Effects of Diisocyanate Structure and Disulfide Chain Extender on Hard Segmental Packing and Self-Healing Property of Polyurea Elastomers

 ,
,  and
and
Abstract
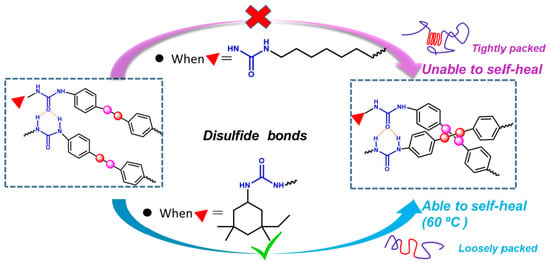
:
1. Introduction
2. Materials and Methods
2.1. Materials
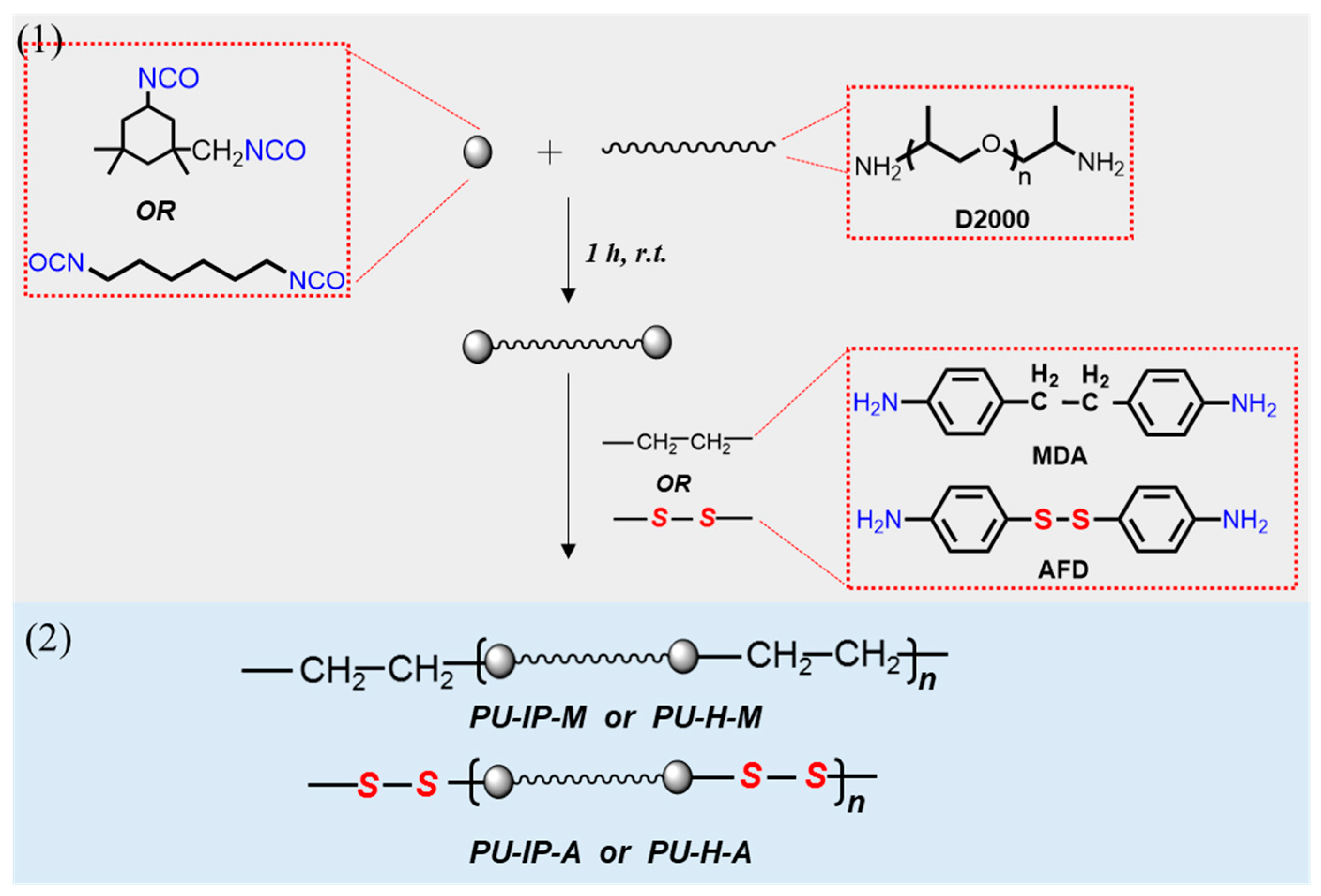
2.2. Sample Preparation
2.3. Characterizations
2.3.1. Fourier Transform Infrared Spectroscopy (FTIR) and Nuclear Magnetic Resonance (NMR)
