Mechanical Recycling of Partially Bio-Based and Recycled Polyethylene Terephthalate Blends by Reactive Extrusion with Poly(styrene-co-glycidyl methacrylate)

 , ,
, ,  ,
, 
 , and
, and 
Abstract
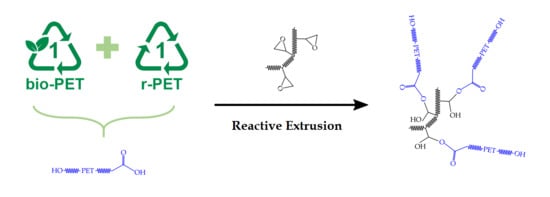
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of the Bio-PET/r-PET Blends
2.3. Mechanical Characterization
2.4. Color Measurements
2.5. Microscopy
2.6. Thermal Characterization
2.7. Thermomechanical Characterization
3. Results and Discussion
3.1. Mechanical Properties of the Bio-PET/r-PET Blends
3.2. Morphologgy and Appareance of the Bio-PET/r-PET Blends
3.3. Thermal Characterization of the Bio-PET/r-PET Blends
3.4. Thermomechanical Properties of the Bio-PET/r-PET Blends
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- European Commission. A European Strategy for Plastics in a Circular Economy; European Comission: Brussels, Belgium, 2018. [Google Scholar]
- Payne, J.; McKeown, P.; Jones, M.D. A circular economy approach to plastic waste. Polym. Degrad. Stab. 2019, 165, 170–181. [Google Scholar] [CrossRef]
- Niaounakis, M. Recycling of biopolymers—The patent perspective. Eur. Polym. J. 2019, 114, 464–475. [Google Scholar] [CrossRef]
- Torres-Giner, S.; Gil, L.; Pascual-Ramírez, L.; Garde-Belza, J.A. Packaging: Food waste reduction. Encycl. Polym. Appl. 2018, 3, 1990–2009. [Google Scholar]
- Tavares, A.A.; Silva, D.F.; Lima, P.S.; Andrade, D.L.; Silva, S.M.; Canedo, E.L. Chain extension of virgin and recycled polyethylene terephthalate. Polym. Test. 2016, 50, 26–32. [Google Scholar] [CrossRef]
- Leblanc, R. Recycling Polyethylene Terephthalate. Available online: https://www.thebalancesmb.com/recycling-polyethylene-terephthalate-pet-2877869 (accessed on 5 December 2019).
- Reuters. Coca-Cola Switches to Recycled Plastic for PET Bottles in Sweden. Available online: https://www.reuters.com/article/us-coca-cola-plastic/coca-cola-switches-to-recycled-plastic-for-pet-bottles-in-sweden-idUSKBN1XT1QS (accessed on 5 December 2019).
- European Bioplastics. Bio-Based Plastics—Fostering a Resource Efficient Circular Economy; European Bioplastics: Berlin, Germany, 2016. [Google Scholar]
- Van Den Oever, M.; Molenveld, K.; Van Der Zee, M.; Bos, H. Bio-based and biodegradable plastics: Facts and figures: Focus on food packaging in the Netherlands—Rapport nr. 1722. In Bio-Based and Biodegradable Plastics: Facts and Figures: Focus on Food Packaging in the Netherlands (No. 1722); Wageningen Food & Biobased Research: Wageningen, The Netherlands, 2017. [Google Scholar]
- European Bioplastics. Global Production Capacities of Bioplastics 2017–2022—Bioplastics Market Data 2017; European Bioplastics: Berlin, Germany, 2017. [Google Scholar]
- European Bioplastics. Available online: https://www.european-bioplastics.org/ (accessed on 28 October 2019).
