOH solution three times and dried through anhydrous magnesium sulfate. After removal of the solvent under reduced pressure, the crude product was purified by column chromatography on silica gel using ethyl acetate/hexane as eluent to yield 2.42 g (82.5%) of light yellow liquid. 1H-NMR (300 MHz, CDCl3, δ, ppm): 1.30 (s, 3H, –CH3 on the oxetane ring), 1.40–1.82 (m, 8H, –OCH2(CH2)4CH2–), 3.46 (m, 4H, –CH2O–), 4.04 (t, 2H, –CH2–OPh), 4.33, 4.49 (AB quartet, each 2H, –CH2–O on the oxetane ring), 6.96 (d, 2H, aromatic protons), 7.81 (d, 2H, aromatic protons), 9.87 (s, 1H, aldehyde protons).
4-Butoxy-benzyl alcohol (4a)
4-Pentoxy-benzyl alcohol (4b)
4-Hexoxy-benzyl alcohol (4c)
4-Heptoxy-benzyl alcohol (4d)
4-Octoxy-benzyl alcohol (4e)
Taking compound 4c as an example, the synthesis is described below.
A solution of sodium tetrahydridoborate (3.28 g, 0.087 mol) in 2 mL of 0.45 N sodium hydroxide with 28 mL water was slowly added dropwise to 4-Hexoxy-benzaldehyde (14.9 g, 0.072 mol) with methanol (150 mL) solution. The solution was stirred 3 h at room temperature. Remove most of the methanol by distillation. The solution was extracted with ether and aqueous of dilute acid solution (50 mL). The organic phase was washed with 2% aqueous of sodium bicarbonate, saturated aqueous of sodium chloride, and dried through anhydrous magnesium sulfate. The crude product was purified by column chromatography on silica gel using n-hexane/ethyl acetate as eluent to yield 13.8 g (92.1%) of colorless liquid. 1H-NMR (300 MHz, CDCl3, δ, ppm): 0.89 (t, 3H, –CH3), 1.31–1.80 (m, 8H, –OCH2(CH2)4CH3), 3.96 (t, 2H, –OCH2(CH2)4–), 4.66 (s, 2H, Ph–CH2Cl), 6.89 (d, 2H, aromatic protons), 7.30 (d, 2H, aromatic protons).
4-Butoxy-benzyl chloride (5a)
4-Pentoxy-benzyl chloride (5b)
4-Hexoxy-benzyl chloride (5c)
4-Heptoxy-benzyl chloride (5d)
4-Octoxy-benzyl chloride (5e)
All five compounds were prepared by the same method. Taking compound 5c as an example, the synthesis is described below.
4-Hexoxy-benzyl alcohol (5 g, 0.024 mol) was reacted with excess thionyl chloride (15 mL, 0.0206 mol) in 50 mL of methylene chloride. The solution was stirred at ice bath for 6 h. Then, added with water (30 mL) into the solution. The organic phase was washed with 10% aqueous of sodium bicarbonate, saturated aqueous of sodium chloride, and dried through anhydrous magnesium sulfate. The crude product was purified by column chromatography on silica gel using n-hexane/ethyl acetate as eluent to yield 4.58 g (84.3%) of light-yellow liquid. 1H-NMR (300 MHz, CDCl3, δ, ppm): 0.89 (t, 3H, –CH3), 1.31–1.80 (m, 8H, –OCH2(CH2)4CH3), 3.96 (t, 2H, –OCH2(CH2)4–), 4.66 (s, 2H, Ph–CH2Cl), 6.89 (d, 2H, aromatic protons), 7.30 (d, 2H, aromatic protons).
Diethyl[(4-butoxy)benzyl]phosphonate (6a)
Diethyl[(4-pentoxy)benzyl]phosphonate (6b)
Diethyl[(4-hexoxy)benzyl]phosphonate (6c)
Diethyl[(4-heptoxy)benzyl]phosphonate (6d)
Diethyl[(4-octoxy)benzyl]phosphonate (6e)
All five compounds were prepared by the same method. Taking compound 6c as an example, the synthesis is described below.