2.3.2. Gel Permeation Chromatography (GPC)
2.3.3. Dynamic Mechanical Analysis (DMA)
2.3.4. Stress-Relaxation Experiments
2.3.5. Mechanical Evaluation
2.3.6. Small Angle X-ray Scattering (SAXS) and X-Ray Diffraction (XRD)
2.3.7. Thermal Analysis
2.3.8. Optical Analysis
2.3.9. Broadband Dielectric Spectrometer (BDS)
3. Results and Discussion
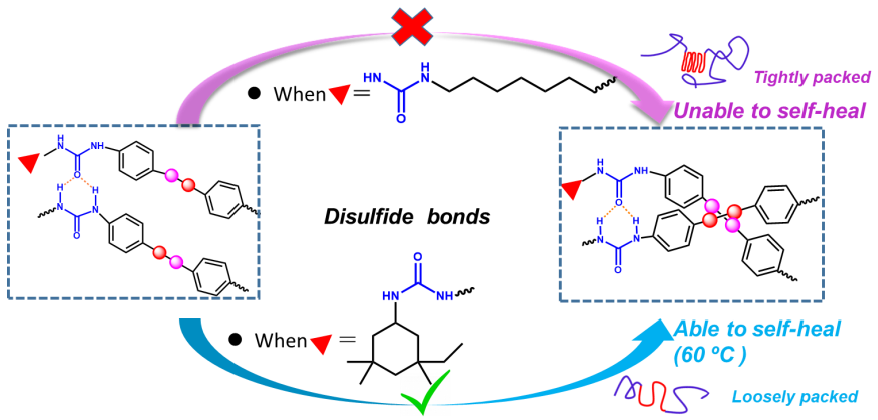
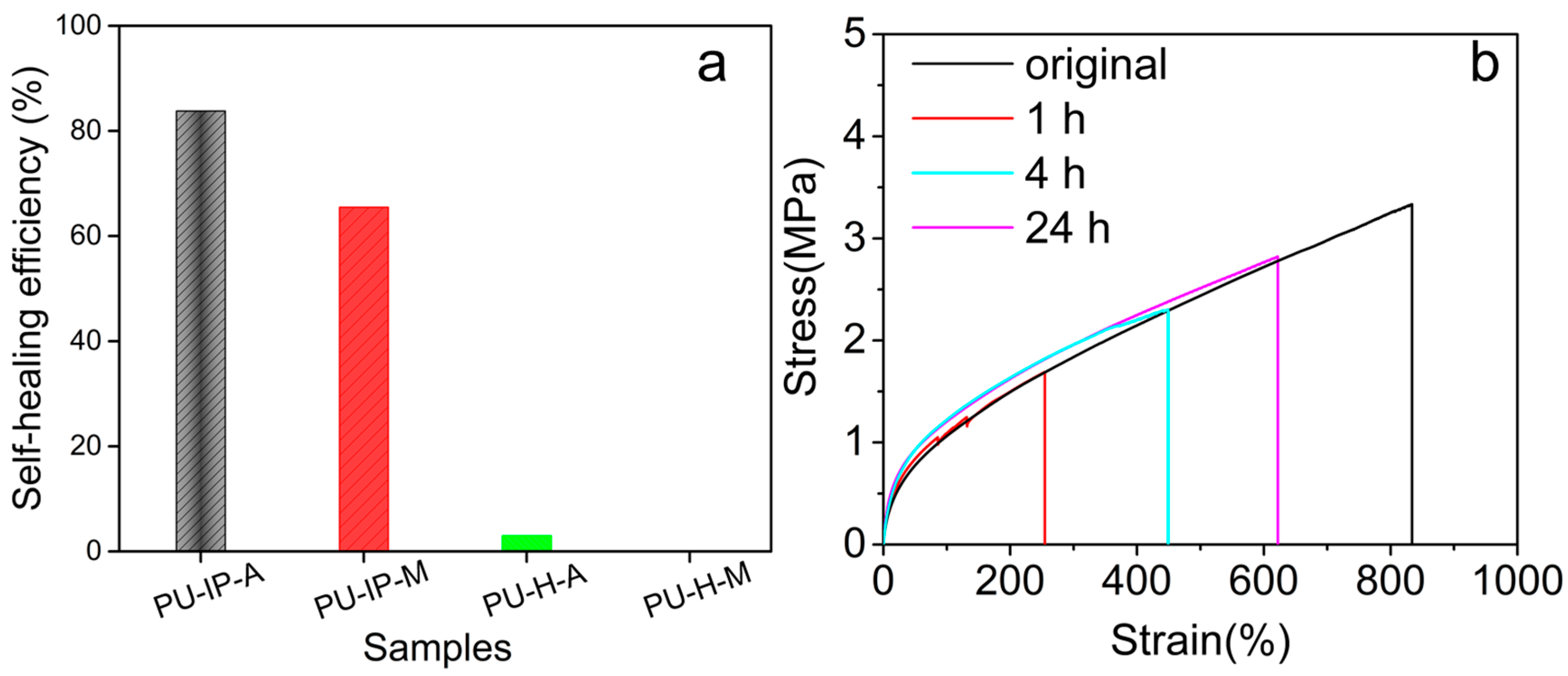