- Park, S.H.; Kim, S.H. Poly(ethylene terephthalate) recycling for high value added textiles. Fash. Text. 2014, 1, 1. [Google Scholar] [CrossRef]
- Okuwaki, A. Feedstock recycling of plastics in Japan. Polym. Degrad. Stab. 2004, 85, 981–988. [Google Scholar] [CrossRef]
- Patel, M.; von Thienen, N.; Jochem, E.; Worrell, E. Recycling of plastics in Germany. Resour. Conserv. Recycl. 2000, 29, 65–90. [Google Scholar] [CrossRef]
- Dullius, J.; Ruecker, C.; Oliveira, V.; Ligabue, R.; Einloft, S. Chemical recycling of post-consumer PET: Alkyd resins synthesis. Prog. Org. Coat. 2006, 57, 123–127. [Google Scholar] [CrossRef]
- Shukla, S.; Harad, A.M.; Jawale, L.S. Recycling of waste PET into useful textile auxiliaries. Waste Manag. 2008, 28, 51–56. [Google Scholar] [CrossRef]
- Sogancioglu, M.; Yucel, A.; Yel, E.; Ahmetli, G. Production of Epoxy Composite from the Pyrolysis Char of Washed PET Wastes. Energy Procedia 2017, 118, 216–220. [Google Scholar] [CrossRef]
- Lee, J.H.; Lim, K.S.; Hahm, W.G.; Kim, S.H. Properties of recycled and virgin poly(ethylene terephthalate) blend fibers. J. Appl. Polym. Sci. 2013, 128, 1250–1256. [Google Scholar] [CrossRef]
- Srithep, Y.; Javadi, A.; Pilla, S.; Turng, L.-S.; Gong, S.; Clemons, C.; Peng, J. Processing and characterization of recycled poly(ethylene terephthalate) blends with chain extenders, thermoplastic elastomer, and/or poly(butylene adipate-co-terephthalate). Polym. Eng. Sci. 2011, 51, 1023–1032. [Google Scholar] [CrossRef]
- Garforth, A.A.; Ali, S.; Hernández-Martínez, J.; Akah, A. Feedstock recycling of polymer wastes. Curr. Opin. Solid State Mater. Sci. 2004, 8, 419–425. [Google Scholar] [CrossRef]
- Pawlak, A.; Pluta, M.; Morawiec, J.; Galeski, A.; Pracella, M. Characterization of scrap poly(ethylene terephthalate). Eur. Polym. J. 2000, 36, 1875–1884. [Google Scholar] [CrossRef]
- Pirzadeh, E.; Zadhoush, A.; Haghighat, M. Hydrolytic and thermal degradation of PET fibers and PET granule: The effects of crystallization, temperature, and humidity. J. Appl. Polym. Sci. 2007, 106, 1544–1549. [Google Scholar] [CrossRef]
- Elamri, A.; Lallam, A.; Harzallah, O.; Bencheikh, L. Mechanical characterization of melt spun fibers from recycled and virgin PET blends. J. Mater. Sci. 2007, 42, 8271–8278. [Google Scholar] [CrossRef]
- Scaffaro, R.; La Mantia, F. Characterization of monopolymer blend of virgin and recycled polyamide 6. Polym. Eng. Sci. 2002, 42, 2412–2417. [Google Scholar] [CrossRef]
- Elamri, A.; Abid, K.; Harzallah, O.; Lallam, A. Characterization of recycled/virgin PET polymers and their composites. Nano 2015, 3, 11–16. [Google Scholar]
- Coltelli, M.B.; Savi, S.; Della Maggiore, I.; Liuzzo, V.; Aglietto, M.; Ciardelli, F. A model study of Ti (OBu) 4 catalyzed reactions during reactive blending of polyethylene (PE) and poly(ethylene terephthalate) (PET). Macromol. Mater. Eng. 2004, 289, 400–412. [Google Scholar] [CrossRef]
- Duarte, I.S.; Tavares, A.A.; Lima, P.S.; Andrade, D.L.; Carvalho, L.H.; Canedo, E.L.; Silva, S.M. Chain extension of virgin and recycled poly(ethylene terephthalate): Effect of processing conditions and reprocessing. Polym. Degrad. Stab. 2016, 124, 26–34. [Google Scholar] [CrossRef]