4-Hexoxy-benzyl chloride (5.00 g, 0.022 mol) was injected by triethyl phosphite (11.0 g, 0.066 mol) with a syringe under nitrogen. The reaction mixture was refluxed for 12 h, then, was purified by distillation under reduced pressure to yield 5.89 g (81.6%) of colorless liquid. 1H-NMR (300 MHz, CDCl3, δ, ppm): 0.89 (t, 3H, –CH3), 1.22 (m, 6H, P(OCH2CH3)2, 1.31–1.79 (m, 8H, –OCH2(CH2)4CH3), 3.05 (d, 2H, PCH2Ph), 3.93–4.01 (m, 6H, –OCH2(CH2)4– and P(OCH2CH3)2), 6.89 (d, 2H, aromatic protons), 7.30 (d, 2H, aromatic protons).
3-[3-(Trans-4′-methoxystilben-4-yloxy)propoxymethyl]-3-methyl oxetane (1M)
3-[4-(Trans-4′-methoxystilben-4-yloxy)butoxymethyl]-3-methyl oxetane (2M)
3-[5-(Trans-4′-methoxystilben-4-yloxy)pentoxymethyl]-3-methyl oxetane (3M)
3-[6-(Trans-4′-methoxystilben-4-yloxy)hexoxymethyl]-3-methyl oxetane (4M)
3-[6-(Trans-4′-butoxystilben-4-yloxy)hexoxymethyl]-3-methyl oxetane (5M)
3-[6-(Trans-4′-pentoxystilben-4-yloxy)hexoxymethyl]-3-methyl oxetane (6M)
3-[6-(Trans-4′-hexoxystilben-4-yloxy)hexoxymethyl]-3-methyl oxetane (7M)
3-[6-(Trans-4′-heptoxystilben-4-yloxy)hexoxymethyl]-3-methyl oxetane (8M)
3-[6-(Trans-4′-octoxystilben-4-yloxy)hexoxymethyl]-3-methyl oxetane (9M)
3-[12-(Trans-4′-butoxystilben-4-yloxy)dodecoxymethyl]-3-methyl oxetane (10M)
3-[12-(Trans-4′-pentoxystilben-4-yloxy)dodecoxymethyl]-3-methyl oxetane (11M)
3-[12-(Trans-4′-hexoxystilben-4-yloxy)dodecoxymethyl]-3-methyl oxetane (12M)
3-[12-(Trans-4′-heptoxystilben-4-yloxy)dodecoxymethyl]-3-methyl oxetane (13M)
3-[12-(Trans-4′-octoxystilben-4-yloxy)dodecoxymethyl]-3-methyl oxetane (14M)
All fourteen monomers 1M~14M were prepared by the same method. Taking monomer 7M as an example, the synthesis is described below.
Sodium hydride (0.33 g, 0.013 mol) dissolved in dry THF (50 mL) on the brown flask, 13-crown-5-ether (30 mg) was added to react under nitrogen in the ice bath. Then, a solution of compound 2d (2.52 g, 0.008 mol) and compound 6d (2.6 g, 0.008 mol) was added dropwise to a stirred mixture. The reaction mixture was stirred for 12 h at room temperature. The reaction solution mixture was poured into ice water. The solution was filtered, the remaining yellow solid was recrystallized from dimethyl formamide to yield 1.62 g of light-yellow solid.
The lH-NMR spectrometer, the product yield, and element analysis of monomers 1M~14M were as follows.
Compound 1M: Yield: 40.7%; 1H-NMR (300 MHz, CDCl3, δ, ppm): 1.28 (s, 3H, –CH3 on the oxetane ring), 2.03 (t, 2H, –OCH2CH2–), 3.48 (s, 2H, –CH2O(CH2)3–), 3.62 (t, 2H, –CH2OCH2–), 3.80 (s, 3H, –PhOCH3), 4.04 (t, 2H, –CH2CH2OPh), 4.32, 4.48 (AB quartet, each 2H, –CH2– on the oxetane ring), 6.84, 7.38 (m, 6H, 4H, stilbene protons); element analysis: Calc. for C23H28O4: C 75.00, H 7.61, O 17.39; found C 75.00, H 7.75, O 17.25%.
