Machine-Learning-Assisted De Novo Design of Organic Molecules and Polymers: Opportunities and Challenges

 ,
,  ,
, 
Abstract
1. Introduction
2. Case Studies of ML-Assisted Materials Design
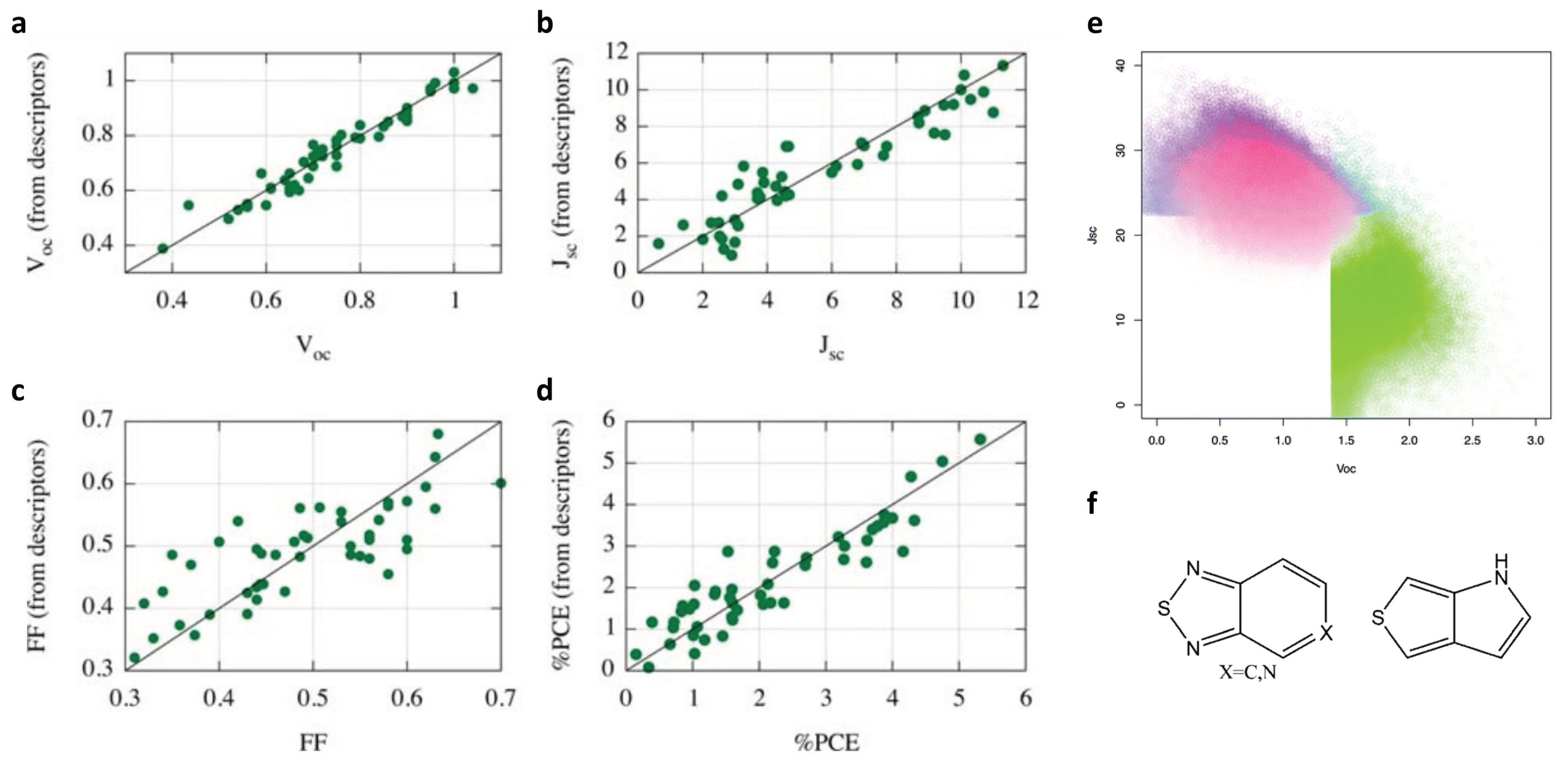
2.1. Molecular Design of Organic Photovoltaics (OPV)
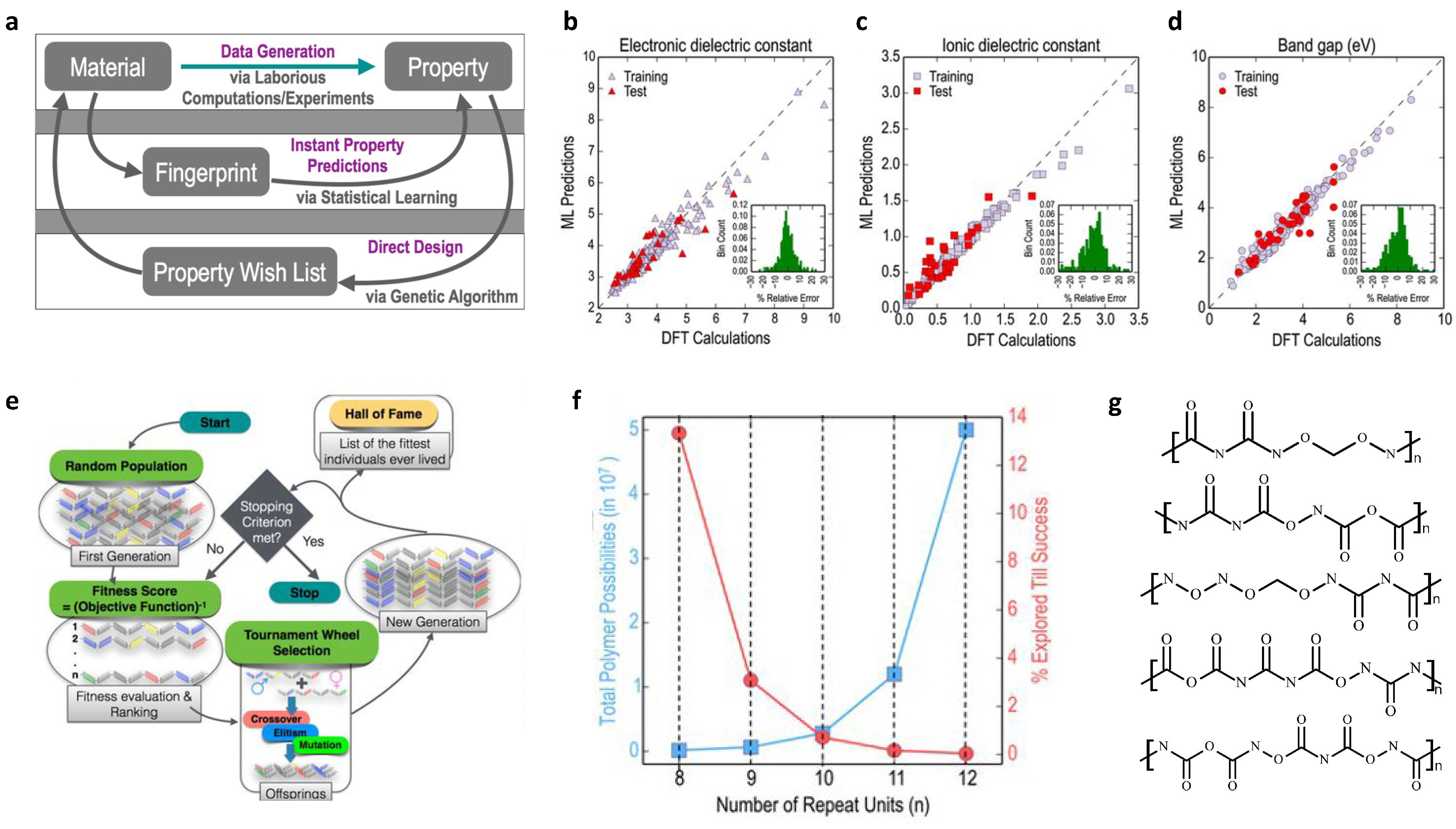
2.2. Design of Polymer Dielectrics
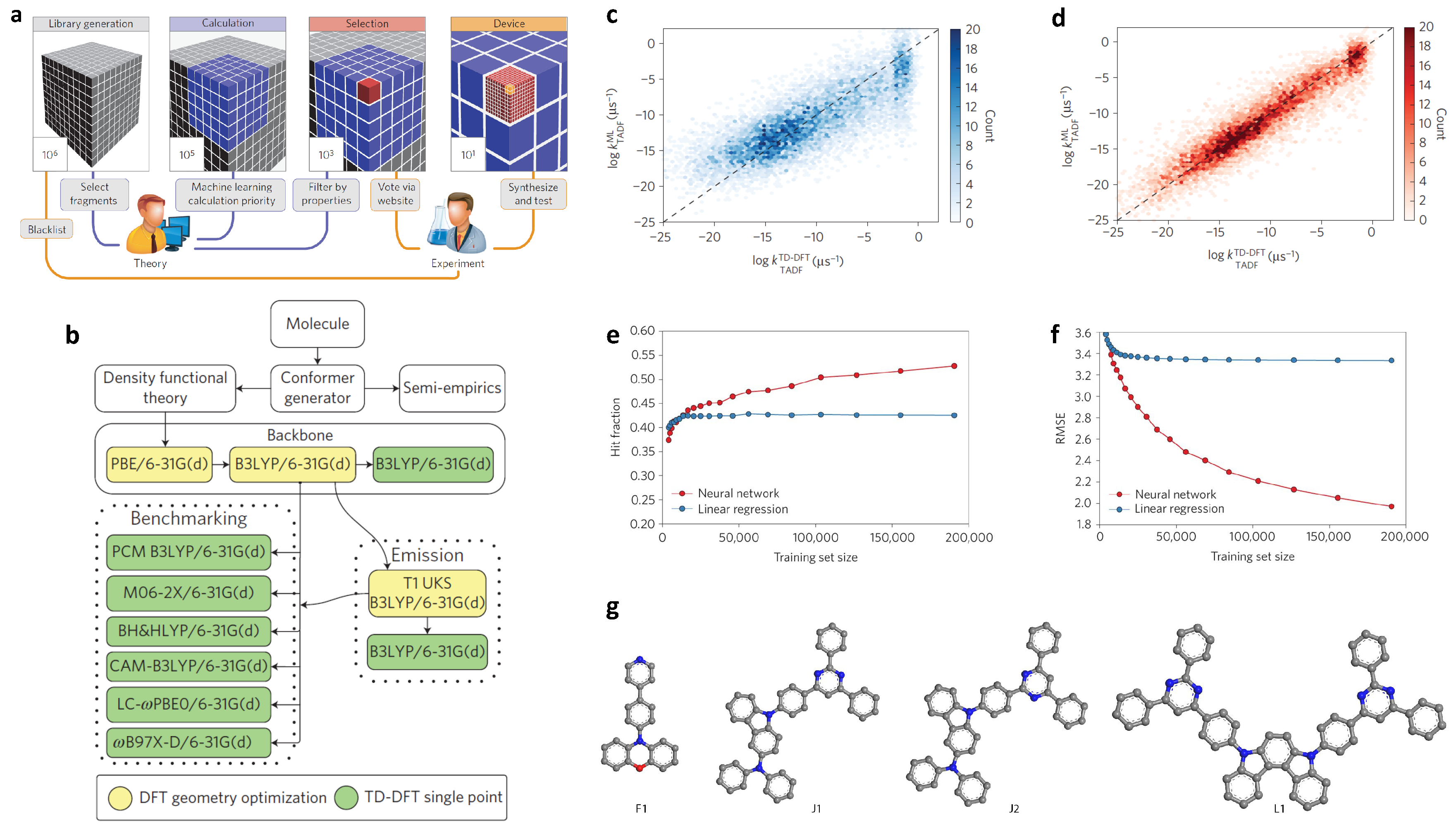
2.3. Molecular Design of Organic Light-Emitting Diodes
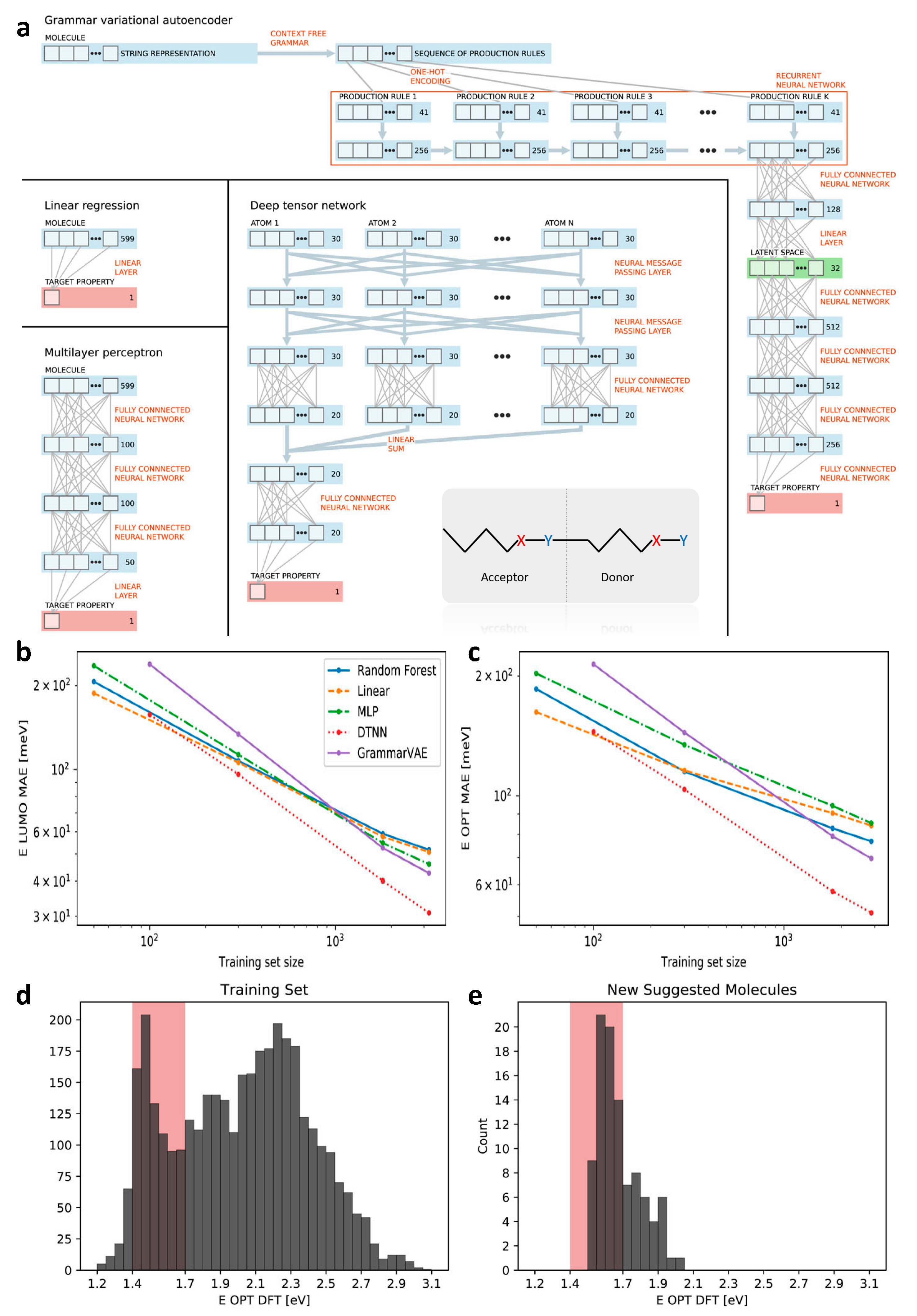
2.4. Design of Polymeric Solar Cell