- Li, J.; Tang, S.; Wu, Z.; Zheng, A.; Guan, Y.; Wei, D. Branching and cross-linking of poly(ethylene terephthalate) and its foaming properties. Polym. Sci. Ser. B 2017, 59, 164–172. [Google Scholar] [CrossRef]
- Torres, N.; Robin, J.; Boutevin, B. Study of thermal and mechanical properties of virgin and recycled poly(ethylene terephthalate) before and after injection molding. Eur. Polym. J. 2000, 36, 2075–2080. [Google Scholar] [CrossRef]
- Awaja, F.; Daver, F.; Kosior, E. Recycled poly(ethylene terephthalate) chain extension by a reactive extrusion process. Polym. Eng. Sci. 2004, 44, 1579–1587. [Google Scholar] [CrossRef]
- Incarnato, L.; Scarfato, P.; Di Maio, L.; Acierno, D. Structure and rheology of recycled PET modified by reactive extrusion. Polymer 2000, 41, 6825–6831. [Google Scholar] [CrossRef]
- Jacques, B.; Devaux, J.; Legras, R.; Nield, E. Reactions induced by triphenyl phosphite addition during melt mixing of poly(ethylene terephthalate)/poly(butylene terephthalate) blends: Influence on polyester molecular structure and thermal behaviour. Polymer 1996, 37, 1189–1200. [Google Scholar] [CrossRef]
- Cavalcanti, F.; Teofilo, E.; Rabello, M.; Silva, S. Chain extension and degradation during reactive processing of PET in the presence of triphenyl phosphite. Polym. Eng. Sci. 2007, 47, 2155–2163. [Google Scholar] [CrossRef]
- Torres, N.; Robin, J.; Boutevin, B. Chemical modification of virgin and recycled poly(ethylene terephthalate) by adding of chain extenders during processing. J. Appl. Polym. Sci. 2001, 79, 1816–1824. [Google Scholar] [CrossRef]
- Nascimento, C.R.; Azuma, C.; Bretas, R.; Farah, M.; Dias, M.L. Chain extension reaction in solid-state polymerization of recycled PET: The influence of 2, 2′-bis-2-oxazoline and pyromellitic anhydride. J. Appl. Polym. Sci. 2010, 115, 3177–3188. [Google Scholar] [CrossRef]
- Karayannidis, G.P.; Psalida, E.A. Chain extension of recycled poly(ethylene terephthalate) with 2, 2′-(1, 4-phenylene) bis (2-oxazoline). J. Appl. Polym. Sci. 2000, 77, 2206–2211. [Google Scholar] [CrossRef]
- Xu, X.; Ding, Y.; Qian, Z.; Wang, F.; Wen, B.; Zhou, H.; Zhang, S.; Yang, M. Degradation of poly(ethylene terephthalate)/clay nanocomposites during melt extrusion: Effect of clay catalysis and chain extension. Polym. Degrad. Stab. 2009, 94, 113–123. [Google Scholar] [CrossRef]
- Haralabakopoulos, A.; Tsiourvas, D.; Paleos, C. Chain extension of poly(ethylene terephthalate) by reactive blending using diepoxides. J. Appl. Polym. Sci. 1999, 71, 2121–2127. [Google Scholar] [CrossRef]
- Makkam, S.; Harnnarongchai, W. Rheological and mechanical properties of recycled PET modified by reactive extrusion. Energy Procedia 2014, 56, 547–553. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, W.; Zhang, H.; Wu, C. Influence of chain extension on the compatibilization and properties of recycled poly(ethylene terephthalate)/linear low density polyethylene blends. Polym. Degrad. Stab. 2009, 94, 1135–1141. [Google Scholar] [CrossRef]