Compound 2M: Yield: 38.6%; 1H-NMR (300 MHz, CDCl3, δ, ppm): 1.31 (s, 3H, –CH3 on the oxetane ring), 1.79 (m, 4H, –OCH2(CH2)2–), 3.49 (t, 4H, –CH2OCH2–), 3.82 (s, 3H, –PhOCH3), 3.98 (t, 2H, –CH2CH2OPh), 4.34, 4.50 (AB quartet, each 2H, –CH2– on the oxetane ring), 6.85, 7.39 (m, 6H, 4H, stilbene protons); element analysis: Calc. for C24H30O4: C 75.39, H 7.85, O 16.75; found C 75.46, H 7.99, O 16.45%.
Compound 3M: Yield: 33.4%; 1H-NMR (300 MHz, CDCl3, δ, ppm): 1.34 (s, 3H, –CH3 on the oxetane ring), 1.58–1.87 (m, 6H, –OCH2(CH2)3–), 3.51 (t, 4H, –CH2OCH2–), 3.85 (s, 3H, –PhOCH3), 3.99 (t, 2H, –CH2CH2OPh), 4.38, 4.53 (AB quartet, each 2H, –CH2– on the oxetane ring), 6.85, 7.43 (m, 6H, 4H, stilbene protons); element analysis: Calc. for C25H32O4: C 75.76, H 8.08, O 16.16; found C 75.52, H 8.15, O 16.33%.
Compound 4M: Yield: 30.8%; 1H-NMR (300 MHz, CDCl3, δ, ppm): 1.31 (s, 3H, –CH3 on the oxetane ring), 1.47–1.85 (m, 8H, –OCH2(CH2)4–), 3.46 (t, 4H, –CH2OCH2–), 3.81 (s, 3H, –PhOCH3), 3.99 (t, 2H, –CH2CH2OPh), 4.34, 4.50 (AB quartet, each 2H, –CH2– on the oxetane ring), 6.87, 7.26 (m, 6H, 4H, stilbene protons); element analysis: Calc. for C26H34O4: C 76.10, H 8.29, O 15.61; found C 75.82, H 8.47, O 15.71%.
Compound 5M: Yield: 40.6%; 1H-NMR (300 MHz, CDCl3, δ, ppm): 0.96 (t, 3H, –CH2–CH3), 1.31 (s, 3H, –CH3 on the oxetane ring), 1.47–1.80 (m, 12H, –OCH2(CH2)2CH3; –OCH2(CH2)4CH2–), 3.46 (t, 4H, –CH2OCH2–), 3.94 (t, 4H, –CH2–OPh–), 4.35, 4.51 (AB quartet, each 2H, –CH2– on the oxetane ring), 6.86, 7.39 (m, 6H, 4H, stilbene protons); element analysis: Calc. for C29H40O4: C 76.99, H 8.85, O 14.16; found C 77.03, H 9.02, O 13.95%.
Compound 6M: Yield: 36.7%; 1H-NMR (300 MHz, CDCl3, δ, ppm): 0.91 (t, 3H, –CH2–CH3), 1.31 (s, 3H, –CH3 on the oxetane ring), 1.41–1.79 (m, 14H, –OCH2(CH2)3CH3; –OCH2(CH2)4CH2–), 3.45 (t, 4H, –CH2OCH2–), 3.94 (t, 4H, –CH2–OPh–), 4.35, 4.50 (AB quartet, each 2H, –CH2– on the oxetane ring), 6.86, 7.39 (m, 6H, 4H, stilbene protons); element analysis: Calc. for C30H42O4: C 77.25, H 9.01, O 13.73; found C 77.21, H 9.15, O 13.64%.
Compound 7M: Yield: 41.0%; 1H-NMR (300 MHz, CDCl3, δ, ppm): 0.91 (t, 3H, –CH2–CH3), 1.31–1.80 (m, 19H, –CH3 on the oxetane ring, –OCH2(CH2)4CH3, –OCH2(CH2)4CH2–), 3.46 (t, 4H, –CH2OCH2–), 3.94 (t, 4H, –CH2–OPh–), 4.35, 4.50 (AB quartet, each 2H, –CH2– on the oxetane ring), 6.86, 7.39 (m, 6H, 4H, stilbene protons); element analysis: Calc. for C31H44O4: C 77.50, H 9.17, O 13.33; found C 77.47, H 9.26, O 13.27%.