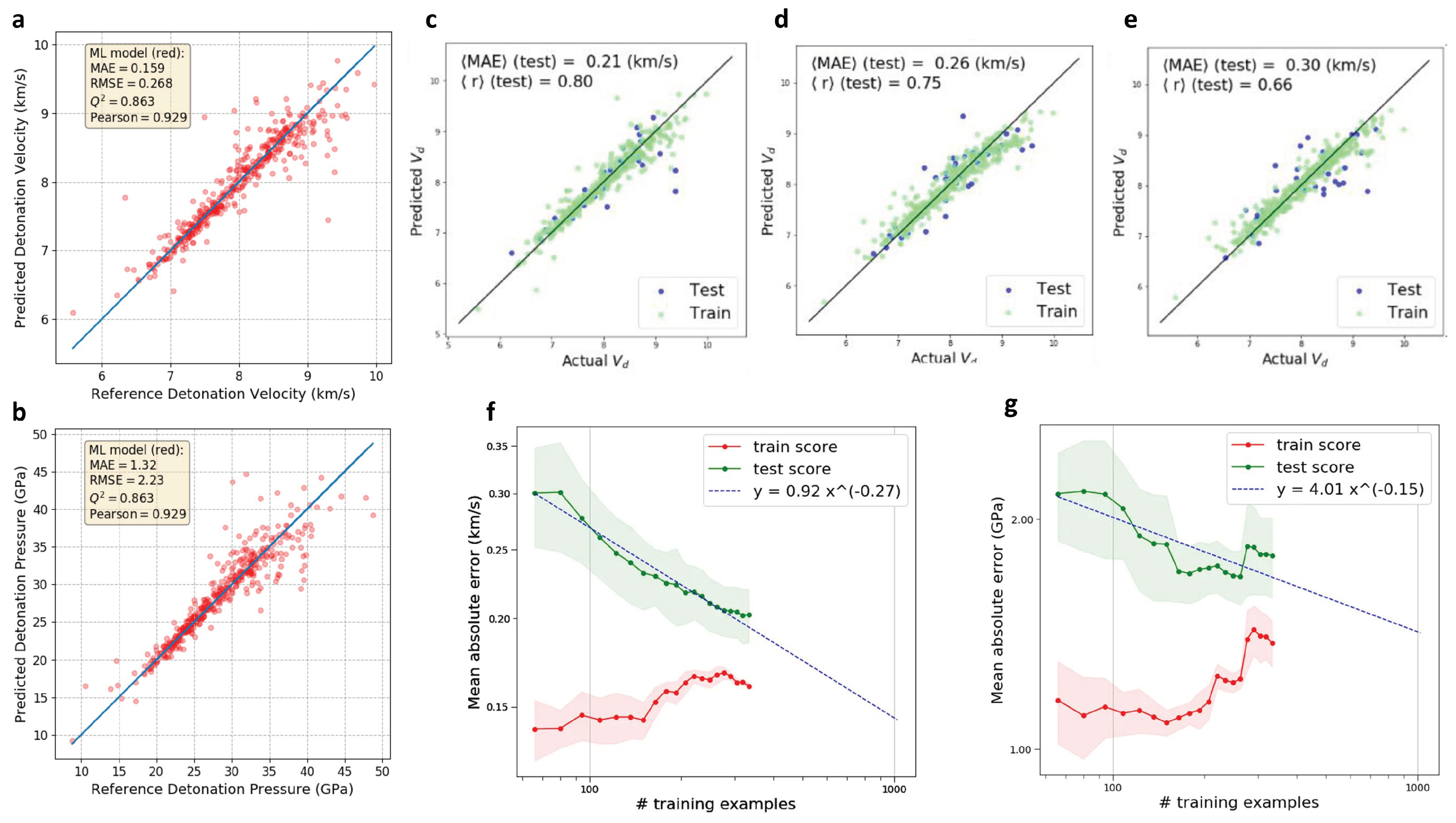
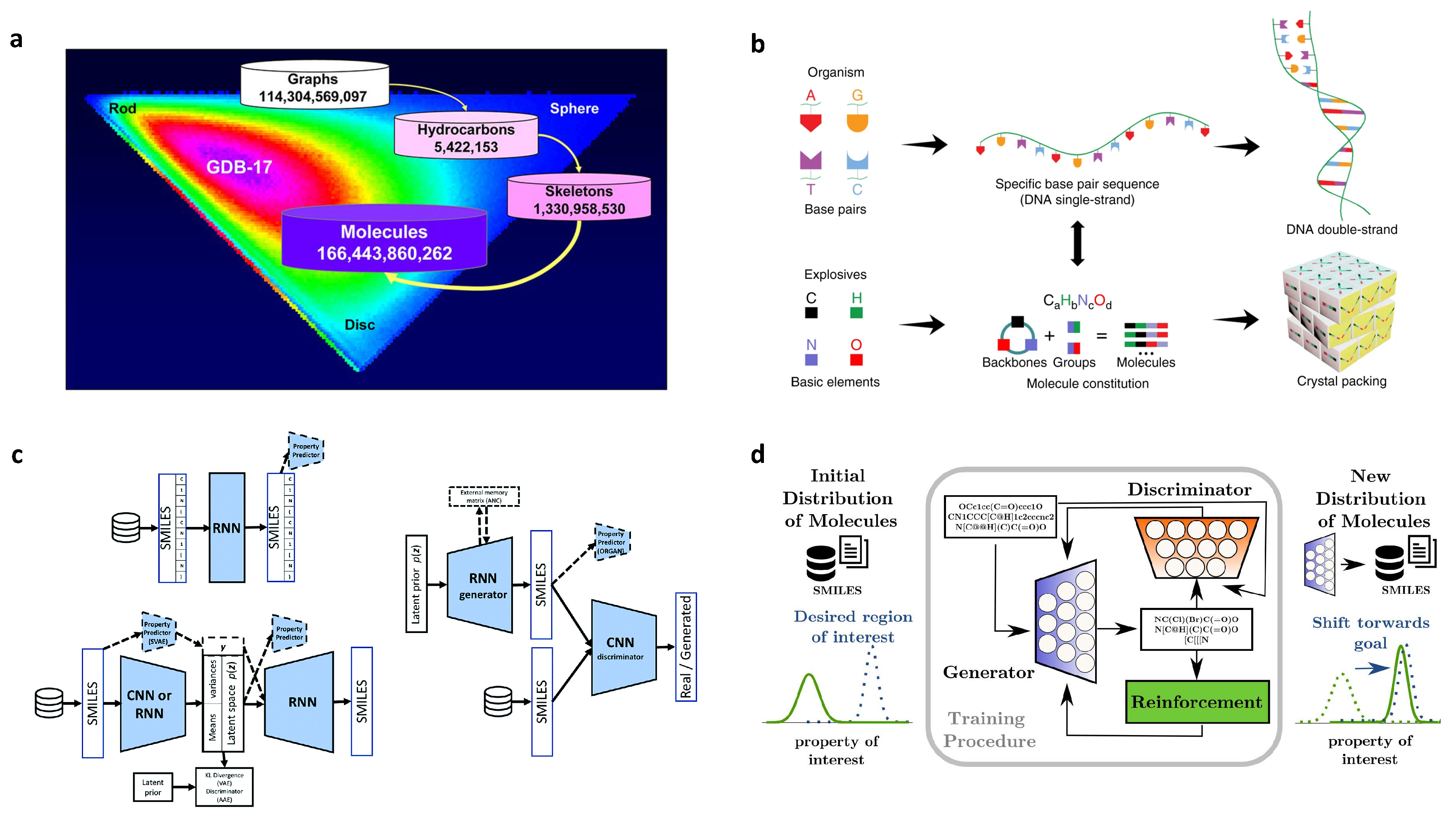
2.5. Design of High Energetic Materials
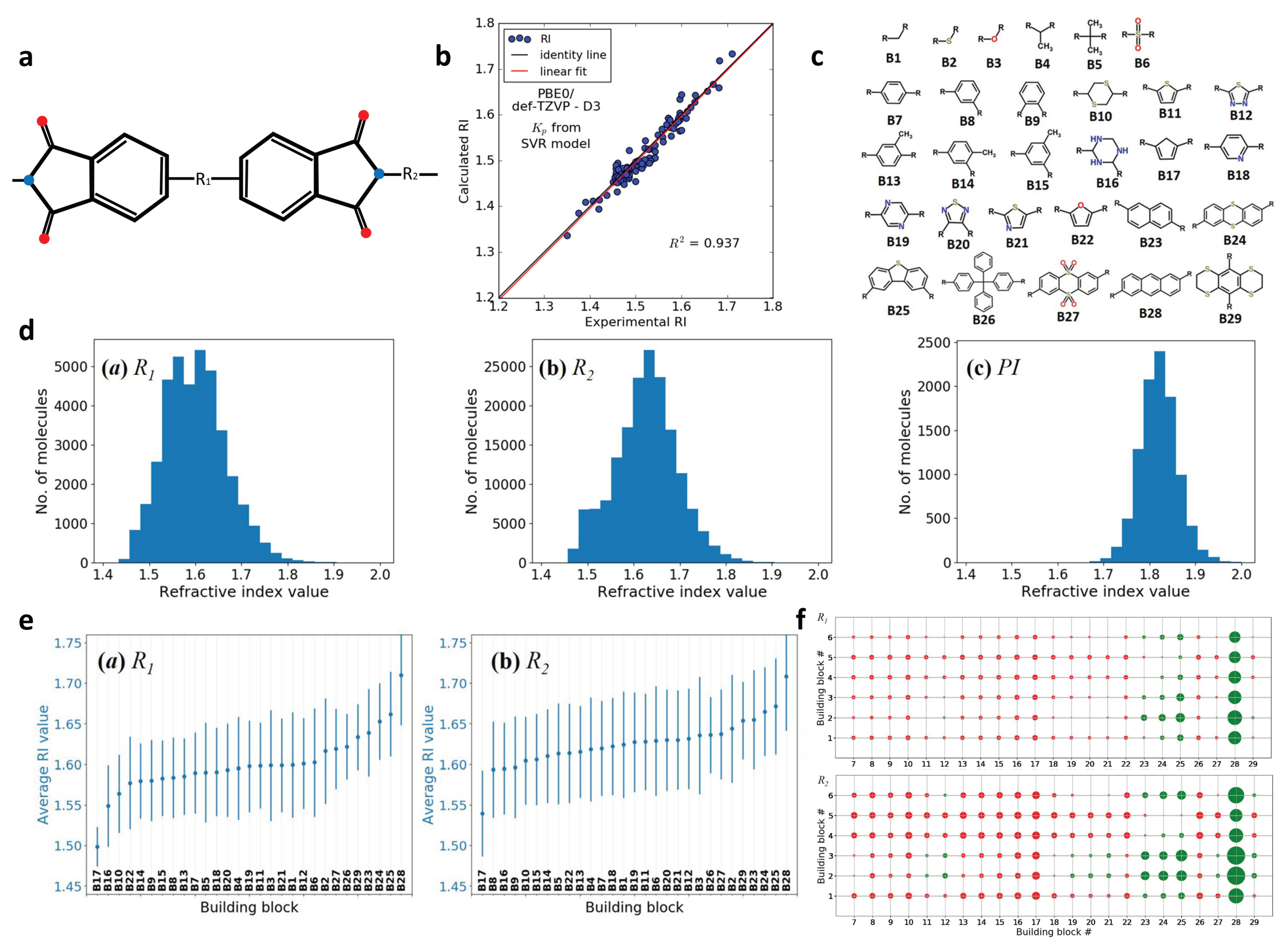
2.6. Design of Polyimides with High Refractive Index (RI)
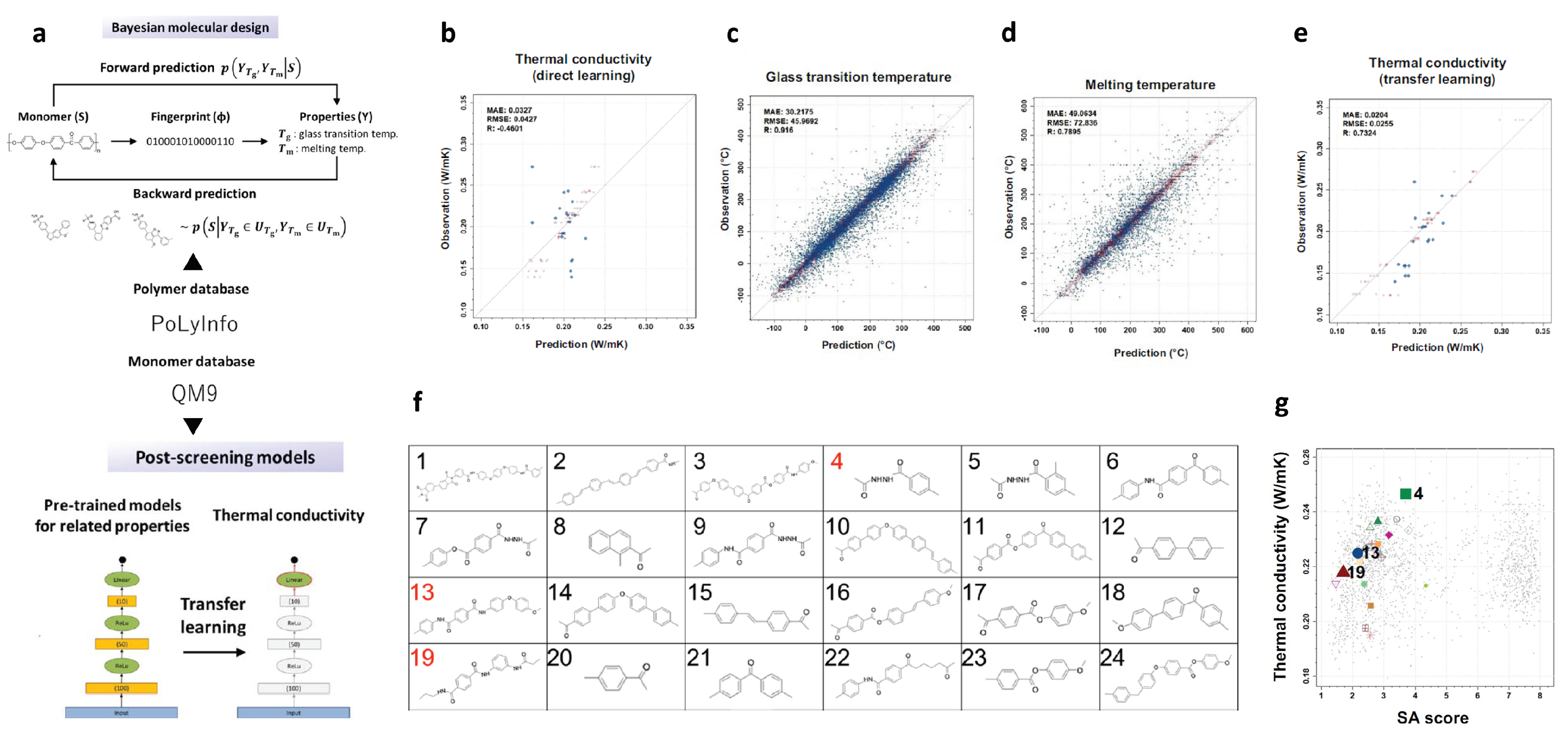
2.7. Polymers with High Thermal Conductivity
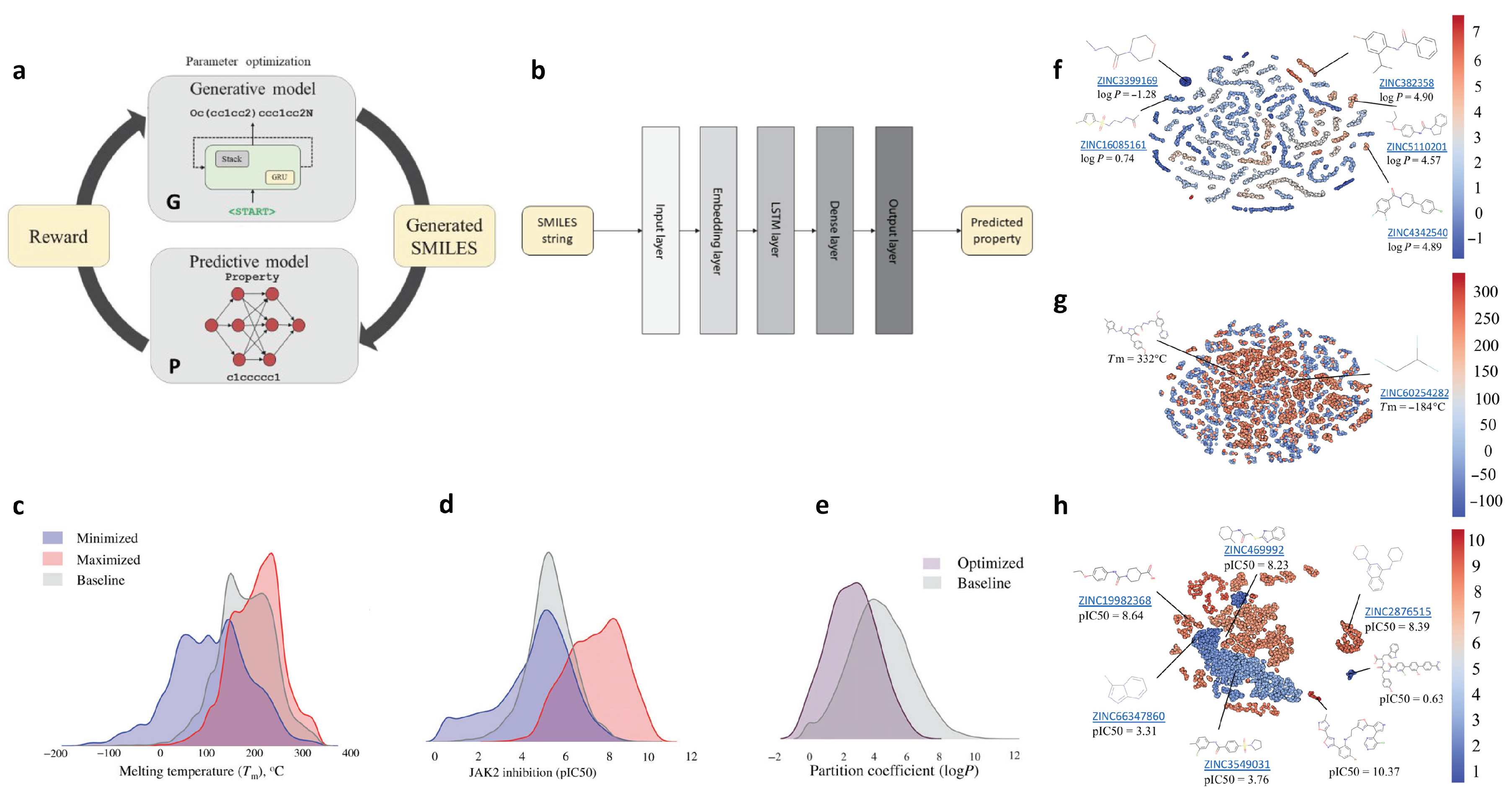
2.8. De Novo Drug-Like Molecular Design
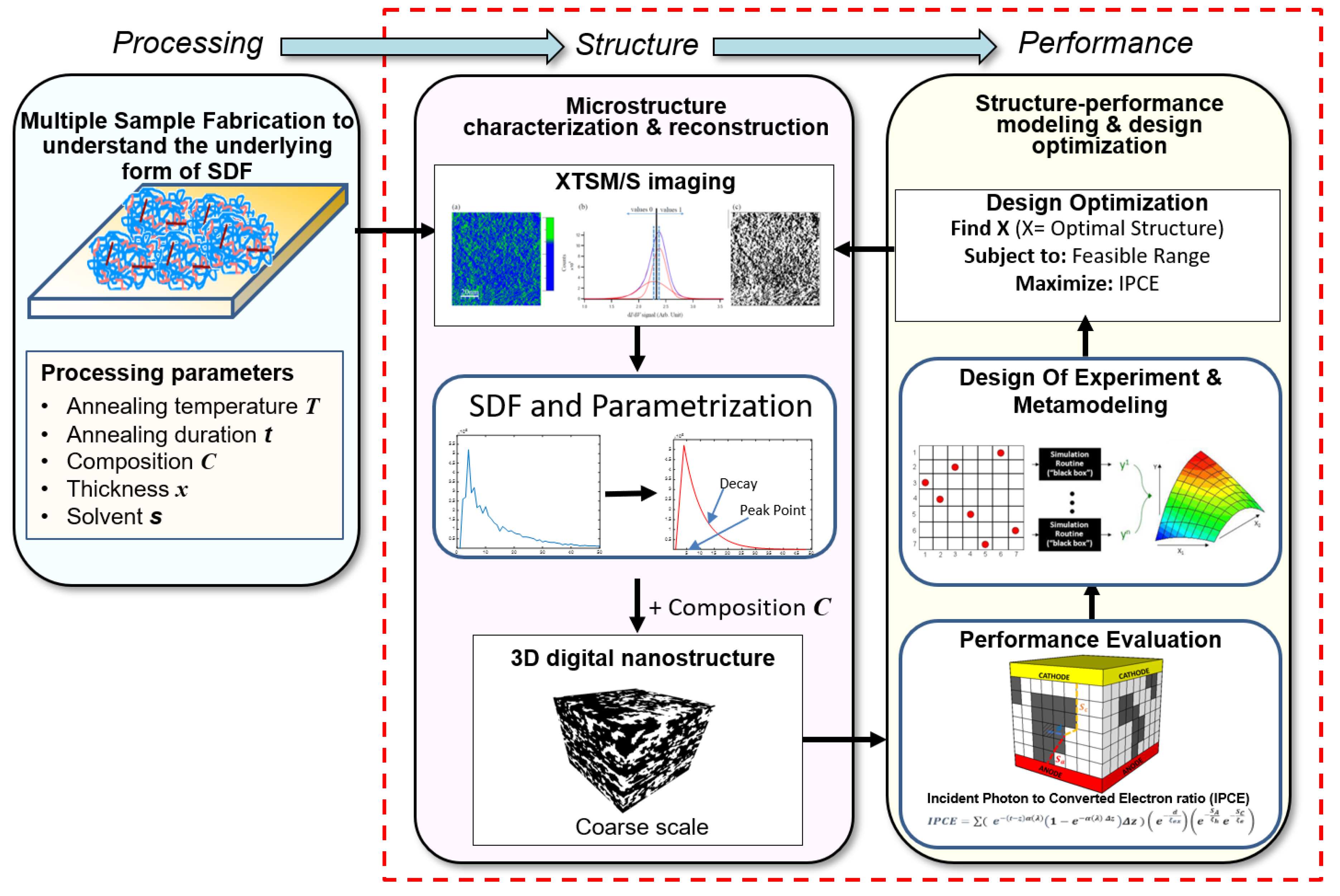
2.9. Microstructure Design of Organic Photovoltaic Solar Cells (OPVCs)
3. Discussion
3.1. Materials Database
3.2. Machine Learning Model