- Huang, J.M. Polymer blends of poly(trimethylene terephthalate) and polystyrene compatibilized by styrene-glycidyl methacrylate copolymers. J. Appl. Polym. Sci. 2003, 88, 2247–2252. [Google Scholar] [CrossRef]
- Pracella, M.; Chionna, D.; Pawlak, A.; Galeski, A. Reactive mixing of PET and PET/PP blends with glycidyl methacrylate–modified styrene-b-(ethylene-co-olefin) block copolymers. J. Appl. Polym. Sci. 2005, 98, 2201–2211. [Google Scholar] [CrossRef]
- Tapia, J.J.B.; Tenorio-López, J.A.; Martínez-Estrada, A.; Guerrero-Sánchez, C. Application of RAFT-synthesized reactive tri-block copolymers for the recycling of post-consumer r-PET by melt processing. Mater. Chem. Phys. 2019, 229, 474–481. [Google Scholar] [CrossRef]
- Al-Sabagh, A.; Yehia, F.; Eshaq, G.; Rabie, A.; ElMetwally, A. Greener routes for recycling of polyethylene terephthalate. Egypt. J. Pet. 2016, 25, 53–64. [Google Scholar] [CrossRef]
- Montava-Jordà, S.; Torres-Giner, S.; Ferrandiz-Bou, S.; Quiles-Carrillo, L.; Montanes, N. Development of sustainable and cost-competitive injection-molded pieces of partially bio-based polyethylene terephthalate through the valorization of cotton textile waste. Int. J. Mol. Sci. 2019, 20, 1378. [Google Scholar] [CrossRef]
- Agüero, A.; Morcillo, M.d.C.; Quiles-Carrillo, L.; Balart, R.; Boronat, T.; Lascano, D.; Torres-Giner, S.; Fenollar, O. Study of the Influence of the Reprocessing Cycles on the Final Properties of Polylactide Pieces Obtained by Injection Molding. Polymers 2019, 11, 1908. [Google Scholar] [CrossRef]
- Negoro, T.; Thodsaratpreeyakul, W.; Takada, Y.; Thumsorn, S.; Inoya, H.; Hamada, H. Role of crystallinity on moisture absorption and mechanical performance of recycled PET compounds. Energy Procedia 2016, 89, 323–327. [Google Scholar] [CrossRef]
- Badia, J.; Vilaplana, F.; Karlsson, S.; Ribes-Greus, A. Thermal analysis as a quality tool for assessing the influence of thermo-mechanical degradation on recycled poly(ethylene terephthalate). Polym. Test. 2009, 28, 169–175. [Google Scholar] [CrossRef]
- Jabarin, S.A.; Lofgren, E.A. Effects of water absorption on physical properties and degree of molecular orientation of poly(ethylene terephthalate). Polym. Eng. Sci. 1986, 26, 620–625. [Google Scholar] [CrossRef]
- Mancini, S.D.; Zanin, M. Consecutive steps of PET recycling by injection: Evaluation of the procedure and of the mechanical properties. J. Appl. Polym. Sci. 2000, 76, 266–275. [Google Scholar] [CrossRef]
- Oromiehie, A.; Mamizadeh, A. Recycling PET beverage bottles and improving properties. Polym. Int. 2004, 53, 728–732. [Google Scholar] [CrossRef]
- Quiles-Carrillo, L.; Blanes-Martínez, M.M.; Montanes, N.; Fenollar, O.; Torres-Giner, S.; Balart, R. Reactive toughening of injection-molded polylactide pieces using maleinized hemp seed oil. Eur. Polym. J. 2018, 98, 402–410. [Google Scholar] [CrossRef]
- Piergiovanni, L.; Limbo, S. Plastic packaging materials. In Food Packaging Materials; Springer: Berlin, Germany, 2016; pp. 33–49. [Google Scholar]