Compound 8M: Yield: 40.6%; 1H-NMR (300 MHz, CDCl3, δ, ppm): 0.88 (t, 3H, –CH2–CH3), 1.26–1.80 (m, 21H, –CH3 on the oxetane ring, –OCH2(CH2)5CH3, –OCH2(CH2)4CH2–), 3.46 (t, 4H, –CH2OCH2–), 3.95 (t, 4H, –CH2–OPh–), 4.35, 4.50 (AB quartet, each 2H, –CH2– on the oxetane ring), 6.86, 7.39 (m, 6H, 4H, stilbene protons); element analysis: Calc. for C32H46O4: C 77.73, H 9.31, O 12.96; found C 77.60, H 9.40, O 13.00%.
Compound 9M: Yield: 38.6%; 1H-NMR (300 MHz, CDCl3, δ, ppm): 0.87 (t, 3H, –CH2–CH3), 1.26–1.82 (m, 23H, –CH3 on the oxetane ring, –OCH2(CH2)6CH3, –OCH2(CH2)4CH2–), 3.46 (t, 4H, –CH2OCH2–), 3.94 (t, 4H, –CH2–OPh–), 4.35, 4.50 (AB quartet, each 2H, –CH2– on the oxetane ring), 6.86, 7.39 (m, 6H, 4H, stilbene protons); element analysis: Calc. for C33H48O4: C 77.95, H 9.45, O 12.60; found C 77.92, H 9.54, O 12.54%.
Compound 10M: Yield: 34.7%; 1H-NMR (300 MHz, CDCl3, δ, ppm): 0.96 (t, 3H, –CH2–CH3), 1.29–1.79 (m, 27H, –CH3 on the oxetane ring, –OCH2(CH2)2CH3, –OCH2(CH2)10CH2–), 3.43 (t, 4H, –CH2OCH2–), 3.96 (t, 4H, –CH2–OPh–), 4.35, 4.50 (AB quartet, each 2H, –CH2– on the oxetane ring), 6.86, 7.39 (m, 6H, 4H, stilbene protons); element analysis: Calc. for C35H52O4: C 78.36, H 9.70, O 11.94; found C 78.22, H 9.80, O 11.98%.
Compound 11M: Yield: 39.1%; 1H-NMR (300 MHz, CDCl3, δ, ppm): 0.91 (t, 3H, –CH2–CH3), 1.27–1.78 (m, 29H, –CH3 on the oxetane ring, –OCH2(CH2)3CH3, –OCH2(CH2)10CH2–), 3.43 (t, 4H, –CH2OCH2–), 3.94 (t, 4H, –CH2–OPh–), 4.34, 4.50 (AB quartet, each 2H, –CH2– on the oxetane ring), 6.86, 7.39 (m, 6H, 4H, stilbene protons); element analysis: Calc. for C36H54O4: C 78.54, H 9.82, O 11.64; found C 78.26, H 9.90, O 11.84%.
Compound 12M: Yield: 24.1%; 1H-NMR (300 MHz, CDCl3, δ, ppm): 0.90 (t, 3H, –CH2–CH3), 1.26–1.79 (m, 31H, –CH3 on the oxetane ring, –OCH2(CH2)4CH3, –OCH2(CH2)10CH2–), 3.43 (t, 4H, –CH2OCH2–), 3.94 (t, 4H, –CH2–OPh–), 4.34, 4.50 (AB quartet, each 2H, –CH2– on the oxetane ring), 6.86, 7.39 (m, 6H, 4H, stilbene protons); element analysis: Calc. for C37H56O4: C 78.72, H 9.93, O 11.35; found C 78.40, H 10.05, O 11.55%.
Compound 13M: Yield: 27.4%; 1H-NMR (300 MHz, CDCl3, δ, ppm): 0.87 (t, 3H, –CH2–CH3), 1.26–1.77 (m, 33H, –CH3 on the oxetane ring, –OCH2(CH2)5CH3, –OCH2(CH2)10CH2–), 3.44 (t, 4H, –CH2OCH2–), 3.95 (t, 4H, –CH2–OPh–), 4.35, 4.49 (AB quartet, each 2H, –CH2– on the oxetane ring), 6.85, 7.38 (m, 6H, 4H, stilbene protons); element analysis: Calc. for C38H58O4: C 78.90, H 10.03, O 11.07; found C 78.73, H 10.11, O 11.16%.