3.2.1. Feature Selection and Extraction
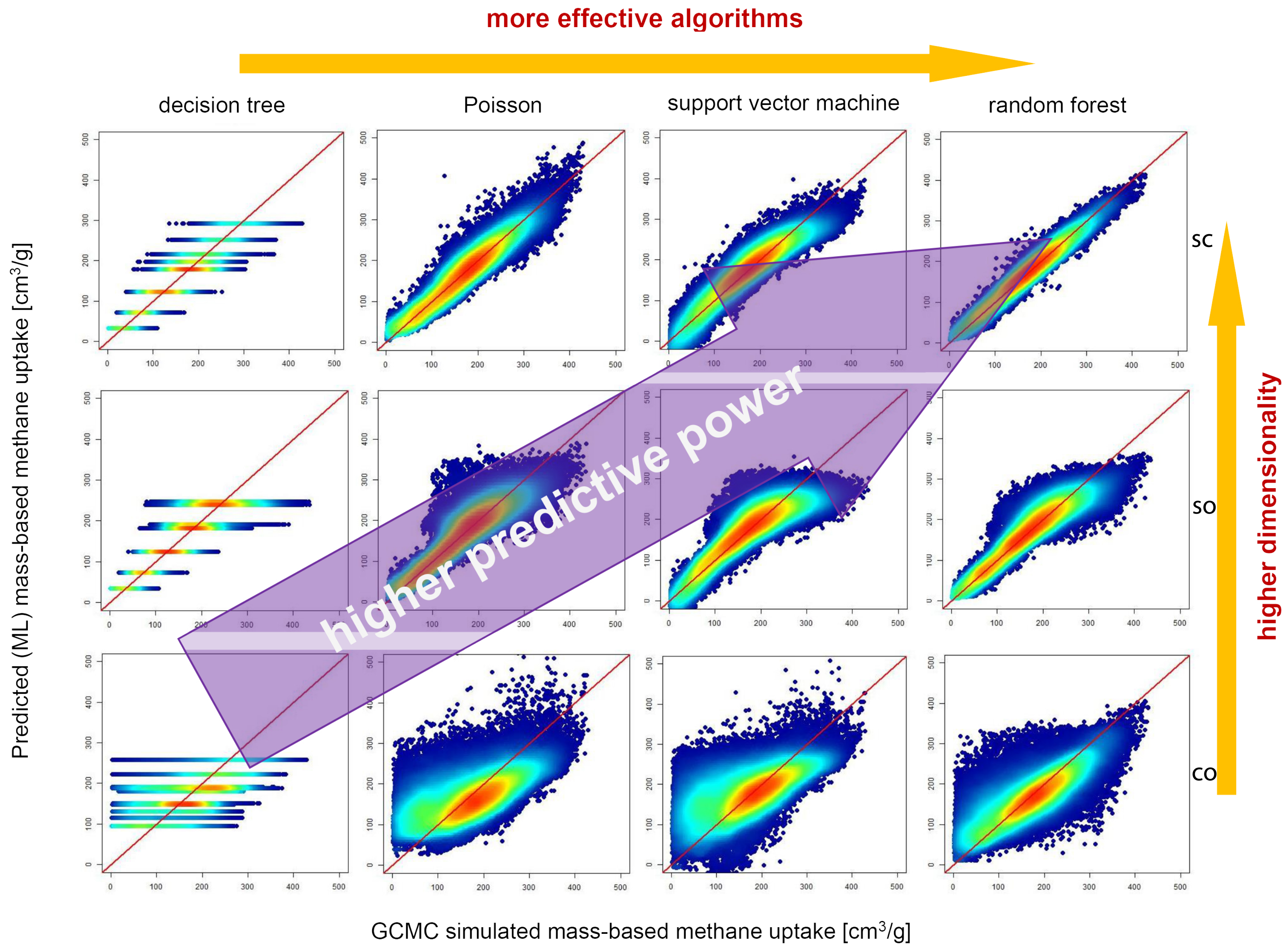
3.2.2. ML Methods and Model Validation
3.3. Molecular Generation
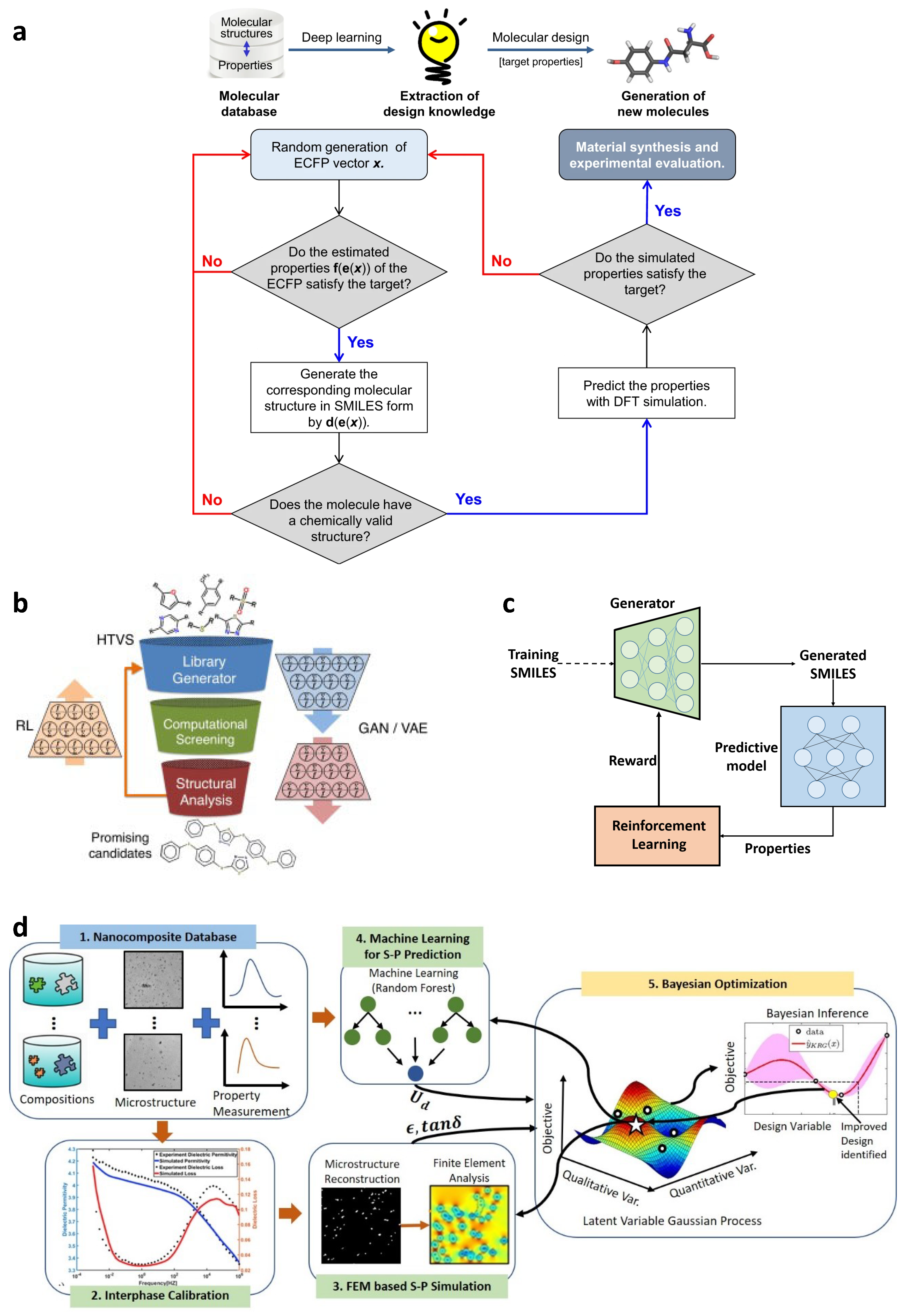
3.4. Inverse Materials Design
3.4.1. Materials Design by High-Throughput Screening
3.4.2. Reinforcement Learning for Materials Design
3.4.3. Bayesian Optimization for Materials Design
- An ML model is trained on available data to predict material property of interest from the design variables and supply uncertainty quantification over the design space.