- Quiles-Carrillo, L.; Montanes, N.; Garcia-Garcia, D.; Carbonell-Verdu, A.; Balart, R.; Torres-Giner, S. Effect of different compatibilizers on injection-molded green composite pieces based on polylactide filled with almond shell flour. Compos. Part B Eng. 2018, 147, 76–85. [Google Scholar] [CrossRef]
- Badia, J.; Strömberg, E.; Karlsson, S.; Ribes-Greus, A. The role of crystalline, mobile amorphous and rigid amorphous fractions in the performance of recycled poly (ethylene terephthalate)(PET). Polym. Degrad. Stab. 2012, 97, 98–107. [Google Scholar] [CrossRef]
- Vilaplana, F.; Karlsson, S.; Ribes-Greus, A. Changes in the microstructure and morphology of high-impact polystyrene subjected to multiple processing and thermo-oxidative degradation. Eur. Polym. J. 2007, 43, 4371–4381. [Google Scholar] [CrossRef]
- Strömberg, E.; Karlsson, S. The design of a test protocol to model the degradation of polyolefins during recycling and service life. J. Appl. Polym. Sci. 2009, 112, 1835–1844. [Google Scholar] [CrossRef]
- Quiles-Carrillo, L.; Duart, S.; Montanes, N.; Torres-Giner, S.; Balart, R. Enhancement of the mechanical and thermal properties of injection-molded polylactide parts by the addition of acrylated epoxidized soybean oil. Mater. Des. 2018, 140, 54–63. [Google Scholar] [CrossRef]
- Frounchi, M. Studies on degradation of PET in mechanical recycling. In Proceedings of the Macromolecular Symposia; Wiley-VCH: Weinheim, Germany, October 1999; pp. 465–469. [Google Scholar]
- Medellin–Rodriguez, F.; Phillips, P.; Lin, J.; Campos, R. The triple melting behavior of poly(ethylene terephthalate): Molecular weight effects. J. Polym. Sci. Part B Polym. Phys. 1997, 35, 1757–1774. [Google Scholar] [CrossRef]
- Awaja, F.; Daver, F.; Kosior, E.; Cser, F. The effect of chain extension on the thermal behaviour and crystallinity of reactive extruded recycled PET. J. Therm. Anal. Calorim. 2004, 78, 865–884. [Google Scholar] [CrossRef]
- Spinacé, M.A.S.; De Paoli, M.A. Characterization of poly(ethylene terephtalate) after multiple processing cycles. J. Appl. Polym. Sci. 2001, 80, 20–25. [Google Scholar] [CrossRef]
- Inata, H.; Matsumura, S. Chain extenders for polyesters. IV. Properties of the polyesters chain-extended by 2, 2′-bis (2-oxazoline). J. Appl. Polym. Sci. 1987, 33, 3069–3079. [Google Scholar] [CrossRef]
- Kiliaris, P.; Papaspyrides, C.D.; Pfaendner, R. Reactive-extrusion route for the closed-loop recycling of poly(ethylene terephthalate). J. Appl. Polym. Sci. 2007, 104, 1671–1678. [Google Scholar] [CrossRef]
- Vannier, A.; Duquesne, S.; Bourbigot, S.; Castrovinci, A.; Camino, G.; Delobel, R. The use of POSS as synergist in intumescent recycled poly(ethylene terephthalate). Polym. Degrad. Stab. 2008, 93, 818–826. [Google Scholar] [CrossRef]
- Levchik, S.V.; Weil, E.D. A review on thermal decomposition and combustion of thermoplastic polyesters. Polym. Adv. Technol. 2004, 15, 691–700. [Google Scholar] [CrossRef]
- Dziȩcioł, M.; Trzeszczyński, J. Temperature and atmosphere influences on smoke composition during thermal degradation of poly(ethylene terephthalate). J. Appl. Polym. Sci. 2001, 81, 3064–3068. [Google Scholar] [CrossRef]