Compound 14M: Yield: 34.7%; 1H-NMR (300 MHz, CDCl3, δ, ppm): 0.89 (t, 3H, –CH2–CH3), 1.29–1.81 (m, 35H, –CH3 on the oxetane ring, –OCH2(CH2)6CH3, –OCH2(CH2)10CH2–), 3.46 (t, 4H, –CH2OCH2–), 3.94 (t, 4H, –CH2–OPh–), 4.35, 4.50 (AB quartet, each 2H, –CH2– on the oxetane ring), 6.86, 7.39 (m, 6H, 4H, stilbene protons); element analysis: Calc. for C39H60O4: C 79.05, H 10.14, O 10.81; found C 78.92, H 10.20, O 10.88%.



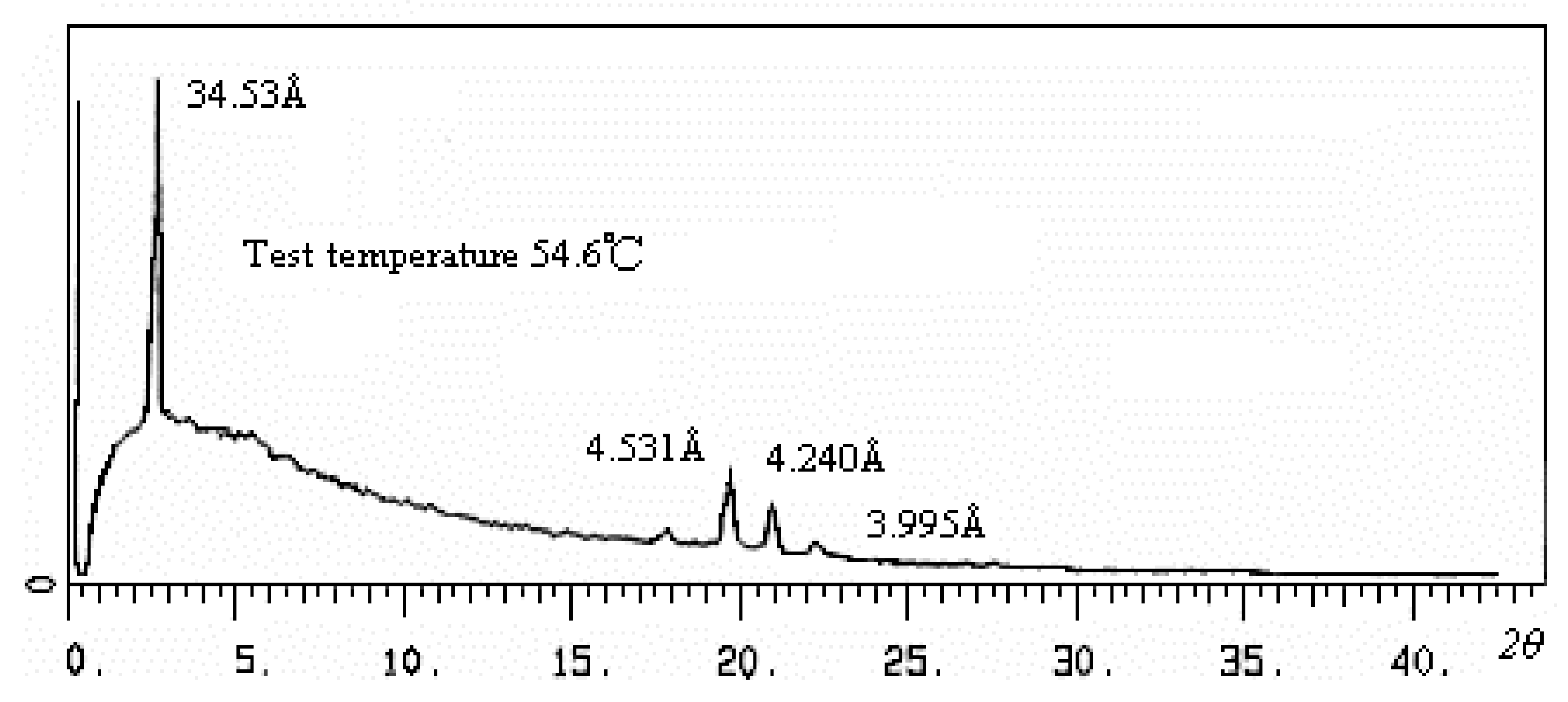


{kind=link}
{kind=link}
{kind=link}
{kind=link}