- An acquisition function uses the prediction and UQ to determine the best design to evaluate next.
- The design recommended by acquisition function is evaluated and added to the dataset.
4. Conclusions
- (i)
- Acquisition of a diverse database. There are many public databases available for various materials, such as the ones summarized in Table 2. If no database of interest is available, we can build one by experiments or simulations. As a result, it is generally not challenging to acquire a database, rather it is challenging to obtain a “good” one. “Good” means that the database is diverse or uniform across the chemical space [140] since this feature of a database significantly affects the capabilities (interpolation, extrapolation, and exploration) of the ML model to be built. With a diverse or uniform database in the chemical space, the ML model guarantees the prediction by interpolation, while with a database in a limited region or class, the prediction is weakened by extrapolation. However, since the whole chemical space is nearly infinite and not clearly known, how can we determine if the database is uniform or not? To overcome this challenge, two areas of algorithmic approaches should be considered [140]: algorithms to perform searches, and more general machine learning and statistical modeling algorithms to predict the chemistry under investigation. The combination of these approaches should be capable of navigating and searching chemical space more efficiently, uniformly, quickly and, importantly, without bias [140].
- (ii)
- Feature representation. Most ML models need all inputs and outputs to be numeric, which requires the data to be represented in a digital form. Many types of representation methods are widely used, such as molecular descriptors, SMILES, and fingerprints, as summarized in Table 3. However, are they universal for all property predictions? Taking fingerprints as an example, it is known that different functional groups (substructures) of a complex structure may have distinct influences on the properties. Therefore, if one fingerprint method with certain bits does demonstrate predictive power in one property prediction, will it have the same capability in another property prediction? In addition, which representation is more suitable to work with specific ML models so that the model can have strong predictive capability? All of these questions require us to be cautious for the feature representation, selection, and extraction by applying the ML models for different materials and properties.
- (iii)
- ML algorithms and training. When conducting a materials design task, the choice of a suitable ML model should be carefully considered. There are many available ML models to choose as reviewed in the Discussion section, but it is not as easy as just to choose any one randomly. Choosing a suitable ML model depends on the database availability and the feature representation method. Which ML model is the best for a certain material property prediction? Does it depend on the type of materials? Can a model that is built with strong predictive power for one material be applicable to other similar but different materials? What about applying to a totally different material? Additionally, when training the selected ML model, there are usually some hyperparameters to be set. It is not trivial to set them without any knowledge of the ML algorithms. In order for the ML model to have better predictive power, the setting of these hyperparameters needs learning efforts, from the user’s point of view.
- (iv)
- Interpretation of results. ML models do show good prediction power in some cases. However, how to explain the constructed model, for example, the DNN model, is still an open question even in the field of computer science. When applying ML models to materials design, is there any unified theory to physically or chemically interpret the relationship established between a chemical structure to its properties? Can the model built increase our understanding of materials? What role should we consider ML models to be in materials design?
- (v)
- Molecular generation. Molecular generation plays an important role in the design of de novo organic molecules and polymers. As we have discussed, there are several deep generative models, including generative adversarial networks, variational autoencoders, and autoregressive models, rapidly growing for the discovery of new organic molecules and materials [24,60,93]. It is very important to benchmark these different deep generative models for their efficiency and accuracy. Very recently, Zhavoronkov and co-workers have proposed MOlecular SEtS (MOSES) as a platform to benchmark different ML techniques for drug discovery [234]. Such a platform is extremely helpful and useful to standardize the research on the molecular generation and facilitate the sharing and comparison of new ML models. Therefore, more efforts are needed to further design and maintain these benchmark platforms for organic molecules and polymers.
- (vi)
- Inverse molecular/materials design. Currently, RL has been widely used for the inverse molecular/materials design, due to its ease of integration with deep generative ML models [25,36,116]. RL usually involves the analysis of possible actions and outcomes, as well as estimation of the statistical relationship between these actions and possible outcomes. By defining the policy or reward function, the RL can be used to bias the generation of organic molecules towards most desirable domain [24,25,116]. Nevertheless, the inverse design of new molecules and materials typically requires multi-objective optimization of several target properties concurrently. For instance, drug-like molecules should be optimized with respect to potency, selectivity, solubility, and drug-likeness properties for drug discovery [116]. Such a multi-objective optimization problem poses significant challenges for the RL technique [235,236,237], combined with the huge design space of organic molecules. Comparing with RL technique, BO is more suitable and effective for multi-objective optimization and multi-point search [238,239,240]. Yet, the design of new molecules and materials involve both continuous/discretized and qualitative/quantitative design variables, representing molecular constituents, material compositions, microstructure morphology, and processing conditions. For these mixed variable design optimization problems, the existing BO approaches are usually restrictive theoretically and fail to capture complex correlations between input variable and output properties [207,208,232]. Therefore, new RL or BO methods should be formulated and developed to resolve these issues.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brazel, C.S.; Rosen, S.L. Fundamental Principles of Polymeric Materials; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Lei, T.; Wang, J.Y.; Pei, J. Roles of flexible chains in organic semiconducting materials. Chem. Mater. 2013, 26, 594–603. [Google Scholar] [CrossRef]
- Afzal, M.A.F. From Virtual High-Throughput Screening and Machine Learning to the Discovery and Rational Design of Polymers for Optical Applications. Ph.D. Thesis, State University of New York at Buffalo, Buffalo, NY, USA, 2018. [Google Scholar]
- Kippelen, B.; Brédas, J.L. Organic photovoltaics. Energy Environ. Sci. 2009, 2, 251–261. [Google Scholar] [CrossRef]
- Schmidt-Mende, L.; Fechtenkötter, A.; Müllen, K.; Moons, E.; Friend, R.H.; MacKenzie, J.D. Self-organized discotic liquid crystals for high-efficiency organic photovoltaics. Science 2001, 293, 1119–1122. [Google Scholar] [CrossRef] [PubMed]
- Brabec, C.J. Organic photovoltaics: Technology and market. Sol. Energy Mater. Sol. Cells 2004, 83, 273–292. [Google Scholar] [CrossRef]
- Sun, W.; Zheng, Y.; Yang, K.; Zhang, Q.; Shah, A.A.; Wu, Z.; Sun, Y.; Feng, L.; Chen, D.; Xiao, Z.; et al. Machine learning–assisted molecular design and efficiency prediction for high-performance organic photovoltaic materials. Sci. Adv. 2019, 5, eaay4275. [Google Scholar] [CrossRef]
- Wang, C.; Pilania, G.; Boggs, S.; Kumar, S.; Breneman, C.; Ramprasad, R. Computational strategies for polymer dielectrics design. Polymer 2014, 55, 979–988. [Google Scholar] [CrossRef]
- Mannodi-Kanakkithodi, A.; Pilania, G.; Huan, T.D.; Lookman, T.; Ramprasad, R. Machine learning strategy for accelerated design of polymer dielectrics. Sci. Rep. 2016, 6, 20952. [Google Scholar] [CrossRef]
- Zhou, H.C.; Long, J.R.; Yaghi, O.M. Introduction to metal–organic frameworks. Chem. Rev. 2012, 112, 673–674. [Google Scholar] [CrossRef]
- James, S.L. Metal-organic frameworks. Chem. Soc. Rev. 2003, 32, 276–288. [Google Scholar] [CrossRef]
- Furukawa, H.; Cordova, K.E.; O’Keeffe, M.; Yaghi, O.M. The chemistry and applications of metal-organic frameworks. Science 2013, 341, 1230444. [Google Scholar] [CrossRef]
- Bucior, B.J.; Rosen, A.S.; Haranczyk, M.; Yao, Z.; Ziebel, M.E.; Farha, O.K.; Hupp, J.T.; Siepmann, J.I.; Aspuru-Guzik, A.; Snurr, R.Q. Identification Schemes for Metal–Organic Frameworks To Enable Rapid Search and Cheminformatics Analysis. Cryst. Growth Des. 2019, 19, 6682–6697. [Google Scholar] [CrossRef]
- Burroughes, J.H.; Bradley, D.D.; Brown, A.; Marks, R.; Mackay, K.; Friend, R.H.; Burns, P.; Holmes, A. Light-emitting diodes based on conjugated polymers. Nature 1990, 347, 539. [Google Scholar] [CrossRef]
- Gross, M.; Müller, D.C.; Nothofer, H.G.; Scherf, U.; Neher, D.; Bräuchle, C.; Meerholz, K. Improving the performance of doped π-conjugated polymers for use in organic light-emitting diodes. Nature 2000, 405, 661. [Google Scholar] [CrossRef]
- Agrawal, J.P. Recent trends in high-energy materials. Prog. Energy Combust. Sci. 1998, 24, 1–30. [Google Scholar] [CrossRef]
- Talawar, M.; Sivabalan, R.; Mukundan, T.; Muthurajan, H.; Sikder, A.; Gandhe, B.; Rao, A.S. Environmentally compatible next generation green energetic materials (GEMs). J. Hazard. Mater. 2009, 161, 589–607. [Google Scholar] [CrossRef]
- Bushuyev, O.S.; Brown, P.; Maiti, A.; Gee, R.H.; Peterson, G.R.; Weeks, B.L.; Hope-Weeks, L.J. Ionic polymers as a new structural motif for high-energy-density materials. J. Am. Chem. Soc. 2012, 134, 1422–1425. [Google Scholar] [CrossRef]
- Kuntz, I.D. Structure-based strategies for drug design and discovery. Science 1992, 257, 1078–1082. [Google Scholar] [CrossRef]
- Silverman, R.B.; Holladay, M.W. The Organic Chemistry of Drug Design and Drug Action; Academic Press: Cambridge, MA, USA, 2014. [Google Scholar]
- Yang, X.; Wang, Y.; Byrne, R.; Schneider, G.; Yang, S. Concepts of Artificial Intelligence for Computer-Assisted Drug Discovery. Chem. Rev. 2019, 119, 10520–10594. [Google Scholar] [CrossRef]
- Polishchuk, P.G.; Madzhidov, T.I.; Varnek, A. Estimation of the size of drug-like chemical space based on GDB-17 data. J. Comput.-Aided Mol. Des. 2013, 27, 675–679. [Google Scholar] [CrossRef]
- Ruddigkeit, L.; Van Deursen, R.; Blum, L.C.; Reymond, J.L. Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17. J. Chem. Inf. Model. 2012, 52, 2864–2875. [Google Scholar] [CrossRef]
- Sanchez-Lengeling, B.; Aspuru-Guzik, A. Inverse molecular design using machine learning: Generative models for matter engineering. Science 2018, 361, 360–365. [Google Scholar] [CrossRef]
- Sanchez-Lengeling, B.; Outeiral, C.; Guimaraes, G.L.; Aspuru-Guzik, A. Optimizing distributions over molecular space. An Objective-Reinforced Generative Adversarial Network for Inverse-design Chemistry (ORGANIC). ChemRxiv 2017. [Google Scholar] [CrossRef]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203. [Google Scholar]
- Parr, R.G. Density functional theory of atoms and molecules. In Horizons of Quantum Chemistry; Springer: Berlin/Heidelberg, Germany, 1980; pp. 5–15. [Google Scholar]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Insights into current limitations of density functional theory. Science 2008, 321, 792–794. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications; Academic Press: Cambridge, MA, USA, 2001. [Google Scholar]