- Dzie cioł, M.; Trzeszczyn’ ski, J. Studies of temperature influence on volatile thermal degradation products of poly(ethylene terephthalate). J. Appl. Polym. Sci. 1998, 69, 2377–2381. [Google Scholar] [CrossRef]
- Wang, B.; Sun, S. An investigation on the influence of molecular structure upon dynamic mechanical properties of polymer materials. Dev. Appl. Mater. 2005, 3, 109–118. [Google Scholar]
- Torres-Giner, S.; Montanes, N.; Fenollar, O.; García-Sanoguera, D.; Balart, R. Development and optimization of renewable vinyl plastisol/wood flour composites exposed to ultraviolet radiation. Mater. Des. 2016, 108, 648–658. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | bio-PET (wt%) | r-PET (wt%) | PS-co-GMA (phr) |
|---|---|---|---|
| B100 | 100 | - | - |
| R100 | - | 100 | - |
| B85-R15 | 85 | 15 | - |
| B70-R30 | 70 | 30 | - |
| B55-R45 | 55 | 45 | - |
| B55-R45-X1 | 55 | 45 | 1 |
| B55-R45-X3 | 55 | 45 | 3 |
| B55-R45-X5 | 55 | 45 | 5 |
| Piece | Et (MPa) | σmax (MPa) | εb (%) | Shore D Hardness | Impact Strength (kJ·m−2) |
|---|---|---|---|---|---|
| B100 | 600.3 ± 19.1 | 56.8 ± 0.8 | 494.6 ± 9.7 | 92.3 ± 0.9 | 2.87 ± 0.42 |
| R100 | 558.8 ± 11.7 | 56.1 ± 0.7 | 23.1 ± 4.7 | 79.8 ± 0.8 | 0.82 ± 0.13 |
| B85-R15 | 665.4 ± 19.2 | 56.6 ± 2.7 | 20.4 ± 4.9 | 77.8 ± 1.2 | 1.73 ± 0.06 |
| B70-R30 | 822.8 ± 25.6 | 57.5 ± 1.0 | 10.5 ± 1.1 | 77.3 ± 1.3 | 1.84 ± 0.29 |
| B55-R45 | 820.5 ± 30.2 | 57.7 ± 2.8 | 10.8 ± 1.4 | 76.0 ± 1.4 | 1.84 ± 0.38 |
| B55-R45-X1 | 742.9 ± 10.9 | 58.1 ± 1.1 | 11.2 ± 0.9 | 79.8 ± 1.5 | 1.89 ± 0.24 |
| B55-R45-X3 | 852.9 ± 21.8 | 58.3 ± 0.3 | 312.9 ± 7.3 | 81.0 ± 1.2 | 2.43 ± 0.29 |
| B55-R45-X5 | 847.8 ± 11.7 | 57.4 ± 0.1 | 378.8 ± 8.4 | 82.8 ±1.8 | 2.52 ± 0.25 |
| Piece | L* | a* | b* | ΔE* |
|---|---|---|---|---|
| B100 | 75.67 ± 0.58 | −2.72 ± 0.16 | −5.27 ± 0.32 | - |
| R100 | 42.86 ± 0.37 | −0.54 ± 0.11 | −2.62 ± 0.10 | 32.99 ± 1.31 |
| B85-R15 | 68.03 ± 0.44 | −3.63 ± 0.07 | 5.27 ± 0.25 | 10.84 ± 0.57 |
| B70-R30 | 64.85 ± 0.48 | −2.95 ± 0.14 | 5.62 ± 0.18 | 15.35 ± 1.18 |
| B55-R45 | 63.44 ± 0.96 | −2.61 ± 0.14 | 5.41 ± 0.50 | 16.24 ± 0.90 |
| B55-R45-X1 | 69.88 ± 0.52 | −1.97 ± 0.17 | 0.57 ± 0.23 | 8.26 ± 1.13 |
| B55-R45-X3 | 72.71 ± 0.86 | −2.56 ± 0.13 | 0.33 ± 0.20 | 6.34 ± 0.77 |
| B55-R45-X5 | 73.73 ± 0.98 | −2.34 ± 0.17 | 1.35 ± 0.31 | 6.91 ± 0.84 |
| Piece | Tg (°C) | Tcc (°C) | Tc (°C) | Tm1 (°C) | Tm2 (°C) | χc (%) |
|---|---|---|---|---|---|---|
| B100 | 82.0 ± 0.7 | 150.8 ± 0.4 | - | 244.9 ± 0.9 | - | 10.6 ± 0.5 |
| R100 | 80. 4± 0.8 | 157.6 ± 0.2 | - | 244.5 ± 1.1 | - | 24.2 ± 0.5 |
| B85-R15 | 81.2 ± 0.6 | - | 182.7 ± 0.3 | 238.4 ± 0.8 | 247.4 ± 1.0 | 29.0 ± 0.8 |
| B70-R30 | 81.5 ± 0.8 | - | 185.1 ± 0.2 | 238.9 ± 0.7 | 247.2 ± 0.8 | 32.3 ± 0.9 |
| B55-R45 | 82.6 ± 0.9 | - | 191.2 ± 0.3 | 237.9 ± 1.0 | 247.8 ± 1.1 | 37.0 ± 0.7 |
| B55-R45-X1 | 81.0 ± 0.7 | - | 191.4 ± 0.1 | 238.0 ± 1.1 | 247.5 ± 0.9 | 38.1 ± 0.8 |
| B55-R45-X3 | 80.9 ± 0.8 | - | 187.4 ± 0.2 | 236.8 ± 0.8 | 247.9 ± 0.8 | 17.7 ± 0.5 |
| B55-R45-X5 | 80.6 ± 0.7 | - | 189.8 ± 0.2 | 237.1 ± 0.7 | 247.9 ± 1.2 | 25.9 ± 0.9 |
| Piece | T5% (°C) | Tdeg1 (°C) | Tdeg2 (°C) | Residual Mass (%) |
|---|---|---|---|---|
| B100 | 405.4 ± 0.2 | 446.8 ± 0.1 | 546.0 ± 0.2 | 1.26 ± 0.04 |
| R100 | 400.8 ± 0.4 | 452.3 ± 0.2 | 552.7 ± 0.1 | 1.43 ± 0.07 |
| B85-R15 | 400.2 ± 0.2 | 452.2 ± 0.3 | 549.4 ± 0.2 | 1.22 ± 0.05 |
| B70-R30 | 393.2 ± 0.3 | 452.3 ± 0.2 | 545.7 ± 0.1 | 1.64 ± 0.18 |
| B55-R45 | 393.0 ± 0.2 | 452.9 ± 0.3 | 559.5 ± 0.2 | 2.18 ± 0.05 |
| B55-R45-X1 | 403.2 ± 0.2 | 452.9 ± 0.2 | 578.3 ± 0.2 | 1.27 ± 0.08 |
| B55-R45-X3 | 403.1 ± 0.1 | 452.4 ± 0.1 | 576.6 ± 0.4 | 1.73 ± 0.15 |
| B55-R45-X5 | 403.0 ± 0.1 | 451.8 ± 0.2 | 576.1 ± 0.2 | 2.29 ± 0.06 |
| Piece | Storage Modulus (MPa) | Tg (°C) | CLTE (µm·m−1·°C−1) | ||
|---|---|---|---|---|---|
| 60 °C | 100 °C | Below Tg | Above Tg | ||
| B100 | 927.4 ± 20.5 | 2.8 ± 0.1 | 81.0 ± 0.1 | 78.9 ± 1.9 | 80.5 ± 1.9 |
| R100 | 1018.5 ± 20.2 | 1.7 ± 0.2 | 80.3 ± 0.1 | 90.9 ± 1.7 | 94.9 ± 0.9 |
| B85-R15 | 943.4 ± 23.2 | 2.5 ± 0.1 | 81.2 ± 0.1 | 80.7 ± 1.1 | 90.1 ± 1.0 |
| B70-R30 | 931.6 ± 32.3 | 2.3 ± 0.1 | 81.3 ± 0.1 | 84.1 ± 1.1 | 103.2 ± 0.2 |
| B55-R45 | 924.3 ± 13.2 | 2.6 ± 0.1 | 81.5 ± 0.1 | 93.1 ± 1.2 | 111.8 ± 0.9 |
| B55-R45-X1 | 977.7 ± 17.3 | 3.4 ± 0.2 | 81.1 ± 0.1 | 90.1 ± 0.5 | 104.0 ± 1.4 |
| B55-R45-X3 | 984.4 ± 27.0 | 4.7 ± 0.2 | 80.9 ± 0.2 | 74.9 ± 0.1 | 101.3 ± 1.1 |
| B55-R45-X5 | 1019.7 ± 18.4 | 4.9 ± 0.2 | 80.8 ± 0.2 | 70.6 ± 0.3 | 99.9 ± 1.8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montava-Jorda, S.; Lascano, D.; Quiles-Carrillo, L.; Montanes, N.; Boronat, T.; Martinez-Sanz, A.V.; Ferrandiz-Bou, S.; Torres-Giner, S. Mechanical Recycling of Partially Bio-Based and Recycled Polyethylene Terephthalate Blends by Reactive Extrusion with Poly(styrene-co-glycidyl methacrylate). Polymers 2020, 12, 174. https://doi.org/10.3390/polym12010174
Montava-Jorda S, Lascano D, Quiles-Carrillo L, Montanes N, Boronat T, Martinez-Sanz AV, Ferrandiz-Bou S, Torres-Giner S. Mechanical Recycling of Partially Bio-Based and Recycled Polyethylene Terephthalate Blends by Reactive Extrusion with Poly(styrene-co-glycidyl methacrylate). Polymers. 2020; 12(1):174. https://doi.org/10.3390/polym12010174
Chicago/Turabian StyleMontava-Jorda, Sergi, Diego Lascano, Luis Quiles-Carrillo, Nestor Montanes, Teodomiro Boronat, Antonio Vicente Martinez-Sanz, Santiago Ferrandiz-Bou, and Sergio Torres-Giner. 2020. "Mechanical Recycling of Partially Bio-Based and Recycled Polyethylene Terephthalate Blends by Reactive Extrusion with Poly(styrene-co-glycidyl methacrylate)" Polymers 12, no. 1: 174. https://doi.org/10.3390/polym12010174
APA StyleMontava-Jorda, S., Lascano, D., Quiles-Carrillo, L., Montanes, N., Boronat, T., Martinez-Sanz, A. V., Ferrandiz-Bou, S., & Torres-Giner, S. (2020). Mechanical Recycling of Partially Bio-Based and Recycled Polyethylene Terephthalate Blends by Reactive Extrusion with Poly(styrene-co-glycidyl methacrylate). Polymers, 12(1), 174. https://doi.org/10.3390/polym12010174