- Rapaport, D.C.; Rapaport, D.C.R. The Art of Molecular Dynamics Simulation; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Tropsha, A. Best practices for QSAR model development, validation, and exploitation. Mol. Inf. 2010, 29, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [PubMed]
- Churchwell, C.J.; Rintoul, M.D.; Martin, S.; Visco, D.P., Jr.; Kotu, A.; Larson, R.S.; Sillerud, L.O.; Brown, D.C.; Faulon, J.L. The signature molecular descriptor: 3. Inverse-quantitative structure–activity relationship of ICAM-1 inhibitory peptides. J. Mol. Graph. Modell. 2004, 22, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.W.; Burkowski, F.J. A constructive approach for discovering new drug leads: Using a kernel methodology for the inverse-QSAR problem. J. Cheminform. 2009, 1, 4. [Google Scholar] [CrossRef]
- Miyao, T.; Kaneko, H.; Funatsu, K. Inverse QSPR/QSAR analysis for chemical structure generation (from y to x). J. Chem. Inf. Model. 2016, 56, 286–299. [Google Scholar] [CrossRef]
- Silver, D.; Huang, A.; Maddison, C.J.; Guez, A.; Sifre, L.; Van Den Driessche, G.; Schrittwieser, J.; Antonoglou, I.; Panneershelvam, V.; Lanctot, M.; et al. Mastering the game of Go with deep neural networks and tree search. Nature 2016, 529, 484. [Google Scholar] [CrossRef]
- Agrawal, A.; Choudhary, A. Perspective: Materials informatics and big data: Realization of the “fourth paradigm” of science in materials science. APL Mater. 2016, 4, 053208. [Google Scholar] [CrossRef]
- Gil, Y.; Greaves, M.; Hendler, J.; Hirsh, H. Amplify scientific discovery with artificial intelligence. Science 2014, 346, 171–172. [Google Scholar] [CrossRef]
- Rajan, K. Informatics for Materials Science and Engineering: Data-driven Discovery for Accelerated Experimentation and Application; Butterworth-Heinemann: Oxford, UK, 2013. [Google Scholar]
- Sarkisov, L.; Kim, J. Computational structure characterization tools for the era of material informatics. Chem. Eng. Sci. 2015, 121, 322–330. [Google Scholar] [CrossRef]
- Adams, N. Polymer informatics. In Polymer Libraries; Springer: Berlin/Heidelberg, Germany, 2010; pp. 107–149. [Google Scholar]
- Audus, D.J.; de Pablo, J.J. Polymer Informatics: Opportunities and Challenges. ACS Macro. Lett. 2017, 6, 1078–1082. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Chandrasekaran, A.; Huan, T.D.; Das, D.; Ramprasad, R. Polymer genome: A data-powered polymer informatics platform for property predictions. J. Phys. Chem. C 2018, 122, 17575–17585. [Google Scholar] [CrossRef]
- Mannodi-Kanakkithodi, A.; Chandrasekaran, A.; Kim, C.; Huan, T.D.; Pilania, G.; Botu, V.; Ramprasad, R. Scoping the polymer genome: A roadmap for rational polymer dielectrics design and beyond. Mater. Today 2018, 21, 785–796. [Google Scholar] [CrossRef]
- Council, N.R. Advanced Energetic Materials; National Academics Press: Washington, DC, USA, 2004. [Google Scholar]
- Pagoria, P. A comparison of the structure, synthesis, and properties of insensitive energetic compounds. Propellants Explos. Pyrotech. 2016, 41, 452–469. [Google Scholar] [CrossRef]
- Nielsen, A.T.; Chafin, A.P.; Christian, S.L.; Moore, D.W.; Nadler, M.P.; Nissan, R.A.; Vanderah, D.J.; Gilardi, R.D.; George, C.F.; Flippen-Anderson, J.L. Synthesis of polyazapolycyclic caged polynitramines. Tetrahedron 1998, 54, 11793–11812. [Google Scholar] [CrossRef]
- White, A. The materials genome initiative: One year on. MRS Bull. 2012, 37, 715–716. [Google Scholar] [CrossRef]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef]
- de Pablo, J.J.; Jones, B.; Kovacs, C.L.; Ozolins, V.; Ramirez, A.P. The materials genome initiative, the interplay of experiment, theory and computation. Curr. Opin. Solid State Mater. Sci. 2014, 18, 99–117. [Google Scholar] [CrossRef]
- Green, M.L.; Choi, C.; Hattrick-Simpers, J.; Joshi, A.; Takeuchi, I.; Barron, S.; Campo, E.; Chiang, T.; Empedocles, S.; Gregoire, J.; et al. Fulfilling the promise of the materials genome initiative with high-throughput experimental methodologies. Appl. Phys. Rev. 2017, 4, 011105. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.; Song, S.; Yang, Z.; Qi, X.; Wang, K.; Liu, Y.; Zhang, Q.; Tian, Y. Accelerating the discovery of insensitive high-energy-density materials by a materials genome approach. Nat. Commun. 2018, 9, 2444. [Google Scholar] [CrossRef] [PubMed]
- Gubernatis, J.; Lookman, T. Machine learning in materials design and discovery: Examples from the present and suggestions for the future. Phys. Rev. Mater. 2018, 2, 120301. [Google Scholar] [CrossRef]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef]
- Van Den Herik, H.J.; Uiterwijk, J.W.; Van Rijswijck, J. Games solved: Now and in the future. Artif. Intell. 2002, 134, 277–311. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, T.; Ju, W.; Shi, S. Materials discovery and design using machine learning. J. Mater. 2017, 3, 159–177. [Google Scholar] [CrossRef]
- Butler, K.T.; Davies, D.W.; Cartwright, H.; Isayev, O.; Walsh, A. Machine learning for molecular and materials science. Nature 2018, 559, 547–555. [Google Scholar] [CrossRef]
- Ramprasad, R.; Batra, R.; Pilania, G.; Mannodi-Kanakkithodi, A.; Kim, C. Machine learning in materials informatics: Recent applications and prospects. Npj Comput. Mater. 2017, 3, 54. [Google Scholar] [CrossRef]
- Kailkhura, B.; Gallagher, B.; Kim, S.; Hiszpanski, A.; Han, T.Y.J. Reliable and explainable machine-learning methods for accelerated material discovery. Npj Comput. Mater. 2019, 5, 1–9. [Google Scholar] [CrossRef]
- Elton, D.C.; Boukouvalas, Z.; Fuge, M.D.; Chung, P.W. Deep learning for molecular design-a review of the state of the art. Mol. Syst. Des. Eng. 2019, 4, 828–849. [Google Scholar] [CrossRef]
- Xu, Y.; Yao, H.; Lin, K. An overview of neural networks for drug discovery and the inputs used. Expert Opin. Drug Discov. 2018, 13, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Engkvist, O.; Wang, Y.; Olivecrona, M.; Blaschke, T. The rise of deep learning in drug discovery. Drug Discov. Today 2018, 23, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, T.; Kreisbeck, C.; Becker, J.S.; Aspuru-Guzik, A.; Saikin, S.K. Autonomous molecular design: Then and now. ACS Appl. Mater. Interfaces 2019, 11, 24825–24836. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.N.; Li, Q.; Jun, Y. Challenges and opportunities of polymer design with machine learning and high throughput experimentation. MRS Commun. 2019, 1–8. [Google Scholar] [CrossRef]
- Outlook, A.E. Energy information administration. Dep. Energy 2010, 92010, 1–15. [Google Scholar]
- Gaudiana, R. Third-generation photovoltaic technology- the potential for low-cost solar energy conversion. J. Phys. Chem. Lett. 2010, 1, 1288–1289. [Google Scholar] [CrossRef]
- Imamzai, M.; Aghaei, M.; Thayoob, Y.H.M.; Forouzanfar, M. A review on comparison between traditional silicon solar cells and thin-film CdTe solar cells. In Proceedings of the National Graduate Conference (Nat-Grad, 2012), Kajang, Malaya, 8–10 November 2012; pp. 1–5. [Google Scholar]
- Heeger, A.J. Semiconducting polymers: The third generation. Chem. Soc. Rev. 2010, 39, 2354–2371. [Google Scholar] [CrossRef]
- Hachmann, J.; Olivares-Amaya, R.; Atahan-Evrenk, S.; Amador-Bedolla, C.; Sánchez-Carrera, R.S.; Gold-Parker, A.; Vogt, L.; Brockway, A.M.; Aspuru-Guzik, A. The Harvard clean energy project: Large-scale computational screening and design of organic photovoltaics on the world community grid. J. Phys. Chem. Lett. 2011, 2, 2241–2251. [Google Scholar] [CrossRef]
- Olivares-Amaya, R.; Amador-Bedolla, C.; Hachmann, J.; Atahan-Evrenk, S.; Sanchez-Carrera, R.S.; Vogt, L.; Aspuru-Guzik, A. Accelerated computational discovery of high-performance materials for organic photovoltaics by means of cheminformatics. Energy Environ. Sci. 2011, 4, 4849–4861. [Google Scholar] [CrossRef]
- ChemAxon. Available online: https://www.chemaxon.com// (accessed on 3 September 2019).
- Kim, C.; Wang, Z.; Choi, H.J.; Ha, Y.G.; Facchetti, A.; Marks, T.J. Printable cross-linked polymer blend dielectrics. Design strategies, synthesis, microstructures, and electrical properties, with organic field-effect transistors as testbeds. J. Am. Chem. Soc. 2008, 130, 6867–6878. [Google Scholar] [CrossRef]
- Müller, K.; Paloumpa, I.; Henkel, K.; Schmeisser, D. A polymer high-k dielectric insulator for organic field-effect transistors. J. Appl. Phys. 2005, 98, 056104. [Google Scholar] [CrossRef]
- Mannodi-Kanakkithodi, A.; Treich, G.M.; Huan, T.D.; Ma, R.; Tefferi, M.; Cao, Y.; Sotzing, G.A.; Ramprasad, R. Rational Co-Design of Polymer Dielectrics for Energy Storage. Adv. Mater. 2016, 28, 6277–6291. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Wang, C.; Lorenzini, R.G.; Ma, R.; Zhu, Q.; Sinkovits, D.W.; Pilania, G.; Oganov, A.R.; Kumar, S.; Sotzing, G.A.; et al. Rational design of all organic polymer dielectrics. Nat. Commun. 2014, 5, 4845. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.L. Crystallographic data and melting points for various polymers. Wiley Database Polym. Prop. 2003. [Google Scholar] [CrossRef]
- Goedecker, S. Minima hopping: An efficient search method for the global minimum of the potential energy surface of complex molecular systems. J. Chem. Phys. 2004, 120, 9911–9917. [Google Scholar] [CrossRef]
- Pilania, G.; Wang, C.; Jiang, X.; Rajasekaran, S.; Ramprasad, R. Accelerating materials property predictions using machine learning. Sci. Rep. 2013, 3, 2810. [Google Scholar] [CrossRef]
- Deb, K.; Pratap, A.; Agarwal, S.; Meyarivan, T. A fast and elitist multiobjective genetic algorithm: NSGA-II. IEEE Trans. Dielectr. Electr. Insul. 2002, 6, 182–197. [Google Scholar] [CrossRef]
- Jou, J.H.; Kumar, S.; Agrawal, A.; Li, T.H.; Sahoo, S. Approaches for fabricating high efficiency organic light emitting diodes. J. Mater. Chem. C 2015, 3, 2974–3002. [Google Scholar] [CrossRef]
- Tao, Y.; Yuan, K.; Chen, T.; Xu, P.; Li, H.; Chen, R.; Zheng, C.; Zhang, L.; Huang, W. Thermally activated delayed fluorescence materials towards the breakthrough of organoelectronics. Adv. Mater. 2014, 26, 7931–7958. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, B.; Huang, S.; Nomura, H.; Tanaka, H.; Adachi, C. Efficient blue organic light-emitting diodes employing thermally activated delayed fluorescence. Nat. Photonics 2014, 8, 326. [Google Scholar] [CrossRef]
- Gómez-Bombarelli, R.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Duvenaud, D.; Maclaurin, D.; Blood-Forsythe, M.A.; Chae, H.S.; Einzinger, M.; Ha, D.G.; Wu, T.; et al. Design of efficient molecular organic light-emitting diodes by a high-throughput virtual screening and experimental approach. Nat. Mater. 2016, 15, 1120. [Google Scholar] [CrossRef]
- Olsson, T.; Oprea, T.I. Cheminformatics: A tool for decision-makers in drug discovery. Curr. Opin. Drug Discov. Dev. 2001, 4, 308–313. [Google Scholar]
- Akella, L.B.; DeCaprio, D. Cheminformatics approaches to analyze diversity in compound screening libraries. Curr. Opin. Chem. Biol. 2010, 14, 325–330. [Google Scholar] [CrossRef]
- Landrum, G. Rdkit documentation. Release 2013, 1, 1–79. [Google Scholar]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Jørgensen, P.B.; Mesta, M.; Shil, S.; García Lastra, J.M.; Jacobsen, K.W.; Thygesen, K.S.; Schmidt, M.N. Machine learning-based screening of complex molecules for polymer solar cells. J. Chem. Phys. 2018, 148, 241735. [Google Scholar] [CrossRef]
- Rupp, M.; Tkatchenko, A.; Müller, K.R.; Von Lilienfeld, O.A. Fast and accurate modeling of molecular atomization energies with machine learning. Phys. Rev. Lett. 2012, 108, 058301. [Google Scholar] [CrossRef]
- Elton, D.C.; Boukouvalas, Z.; Butrico, M.S.; Fuge, M.D.; Chung, P.W. Applying machine learning techniques to predict the properties of energetic materials. Sci. Rep. 2018, 8, 9059. [Google Scholar] [CrossRef]
- Hansen, K.; Biegler, F.; Ramakrishnan, R.; Pronobis, W.; Von Lilienfeld, O.A.; Müller, K.R.; Tkatchenko, A. Machine learning predictions of molecular properties: Accurate many-body potentials and nonlocality in chemical space. J. Phys. Chem. Lett. 2015, 6, 2326–2331. [Google Scholar] [CrossRef]
- Xu, Y.; Lin, K.; Wang, S.; Wang, L.; Cai, C.; Song, C.; Lai, L.; Pei, J. Deep learning for molecular generation. Future Med. Chem. 2019, 11, 567–597. [Google Scholar] [CrossRef]
- Barnes, B.C.; Elton, D.C.; Boukouvalas, Z.; Taylor, D.E.; Mattson, W.D.; Fuge, M.D.; Chung, P.W. Machine learning of energetic material properties. arXiv 2018, arXiv:1807.06156. [Google Scholar]
- Huang, L.; Massa, L. Applications of energetic materials by a theoretical method (discover energetic materials by a theoretical method). Int. J. Energ. Mater. Chem. Propul. 2013, 12, 197–262. [Google Scholar] [CrossRef]
- Mathieu, D. Sensitivity of energetic materials: Theoretical relationships to detonation performance and molecular structure. Ind. Eng. Chem. Res. 2017, 56, 8191–8201. [Google Scholar] [CrossRef]
- Ravi, P.; Gore, G.M.; Tewari, S.P.; Sikder, A.K. DFT study on the structure and explosive properties of nitropyrazoles. Mol. Simul. 2012, 38, 218–226. [Google Scholar] [CrossRef]
- Liu, J.g.; Ueda, M. High refractive index polymers: Fundamental research and practical applications. J. Mater. Chem. 2009, 19, 8907–8919. [Google Scholar] [CrossRef]
- Odian, G. Principles of Polymerization; John Wiley & Sons: Hoboken, NJ, USA, 2004. [Google Scholar]
- Afzal, M.A.F.; Haghighatlari, M.; Prasad Ganesh, S.; Cheng, C.; Hachmann, J. Accelerated Discovery of High-Refractive-Index Polyimides via First-Principles Molecular Modeling, Virtual High-Throughput Screening, and Data Mining. J. Phys. Chem. C 2019, 123, 14610–14618. [Google Scholar] [CrossRef]
- Slonimskii, G.; Askadskii, A.; Kitaigorodskii, A. The packing of polymer molecules. Polym. Sci. USSR 1970, 12, 556–577. [Google Scholar] [CrossRef]
- Afzal, M.A.F.; Cheng, C.; Hachmann, J. Combining first-principles and data modeling for the accurate prediction of the refractive index of organic polymers. J. Chem. Phys. 2018, 148, 241712. [Google Scholar] [CrossRef]
- Hansson, J.; Nilsson, T.M.; Ye, L.; Liu, J. Novel nanostructured thermal interface materials: A review. Int. Mater. Rev. 2018, 63, 22–45. [Google Scholar] [CrossRef]
- Razeeb, K.M.; Dalton, E.; Cross, G.L.W.; Robinson, A.J. Present and future thermal interface materials for electronic devices. Int. Mater. Rev. 2018, 63, 1–21. [Google Scholar] [CrossRef]
- Wan, X.; Feng, W.; Wang, Y.; Wang, H.; Zhang, X.; Deng, C.; Yang, N. Materials Discovery and Properties Prediction in Thermal Transport via Materials Informatics: A Mini Review. Nano Lett. 2019, 19, 3387–3395. [Google Scholar] [CrossRef]
- Wu, S.; Kondo, Y.; Kakimoto, M.A.; Yang, B.; Yamada, H.; Kuwajima, I.; Lambard, G.; Hongo, K.; Xu, Y.; Shiomi, J.; et al. Machine-learning-assisted discovery of polymers with high thermal conductivity using a molecular design algorithm. Npj Comput. Mater. 2019, 5, 5. [Google Scholar] [CrossRef]
- Otsuka, S.; Kuwajima, I.; Hosoya, J.; Xu, Y.; Yamazaki, M. PoLyInfo: Polymer database for polymeric materials design. In Proceedings of the 2011 International Conference on Emerging Intelligent Data and Web Technologies, Tirana, Albania, 7–9 September 2011; pp. 22–29. [Google Scholar]
- Blum, L.C.; Reymond, J.L. 970 million druglike small molecules for virtual screening in the chemical universe database GDB-13. J. Am. Chem. Soc. 2009, 131, 8732–8733. [Google Scholar] [CrossRef]
- Morikawa, J.; Tan, J.; Hashimoto, T. Study of change in thermal diffusivity of amorphous polymers during glass transition. Polymer 1995, 36, 4439–4443. [Google Scholar] [CrossRef]
- Allen, P.B.; Feldman, J.L. Thermal conductivity of disordered harmonic solids. Phys. Rev. B: Condens. Matter 1993, 48, 12581. [Google Scholar] [CrossRef]
- Ikebata, H.; Hongo, K.; Isomura, T.; Maezono, R.; Yoshida, R. Bayesian molecular design with a chemical language model. J. Comput.-Aided Mol. Des. 2017, 31, 379–391. [Google Scholar] [CrossRef]
- Segler, M.H.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci. 2017, 4, 120–131. [Google Scholar] [CrossRef]
- Lim, J.; Ryu, S.; Kim, J.W.; Kim, W.Y. Molecular generative model based on conditional variational autoencoder for de novo molecular design. J. Cheminform. 2018, 10, 31. [Google Scholar] [CrossRef]
- Kadurin, A.; Aliper, A.; Kazennov, A.; Mamoshina, P.; Vanhaelen, Q.; Khrabrov, K.; Zhavoronkov, A. The cornucopia of meaningful leads: Applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget 2017, 8, 10883. [Google Scholar] [CrossRef]
- Putin, E.; Asadulaev, A.; Ivanenkov, Y.; Aladinskiy, V.; Sanchez-Lengeling, B.; Aspuru-Guzik, A.; Zhavoronkov, A. Reinforced adversarial neural computer for de novo molecular design. J. Chem. Inf. Model. 2018, 58, 1194–1204. [Google Scholar] [CrossRef]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef]
- Tetko, I.V.; Sushko, Y.; Novotarskyi, S.; Patiny, L.; Kondratov, I.; Petrenko, A.E.; Charochkina, L.; Asiri, A.M. How accurately can we predict the melting points of drug-like compounds? J. Chem. Inf. Model. 2014, 54, 3320–3329. [Google Scholar] [CrossRef]
- Lee, W.K.; Yu, S.; Engel, C.J.; Reese, T.; Rhee, D.; Chen, W.; Odom, T.W. Concurrent design of quasi-random photonic nanostructures. Proc. Natl. Acad. Sci. USA 2017, 114, 8734–8739. [Google Scholar] [CrossRef]
- Bostanabad, R.; Zhang, Y.; Li, X.; Kearney, T.; Brinson, L.C.; Apley, D.W.; Liu, W.K.; Chen, W. Computational microstructure characterization and reconstruction: Review of the state-of-the-art techniques. Prog. Mater Sci. 2018, 95, 1–41. [Google Scholar] [CrossRef]
- Ghumman, U.F.; Iyer, A.; Dulal, R.; Wang, A.; Munshi, J.; Chien, T.; Balasubramanian, G.; Chen, W. A Spectral Density Function Approach for Design of Organic Photovoltaic Cells. In Proceedings of the ASME 2018 International Design Engineering Technical Conferences and Computers and Information in Engineering Conference. American Society of Mechanical Engineers Digital Collection, Quebec City, QC, Canada, 26–29 August 2018. [Google Scholar]
- Munshi, J.; Ghumman, U.F.; Iyer, A.; Dulal, R.; Chen, W.; Chien, T.; Balasubramanian, G. Effect of polydispersity on the bulk-heterojunction morphology of P3HT: PCBM solar cells. J. Polym. Sci. Part B Polym. Phys. 2019. [Google Scholar] [CrossRef]
- Munshi, J.; Dulal, R.; Chien, T.; Chen, W.; Balasubramanian, G. Solution Processing Dependent Bulk Heterojunction Nanomorphology of P3HT/PCBM Thin Films. ACS Appl. Mater. Interfaces 2019, 11, 17056–17067. [Google Scholar] [CrossRef]
- Olson, G.B. Computational design of hierarchically structured materials. Science 1997, 277, 1237–1242. [Google Scholar] [CrossRef]
- Gleiter, H. Nanostructured materials: Basic concepts and microstructure. Acta Mater. 2000, 48, 1–29. [Google Scholar] [CrossRef]
- Biswas, A.; Bayer, I.S.; Biris, A.S.; Wang, T.; Dervishi, E.; Faupel, F. Advances in top–down and bottom–up surface nanofabrication: Techniques, applications & future prospects. Adv. Colloid Interface Sci. 2012, 170, 2–27. [Google Scholar] [PubMed]
- Brabec, C.; Scherf, U.; Dyakonov, V. Organic Photovoltaics: Materials, Device Physics, and Manufacturing Technologies; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Brabec, C.J.; Dyakonov, V.; Parisi, J.; Sariciftci, N.S. Organic Photovoltaics: Concepts and Realization; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Ghumman, U.F.; Iyer, A.; Dulal, R.; Munshi, J.; Wang, A.; Chien, T.; Balasubramanian, G.; Chen, W. A Spectral Density Function Approach for Active Layer Design of Organic Photovoltaic Cells. J. Mech. Des. 2018, 140, 111408. [Google Scholar] [CrossRef]
- Liu, Y.; Greene, M.S.; Chen, W.; Dikin, D.A.; Liu, W.K. Computational microstructure characterization and reconstruction for stochastic multiscale material design. Comput.-Aided Des. 2013, 45, 65–76. [Google Scholar] [CrossRef]
- Xu, H.; Li, Y.; Brinson, C.; Chen, W. A descriptor-based design methodology for developing heterogeneous microstructural materials system. J. Mech. Des. 2014, 136, 051007. [Google Scholar] [CrossRef]
- Yeong, C.; Torquato, S. Reconstructing random media. II. Three-dimensional media from two-dimensional cuts. Phys. Rev. E: Stat. Nonlinear Soft Matter Phys. 1998, 58, 224. [Google Scholar] [CrossRef]
- Xu, H.; Dikin, D.A.; Burkhart, C.; Chen, W. Descriptor-based methodology for statistical characterization and 3D reconstruction of microstructural materials. Comput. Mater. Sci. 2014, 85, 206–216. [Google Scholar] [CrossRef]
- Yu, S.; Wang, C.; Zhang, Y.; Dong, B.; Jiang, Z.; Chen, X.; Chen, W.; Sun, C. Design of non-deterministic quasi-random nanophotonic structures using Fourier space representations. Sci. Rep. 2017, 7, 3752. [Google Scholar] [CrossRef]
- Yu, S.; Zhang, Y.; Wang, C.; Lee, W.k.; Dong, B.; Odom, T.W.; Sun, C.; Chen, W. Characterization and design of functional quasi-random nanostructured materials using spectral density function. J. Mech. Des. 2017, 139, 071401. [Google Scholar] [CrossRef]
- van Lare, M.C.; Polman, A. Optimized scattering power spectral density of photovoltaic light-trapping patterns. ACS Photonics 2015, 2, 822–831. [Google Scholar] [CrossRef]
- Lee, W.K.; Jung, W.B.; Nagel, S.R.; Odom, T.W. Stretchable superhydrophobicity from monolithic, three-dimensional hierarchical wrinkles. Nano Lett. 2016, 16, 3774–3779. [Google Scholar] [CrossRef]
- Kleijnen, J.P. Kriging metamodeling in simulation: A review. Eur. J. Oper. Res. 2009, 192, 707–716. [Google Scholar] [CrossRef]
- Jin, R.; Chen, W.; Simpson, T.W. Comparative studies of metamodelling techniques under multiple modelling criteria. Struct. Multidiscip. Optim. 2001, 23, 1–13. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Trame, M.; Lesko, L.; Schmidt, S. Sobol sensitivity analysis: A tool to guide the development and evaluation of systems pharmacology models. CPT: Pharmacomet. Syst. Pharmacol. 2015, 4, 69–79. [Google Scholar] [CrossRef]
- Gromski, P.S.; Henson, A.B.; Granda, J.M.; Cronin, L. How to explore chemical space using algorithms and automation. Nat. Rev. Chem. 2019, 3, 119–128. [Google Scholar] [CrossRef]
- Cooper, C.B.; Beard, E.J.; Vázquez-Mayagoitia, Á.; Stan, L.; Stenning, G.B.; Nye, D.W.; Vigil, J.A.; Tomar, T.; Jia, J.; Bodedla, G.B.; et al. Design-to-Device Approach Affords Panchromatic Co-Sensitized Solar Cells. Adv. Energy Mater. 2019, 9, 1802820. [Google Scholar] [CrossRef]
- Swain, M.C.; Cole, J.M. ChemDataExtractor: A toolkit for automated extraction of chemical information from the scientific literature. J. Chem. Inf. Model. 2016, 56, 1894–1904. [Google Scholar] [CrossRef]
- Huan, T.D.; Mannodi-Kanakkithodi, A.; Ramprasad, R. Accelerated materials property predictions and design using motif-based fingerprints. Phys. Rev. B: Condens. Matter 2015, 92, 014106. [Google Scholar] [CrossRef]
- Dong, J.; Cao, D.S.; Miao, H.Y.; Liu, S.; Deng, B.C.; Yun, Y.H.; Wang, N.N.; Lu, A.P.; Zeng, W.B.; Chen, A.F. ChemDes: An integrated web-based platform for molecular descriptor and fingerprint computation. J. Cheminform. 2015, 7, 60. [Google Scholar] [CrossRef]
- Karelson, M. Molecular Descriptors in QSAR/QSPR; Wiley-Interscience: New York, NY, USA, 2000. [Google Scholar]
- Puzyn, T.; Leszczynski, J.; Cronin, M.T. Recent Advances in QSAR Studies: Methods and Applications; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Varmuza, K.; Dehmer, M.; Bonchev, D. Statistical Modelling of Molecular Descriptors in QSAR/QSPR; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Tauler, R.; Walczak, B.; Brown, S.D. Comprehensive Chemometrics: Chemical and Biochemical Data Analysis; Elsevier: Amsterdam, The Netherlands, 2009. [Google Scholar]
- Consonni, V.; Todeschini, R.; Pavan, M.; Gramatica, P. Structure/response correlations and similarity/diversity analysis by GETAWAY descriptors. 2. Application of the novel 3D molecular descriptors to QSAR/QSPR studies. J. Chem. Inf. Comput. Sci. 2002, 42, 693–705. [Google Scholar] [CrossRef]
- Todeschini, R.; Gramatica, P. New 3D molecular descriptors: The WHIM theory and QSAR applications. In 3D QSAR in Drug Design; Springer: Berlin/Heidelberg, Germany, 2002; pp. 355–380. [Google Scholar]
- Devillers, J.; Balaban, A.T. Topological Indices and Related Descriptors in QSAR and QSPAR; CRC Press: Boca Raton, FL, USA, 2000. [Google Scholar]
- Dragon7. Available online: https://chm.kode-solutions.net/index.php// (accessed on 3 September 2019).
- Mauri, A.; Consonni, V.; Pavan, M.; Todeschini, R. Dragon software: An easy approach to molecular descriptor calculations. Match 2006, 56, 237–248. [Google Scholar]
- Muegge, I.; Mukherjee, P. An overview of molecular fingerprint similarity search in virtual screening. Expert Opin. Drug Discov. 2016, 11, 137–148. [Google Scholar] [CrossRef]
- Tabor, D.P.; Roch, L.M.; Saikin, S.K.; Kreisbeck, C.; Sheberla, D.; Montoya, J.H.; Dwaraknath, S.; Aykol, M.; Ortiz, C.; Tribukait, H.; et al. Accelerating the discovery of materials for clean energy in the era of smart automation. Nat. Rev. Mater. 2018, 3, 5. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, L.; Liu, Z. Multi-objective de novo drug design with conditional graph generative model. J. Cheminform. 2018, 10, 33. [Google Scholar] [CrossRef]
- Simonovsky, M.; Komodakis, N. Graphvae: Towards generation of small graphs using variational autoencoders. In Proceedings of the International Conference on Artificial Neural Networks, Rhodes, Greece, 4–7 October 2018; Springer: Berlin/Heidelberg, Germany, 2018; pp. 412–422. [Google Scholar]
- De Cao, N.; Kipf, T. MolGAN: An implicit generative model for small molecular graphs. arXiv 2018, arXiv:1805.11973. [Google Scholar]
- Liu, Q.; Allamanis, M.; Brockschmidt, M.; Gaunt, A. Constrained graph variational autoencoders for molecule design. In Proceedings of the Neural Information Processing Systems 2018, Montréal, QC, Canada, 3–8 December 2018; pp. 7795–7804. [Google Scholar]
- Batra, R.; Tran, H.D.; Kim, C.; Chapman, J.; Chen, L.; Chandrasekaran, A.; Ramprasad, R. A General Atomic Neighborhood Fingerprint for Machine Learning Based Methods. J. Phys. Chem. C 2019, 123, 15859–15866. [Google Scholar] [CrossRef]
- Montavon, G.; Hansen, K.; Fazli, S.; Rupp, M.; Biegler, F.; Ziehe, A.; Tkatchenko, A.; Lilienfeld, A.V.; Müller, K.R. Learning invariant representations of molecules for atomization energy prediction. In Proceedings of the Neural Information Processing Systems 2012, Lake Tahoe, NV, USA, 3–6 December 2012; pp. 440–448. [Google Scholar]
- Faber, F.; Lindmaa, A.; von Lilienfeld, O.A.; Armiento, R. Crystal structure representations for machine learning models of formation energies. Int. J. Quantum Chem. 2015, 115, 1094–1101. [Google Scholar] [CrossRef]
- Himanen, L.; Jäger, M.O.; Morooka, E.V.; Canova, F.F.; Ranawat, Y.S.; Gao, D.Z.; Rinke, P.; Foster, A.S. DScribe: Library of descriptors for machine learning in materials science. Comput. Phys. Commun. 2020, 247, 106949. [Google Scholar] [CrossRef]
- Bartók, A.P.; Kondor, R.; Csányi, G. On representing chemical environments. Phys. Rev. B 2013, 87, 184115. [Google Scholar] [CrossRef]
- De, S.; Bartók, A.P.; Csányi, G.; Ceriotti, M. Comparing molecules and solids across structural and alchemical space. Phys. Chem. Chem. Phys. 2016, 18, 13754–13769. [Google Scholar] [CrossRef] [PubMed]
- Caro, M.A. Optimizing many-body atomic descriptors for enhanced computational performance of machine-learning-based interatomic potentials. arXiv 2019, arXiv:1905.02142. [Google Scholar] [CrossRef]
- Behler, J.; Lorenz, S.; Reuter, K. Representing molecule-surface interactions with symmetry-adapted neural networks. J. Chem. Phys. 2007, 127, 07B603. [Google Scholar] [CrossRef] [PubMed]
- Behler, J. Atom-centered symmetry functions for constructing high-dimensional neural network potentials. J. Chem. Phys. 2011, 134, 074106. [Google Scholar] [CrossRef] [PubMed]
- Gastegger, M.; Schwiedrzik, L.; Bittermann, M.; Berzsenyi, F.; Marquetand, P. wACSF-Weighted atom-centered symmetry functions as descriptors in machine learning potentials. J. Chem. Phys. 2018, 148, 241709. [Google Scholar] [CrossRef]
- Ragoza, M.; Hochuli, J.; Idrobo, E.; Sunseri, J.; Koes, D.R. Protein–ligand scoring with convolutional neural networks. J. Chem. Inf. Model. 2017, 57, 942–957. [Google Scholar] [CrossRef] [PubMed]
- Sunseri, J.; King, J.E.; Francoeur, P.G.; Koes, D.R. Convolutional neural network scoring and minimization in the D3R 2017 community challenge. J. Comput.-Aided Mol. Des. 2019, 33, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Steinbeck, C.; Han, Y.; Kuhn, S.; Horlacher, O.; Luttmann, E.; Willighagen, E. The Chemistry Development Kit (CDK): An open-source Java library for chemo-and bioinformatics. J. Chem. Inf. Comput. Sci. 2003, 43, 493–500. [Google Scholar] [CrossRef]
- Steinbeck, C.; Hoppe, C.; Kuhn, S.; Floris, M.; Guha, R.; Willighagen, E.L. Recent developments of the chemistry development kit (CDK)-an open-source java library for chemo-and bioinformatics. Curr. Pharm. Des. 2006, 12, 2111–2120. [Google Scholar] [CrossRef]
- May, J.W.; Steinbeck, C. Efficient ring perception for the Chemistry Development Kit. J. Cheminform. 2014, 6, 3. [Google Scholar] [CrossRef]
- Willighagen, E.L.; Mayfield, J.W.; Alvarsson, J.; Berg, A.; Carlsson, L.; Jeliazkova, N.; Kuhn, S.; Pluskal, T.; Rojas-Chertó, M.; Spjuth, O.; et al. The Chemistry Development Kit (CDK) v2. 0: Atom typing, depiction, molecular formulas, and substructure searching. J. Cheminform. 2017, 9, 33. [Google Scholar] [CrossRef]
- ChemDes. Available online: http://www.scbdd.com/chemdes/ (accessed on 12 December 2019).
- Backman, T.W.; Cao, Y.; Girke, T. ChemMine tools: An online service for analyzing and clustering small molecules. Nucleic Acids Res. 2011, 39, W486–W491. [Google Scholar] [CrossRef]
- ChemMine Tools. Available online: https://chemminetools.ucr.edu/ (accessed on 12 December 2019).
- OEChem Toolkit. Available online: https://docs.eyesopen.com/toolkits/python/oechemtk/index.html (accessed on 12 December 2019).
- Stahl, M.; Mauser, H. Database clustering with a combination of fingerprint and maximum common substructure methods. J. Chem. Inf. Model. 2005, 45, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Marcou, G.; Rognan, D. Optimizing fragment and scaffold docking by use of molecular interaction fingerprints. J. Chem. Inf. Model. 2007, 47, 195–207. [Google Scholar] [CrossRef]
- Open Babel. Available online: http://openbabel.org/ (accessed on 12 December 2019).
- O’Boyle, N.M.; Morley, C.; Hutchison, G.R. Pybel: A Python wrapper for the OpenBabel cheminformatics toolkit. Chem. Cent. J. 2008, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- PaDEL-Descriptor. Available online: http://www.yapcwsoft.com/dd/padeldescriptor/ (accessed on 12 December 2019).
- PubChemPy. Available online: https://pubchempy.readthedocs.io/en/latest/ (accessed on 12 December 2019).
- RDKit. Available online: https://www.rdkit.org/ (accessed on 12 December 2019).
- Mueller, T.; Kusne, A.G.; Ramprasad, R. Machine learning in materials science: Recent progress and emerging applications. Rev. Comput. Chem. 2016, 29, 186–273. [Google Scholar]
- Pardakhti, M.; Moharreri, E.; Wanik, D.; Suib, S.L.; Srivastava, R. Machine learning using combined structural and chemical descriptors for prediction of methane adsorption performance of metal organic frameworks (MOFs). ACS Comb. Sci. 2017, 19, 640–645. [Google Scholar] [CrossRef]
- Wilmer, C.E.; Leaf, M.; Lee, C.Y.; Farha, O.K.; Hauser, B.G.; Hupp, J.T.; Snurr, R.Q. Large-scale screening of hypothetical metal–organic frameworks. Nat. Chem. 2012, 4, 83. [Google Scholar] [CrossRef]
- Fink, T.; Reymond, J.L. Virtual exploration of the chemical universe up to 11 atoms of C, N, O, F: Assembly of 26.4 million structures (110.9 million stereoisomers) and analysis for new ring systems, stereochemistry, physicochemical properties, compound classes, and drug discovery. J. Chem. Inf. Model. 2007, 47, 342–353. [Google Scholar]
- McKay, B.D. Practical Graph Isomorphism; CRC Press: Boca Raton, FL, USA, 1981. [Google Scholar]
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Bryant, S.H. PubChem: A public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009, 37, W623–W633. [Google Scholar] [CrossRef]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2011, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed]
- Knox, C.; Law, V.; Jewison, T.; Liu, P.; Ly, S.; Frolkis, A.; Pon, A.; Banco, K.; Mak, C.; Neveu, V.; et al. DrugBank 3.0: A comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 2010, 39, D1035–D1041. [Google Scholar] [CrossRef] [PubMed]
- Ruddigkeit, L.; Blum, L.C.; Reymond, J.L. Visualization and virtual screening of the chemical universe database GDB-17. J. Chem. Inf. Model. 2013, 53, 56–65. [Google Scholar] [CrossRef]
- Gupta, A.; Müller, A.T.; Huisman, B.J.; Fuchs, J.A.; Schneider, P.; Schneider, G. Generative recurrent networks for de novo drug design. Mol. Inf. 2018, 37, 1700111. [Google Scholar] [CrossRef]
- O’Boyle, N.M. Towards a Universal SMILES representation-A standard method to generate canonical SMILES based on the InChI. J. Cheminform. 2012, 4, 22. [Google Scholar] [CrossRef]
- Samanta, B.; Abir, D.; Jana, G.; Chattaraj, P.K.; Ganguly, N.; Rodriguez, M.G. Nevae: A deep generative model for molecular graphs. In Proceedings of the AAAI Conference on Artificial Intelligence, Honolulu, HI, USA, 27 January–1 February 2019; Volume 33, pp. 1110–1117. [Google Scholar]
- You, J.; Liu, B.; Ying, Z.; Pande, V.; Leskovec, J. Graph convolutional policy network for goal-directed molecular graph generation. In Proceedings of the 32nd International Conference on Neural Information Processing Systems, Montréal, QC, Canada, 3–8 December 2018; pp. 6410–6421. [Google Scholar]
- Zhavoronkov, A.; Ivanenkov, Y.A.; Aliper, A.; Veselov, M.S.; Aladinskiy, V.A.; Aladinskaya, A.V.; Terentiev, V.A.; Polykovskiy, D.A.; Kuznetsov, M.D.; Asadulaev, A.; et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.R.; Schonlau, M.; Welch, W.J. Efficient global optimization of expensive black-box functions. J. Glob. Optim. 1998, 13, 455–492. [Google Scholar] [CrossRef]
- Kim, K.; Kang, S.; Yoo, J.; Kwon, Y.; Nam, Y.; Lee, D.; Kim, I.; Choi, Y.S.; Jung, Y.; Kim, S.; et al. Deep-learning-based inverse design model for intelligent discovery of organic molecules. NPJ Comput. Mater. 2018, 4, 67. [Google Scholar] [CrossRef]
- Haghighatlari, M.; Hachmann, J. Advances of machine learning in molecular modeling and simulation. Curr. Opin. Chem. Eng. 2019, 23, 51–57. [Google Scholar] [CrossRef]
- Zhang, Y.; Apley, D.; Chen, W. Bayesian Optimization for Materials Design with Mixed Quantitative and Qualitative Variables. arXiv 2019, arXiv:1910.01688. [Google Scholar]
- Iyer, A.; Zhang, Y.; Prasad, A.; Tao, S.; Wang, Y.; Schadler, L.; Brinson, L.C.; Chen, W. Data-Centric Mixed-Variable Bayesian Optimization For Materials Design. arXiv 2019, arXiv:1907.02577. [Google Scholar]
- Rasmussen, C. Gaussian Processes in Machine Learning, Summer School on Machine Learning; Springer: Berlin/Heidelberg, Germany, 2003. [Google Scholar]
- Bostanabad, R.; Kearney, T.; Tao, S.; Apley, D.W.; Chen, W. Leveraging the nugget parameter for efficient Gaussian process modeling. Int. J. Numer. Methods Eng. 2018, 114, 501–516. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Mockus, J.; Tiesis, V.; Zilinskas, A. The application of Bayesian methods for seeking the extremum. In Towards Global Optimisation 2; North-Holand: Amsterdam, The Netherlands, 1978. [Google Scholar]
- Kushner, H.J. A new method of locating the maximum point of an arbitrary multipeak curve in the presence of noise. J. Basic Eng. 1964, 86, 97–106. [Google Scholar] [CrossRef]
- Scott, W.; Frazier, P.; Powell, W. The correlated knowledge gradient for simulation optimization of continuous parameters using gaussian process regression. SIAM J. Optim. 2011, 21, 996–1026. [Google Scholar] [CrossRef]
- Shahriari, B.; Swersky, K.; Wang, Z.; Adams, R.P.; De Freitas, N. Taking the human out of the loop: A review of Bayesian optimization. Proc. IEEE 2015, 104, 148–175. [Google Scholar] [CrossRef]
- Wen, C.; Zhang, Y.; Wang, C.; Xue, D.; Bai, Y.; Antonov, S.; Dai, L.; Lookman, T.; Su, Y. Machine learning assisted design of high entropy alloys with desired property. Acta Mater. 2019, 170, 109–117. [Google Scholar] [CrossRef]
- Li, C.; de Celis Leal, D.R.; Rana, S.; Gupta, S.; Sutti, A.; Greenhill, S.; Slezak, T.; Height, M.; Venkatesh, S. Rapid Bayesian optimisation for synthesis of short polymer fiber materials. Sci. Rep. 2017, 7, 5683. [Google Scholar] [CrossRef]
- Xue, D.; Balachandran, P.V.; Hogden, J.; Theiler, J.; Xue, D.; Lookman, T. Accelerated search for materials with targeted properties by adaptive design. Nat. Commun. 2016, 7, 11241. [Google Scholar] [CrossRef]
- Balachandran, P.V.; Xue, D.; Theiler, J.; Hogden, J.; Lookman, T. Adaptive strategies for materials design using uncertainties. Sci. Rep. 2016, 6, 19660. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.; Tian, Y.; Xue, D.; Xue, D.; Zhou, Y.; Ding, X.; Sun, J.; Lookman, T. Accelerated Search for BaTiO3-Based Ceramics with Large Energy Storage at Low Fields Using Machine Learning and Experimental Design. Adv. Sci. 2019, 6, 1901395. [Google Scholar] [CrossRef] [PubMed]
- Winkler, D.A.; Burden, F.R. Bayesian neural nets for modeling in drug discovery. Drug Discov. Today BIOSILICO 2004, 2, 104–111. [Google Scholar] [CrossRef]
- Lavecchia, A. Machine-learning approaches in drug discovery: Methods and applications. Drug Discov. Today 2015, 20, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Madhukar, N.S.; Khade, P.K.; Huang, L.; Gayvert, K.; Galletti, G.; Stogniew, M.; Allen, J.E.; Giannakakou, P.; Elemento, O. A Bayesian machine learning approach for drug target identification using diverse data types. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lookman, T.; Balachandran, P.V.; Xue, D.; Yuan, R. Active learning in materials science with emphasis on adaptive sampling using uncertainties for targeted design. Npj Comput. Mater. 2019, 5, 21. [Google Scholar] [CrossRef]
- Tanaka, T.; Montanari, G.; Mulhaupt, R. Polymer nanocomposites as dielectrics and electrical insulation-perspectives for processing technologies, material characterization and future applications. IEEE Trans. Dielectr. Electr. Insul. 2004, 11, 763–784. [Google Scholar] [CrossRef]
- Weidner, J.R.; Pohlmann, F.; Gröppel, P.; Hildinger, T. Nanotechnology in high voltage insulation systems for turbine generators-First results. In Proceedings of the 17th ISH, Hannover, Germany, 22–26 August 2011. [Google Scholar]
- Torquato, S.; Haslach, H. Random heterogeneous materials: Microstructure and macroscopic properties. Appl. Mech. Rev. 2002, 55, B62–B63. [Google Scholar] [CrossRef]
- Niblack, W. An Introduction to Digital Image Processing; Strandberg Publishing Company: Birkeroed, Denmark, 1985. [Google Scholar]
- Wang, Y.; Zhang, Y.; Zhao, H.; Li, X.; Huang, Y.; Schadler, L.S.; Chen, W.; Brinson, L.C. Identifying interphase properties in polymer nanocomposites using adaptive optimization. Compos. Sci. Technol. 2018, 162, 146–155. [Google Scholar] [CrossRef]
- Zhao, H.; Li, Y.; Brinson, L.C.; Huang, Y.; Krentz, T.M.; Schadler, L.S.; Bell, M.; Benicewicz, B. Dielectric spectroscopy analysis using viscoelasticity-inspired relaxation theory with finite element modeling. IEEE Trans. Dielectr. Electr. Insul. 2017, 24, 3776–3785. [Google Scholar] [CrossRef]
- Zhang, Y.; Tao, S.; Chen, W.; Apley, D.W. A latent variable approach to Gaussian process modeling with qualitative and quantitative factors. Technometrics 2019, 1–12. [Google Scholar] [CrossRef]
- Goldberg, D.E. Genetic Algorithms; Pearson Education India: Delhi, India, 2006. [Google Scholar]
- Polykovskiy, D.; Zhebrak, A.; Sanchez-Lengeling, B.; Golovanov, S.; Tatanov, O.; Belyaev, S.; Kurbanov, R.; Artamonov, A.; Aladinskiy, V.; Veselov, M.; et al. Molecular sets (moses): A benchmarking platform for molecular generation models. arXiv 2018, arXiv:1811.12823. [Google Scholar]
- Van Moffaert, K.; Drugan, M.M.; Nowé, A. Scalarized multi-objective reinforcement learning: Novel design techniques. In Proceedings of the 2013 IEEE Symposium on Adaptive Dynamic Programming and Reinforcement Learning (ADPRL), Singapore, 16–19 April 2013; pp. 191–199. [Google Scholar]
- Van Moffaert, K.; Nowé, A. Multi-objective reinforcement learning using sets of pareto dominating policies. J. Mach. Learn. Res. 2014, 15, 3483–3512. [Google Scholar]
- Mossalam, H.; Assael, Y.M.; Roijers, D.M.; Whiteson, S. Multi-objective deep reinforcement learning. arXiv 2016, arXiv:1610.02707. [Google Scholar]
- Khan, N.; Goldberg, D.E.; Pelikan, M. Multi-objective Bayesian Optimization Algorithm. In Proceedings of the 4th Annual Conference on Genetic and Evolutionary Computation (GECCO’02), New York, NY, USA, 9–13 July 2002; Morgan Kaufmann Publishers Inc.: San Francisco, CA, USA, 2002. [Google Scholar]
- Laumanns, M.; Ocenasek, J. Bayesian optimization algorithms for multi-objective optimization. In Proceedings of the International Conference on Parallel Problem Solving from Nature, Granada, Spain, 7–11 September 2002; pp. 298–307. [Google Scholar]
- Wada, T.; Hino, H. Bayesian Optimization for Multi-objective Optimization and Multi-point Search. arXiv 2019, arXiv:1905.02370. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | Design Feature | Design Scope | Data Size | Representation | ML Model |
|---|---|---|---|---|---|
| Organic photovoltaics (2011) | Self-built library and screening | Power conversion efficiency (molecular level) | 2.6M | Molecular descriptors | MLR |
| Polymer dielectrics | Self-build library; building blocks for molecular generation; genetic algorithm | bandgap and dielectric constant (molecular level) | 284 | Fingerprints | KRR |
| Organic light-emitting diodes | Self-build library and screening; building blocks for molecular generation | delayed fluorescent rate constant (molecular level) | 40,000 | ECFPs | ANN |
| Polymer solar cell (2018) | Self-build library and screening; building blocks for molecular generation; various combinations of feature representations and ML models are compared | highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) (molecular level) | 3938 | Fixed length vector; string; spatial coordinate | LRR; MLP; RF; DTNN; GrammarVAE |
| High-energetic material | Material design with limited data; various combinations of feature representations and ML models are compared | high energy density and low sensitivity (molecular level) | 109; 309 | CDS; SoB; CM; BoB; fingerprints | KRR; RR; SVR; RF; kNN; LASSO; GPR; ANN |
| Polyimides with high refractive index | Self-build library and screening; building blocks for molecular generation; ML model construction with limited data | polarizability and number density (molecular level) | 196 | Number of monomer units | SVM |
| Polymer with high thermal conductivity | ML model construction with limited data; transfer learning | thermal conductivity (molecular level) | 28; 5917; 3234 | ECFPs | Bayesian model |
| de novo drug-like molecule | Material design with arbitrary target property range; SMILES strings as input for molecular generation | physical/chemical/biological properties (molecular level) | 1.5M | SMILES | DNN; RL |
| Organic photovoltaic solar cells (2019) | Polymer composite design; bottom-up nanofabrication; microstructure characterization and reconstruction | IPCEefficiency (microstructure level) | 45 | Microstructure characterization | SDF |
| Database | Type | Description | URL |
|---|---|---|---|
| AFLOWLIB | Computation | Database of 2,961,744 material compounds with over 527,190,432 calculated properties | http://aflowlib.org |
| BNPAH | Computation | Structures and properties of 77 polycyclic aromatic hydrocarbons and 33,059 B, N substituted compounds | https://moldis.tifrh.res.in/datasets.html |
| ChemDiv | Comp./Exp. | Collection of over 1,500,000 individually crafted, lead-like, drug-like small molecules | http://www.chemdiv.com/complete-list/ |
| ChemSpider | Experiment | A free chemical structure database providing fast text and structure search access to over 67 million structures | https://chemspider.com |
| ChEMBL | Experiment | A manually-curated database of bioactive molecules with drug-like properties | https://www.ebi.ac.uk/chembl |
| Citrination | Experiment | A premier open database and analytics platform for the world’s material and chemical information | https://citrination.com |
| CMR | Computation | A collection of molecules obtained from electron-structure codes | https://cmr.fysik.dtu.dk |
| COD | Experiment | A collection of crystal structures of organic, inorganic, metal-organics compounds, and minerals, excluding biopolymers | http://www.crystallography.net/cod/ |
| CSD | Experiment | A database of over one million small-molecule organic and metal-organic crystal structures | https://www.ccdc.cam.ac.uk |
| DrugBank | Experiments | Drug database with comprehensive drug target information | https://www.drugbank.ca/ |
| eMolecules | N/A | Commercially available with over seven million compounds for drug discovery | https://reaxys.emolecules.com/index.php |
| Energetics | Computation | A database of energetic molecules | https://git.io/energeticmols |
| GDB | Computation | A database containing hypothetical small organic molecules | http://gdb.unibe.ch/downloads |
| HCEP | Computation | Harvard Clean Energy project for solar absorber materials | https://cepdb.molecularspace.org |
| HOPV15 | Comp./Exp. | A collation of experimental photovoltaic data from the literature and calibrated by DFT calculation | https://figshare.com/articles/HOPV15_Dataset/1610063/4 |
| ICSD | Experiment | A database of inorganic crystal structure | https://icsd.fiz-karlsruhe.de |
| MatNavi | Experiment | A materials databases of polymer, ceramic, alloy, superconducting material, composite, and diffusion | http://mits.nims.go.jp |
| MatWeb | Experiment | A database of material properties of polymers, metals, ceramics, and semiconductor | http://matweb.com |
| MP | Computation | Computed information on known and predicted materials | https://materialsproject.org |
| NIST CW | Experiment | A database of thermochemical properties | https://webbook.nist.gov/chemistry |
| NIST MDR | Experiment | A repository of material data being updated | https://materialsdata.nist.gov |
| NOMAD | Computation | A repository to host, organize, and share material data | https://nomad-repository.eu |
| NREL MD | Computation | A computational materials database for renewable energy applications | https://materials.nrel.gov |
| OQMD | Computation | A database of DFT-calculated thermodynamic and structural properties | http://oqmd.org |
| PubChem | Experiment | A chemical database of chemical and physical properties, biological activities, and safety and toxicity information | https://pubchem.ncbi.nlm.nih.gov |
| QM | Computation | Small organic molecules calculated by DFT | http://quantum-machine.org/datasets/ |
| TEDesignLab | Comp./Exp. | Thermoelectric material design | http://tedesignlab.org |
| ZINC | Computation | Database of commercially-available compounds for virtual screening | https://zinc15.docking.org |
| Representation | Description | References |
| SMILES | Line notation for describing a chemical structure using text strings | [87,112,115,116] |
| Fingerprints | A special descriptor using vector of fixed or variable length to represent a chemical structure | [58,143,155,161] |
| Molecular graphs | A representation of chemical structures by graph theory | [157,158,159,160] |
| Coulomb matrix | A matrix representation embedded nuclear coordinates and charges, similar representations include Ewald sum matrix, Sine matrix | [90,91,162,163,164] |
| Smooth overlap of atomic orbitals (SOAP) | A special descriptor encoding atomic structures using local expansion of atomic density | [165,166,167] |
| Atom-centereded symmetry functions (ACSF) | A special descriptor representing the local environment near an atom using two- or three-body functions | [168,169,170] |
| Bag of bonds | A vector enclosing chemical bonds and corresponding numbers | [91,92] |
| Grids of molecules | A visual form of molecules generated by their coordinates | [61,171,172] |
| Tools | Description | References |
| CDK | Chemistry Development Kit: open-source Java libraries for cheminformatics to generate various descriptors, fingerprints, etc. | [173,174,175,176] |
| ChemDes | A free web-based tool for generation of molecular descriptors (3679 types) and fingerprints (59 types) | [144,177] |
| ChemMine | A free online tool for analyzing and clustering small molecules, including similarity search and properties calculations | [178,179] |
| OEChem | Programming library for chemistry and cheminformatics with small molecules | [180,181,182] |
| Open Bable/Pybel | Open-source chemical toolbox to search, convert, analyze, and store data | [183,184,185] |
| PaDEL | A software to generate molecular descriptors (1875 types) and fingerprints (12 types) using CDK | [186,187] |
| PubChemPy | An open-source python library to interact with PubChem | [188] |
| RDKit | A collection of cheminformatics and machine-learning tools | [86,189] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, G.; Shen, Z.; Iyer, A.; Ghumman, U.F.; Tang, S.; Bi, J.; Chen, W.; Li, Y. Machine-Learning-Assisted De Novo Design of Organic Molecules and Polymers: Opportunities and Challenges. Polymers 2020, 12, 163. https://doi.org/10.3390/polym12010163
Chen G, Shen Z, Iyer A, Ghumman UF, Tang S, Bi J, Chen W, Li Y. Machine-Learning-Assisted De Novo Design of Organic Molecules and Polymers: Opportunities and Challenges. Polymers. 2020; 12(1):163. https://doi.org/10.3390/polym12010163
Chicago/Turabian StyleChen, Guang, Zhiqiang Shen, Akshay Iyer, Umar Farooq Ghumman, Shan Tang, Jinbo Bi, Wei Chen, and Ying Li. 2020. "Machine-Learning-Assisted De Novo Design of Organic Molecules and Polymers: Opportunities and Challenges" Polymers 12, no. 1: 163. https://doi.org/10.3390/polym12010163
APA StyleChen, G., Shen, Z., Iyer, A., Ghumman, U. F., Tang, S., Bi, J., Chen, W., & Li, Y. (2020). Machine-Learning-Assisted De Novo Design of Organic Molecules and Polymers: Opportunities and Challenges. Polymers, 12(1), 163. https://doi.org/10.3390/polym12010163