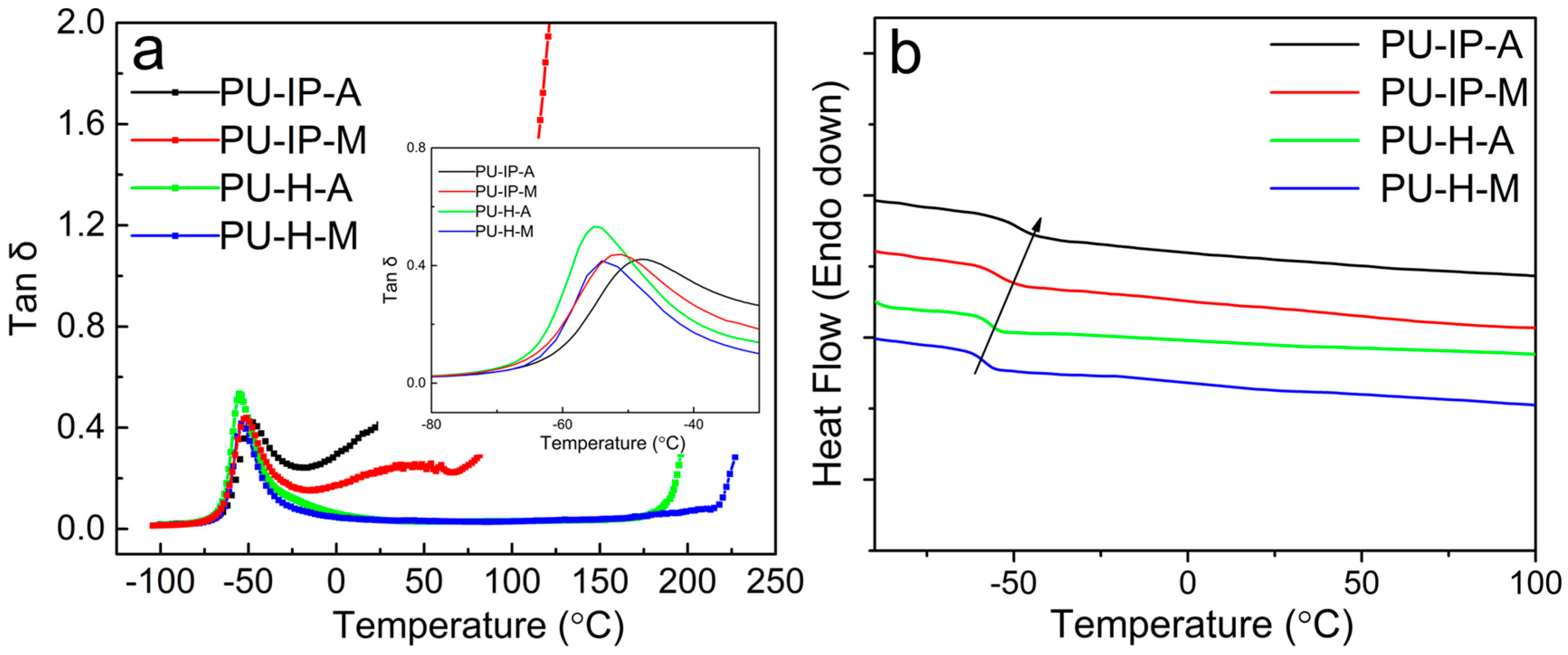
3.1. Self-Healing Ability Evaluation
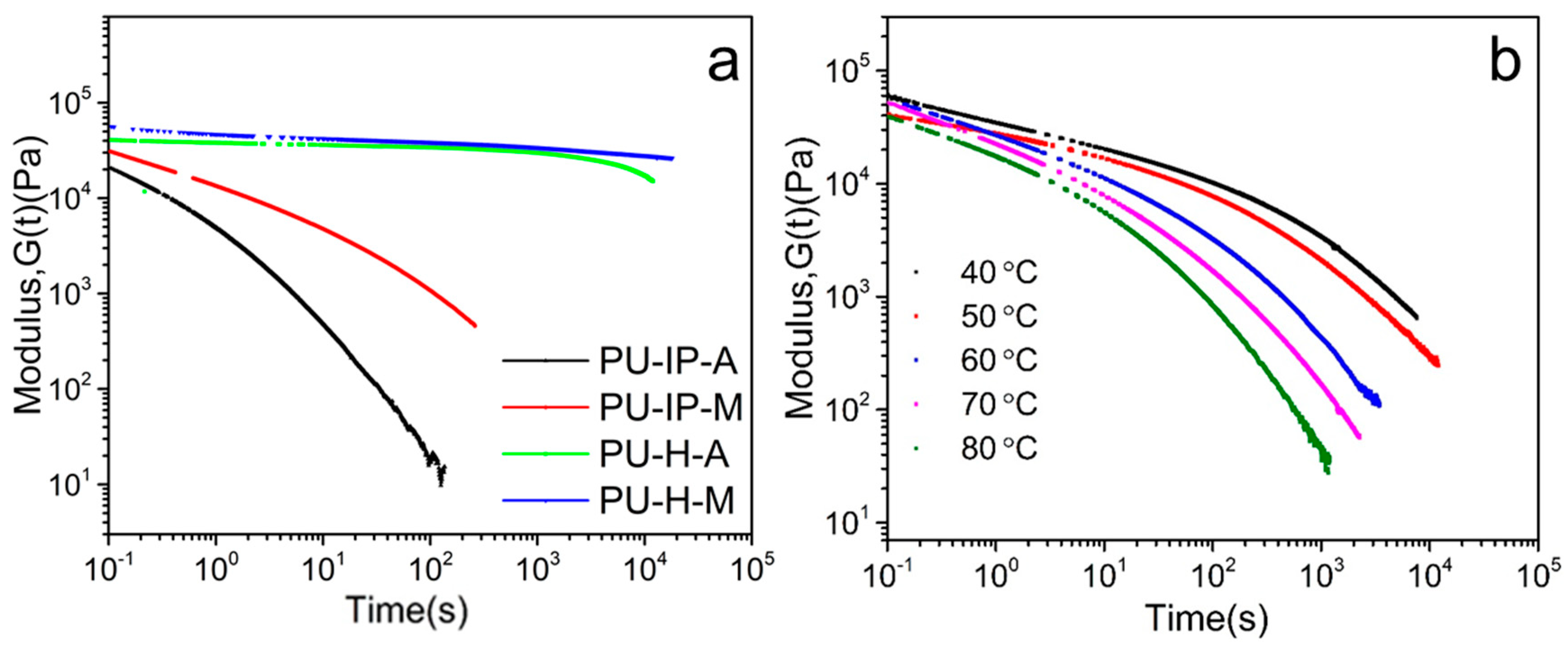
3.2. Chain Mobility Analysis from a Rheological Perspective
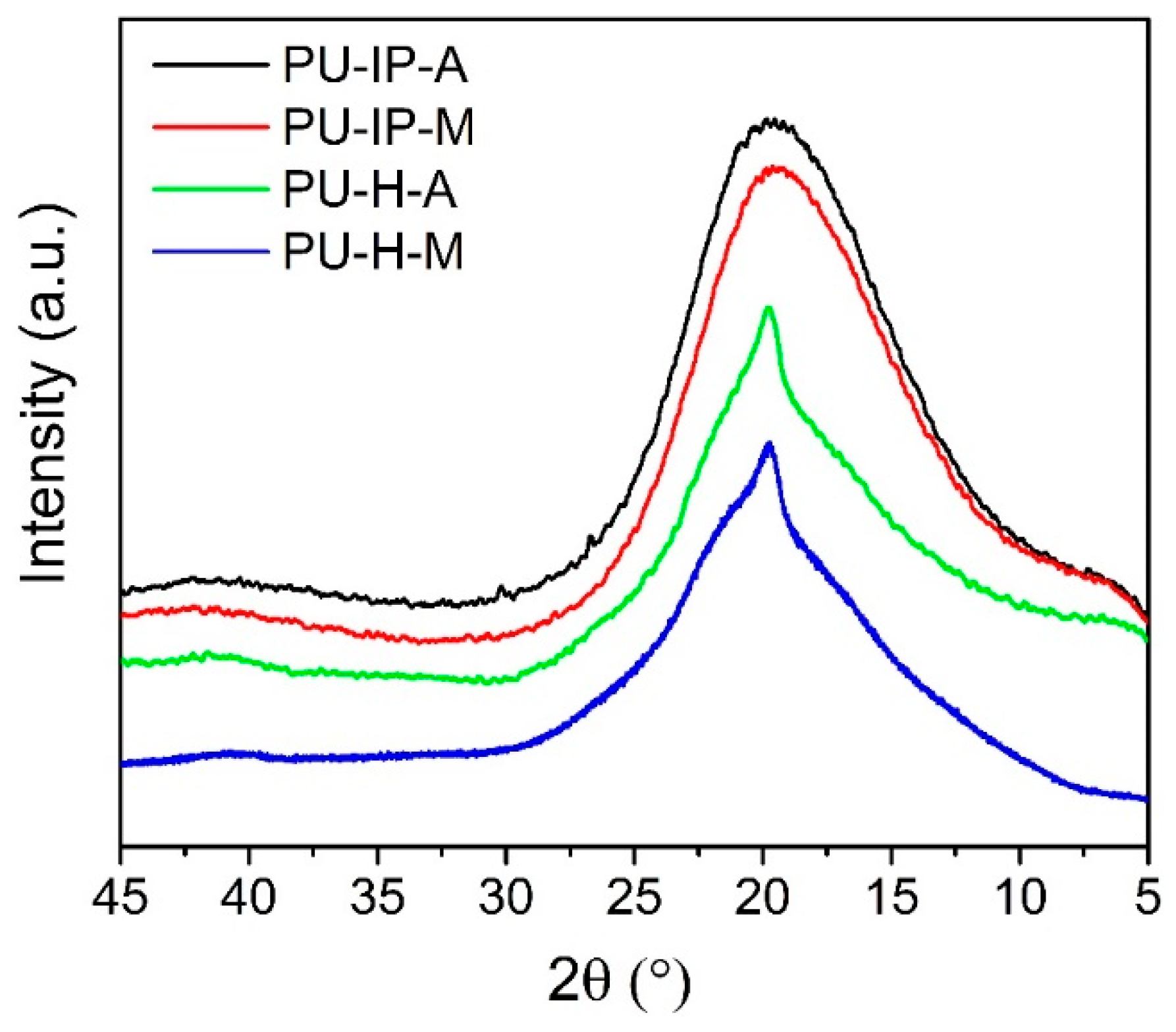
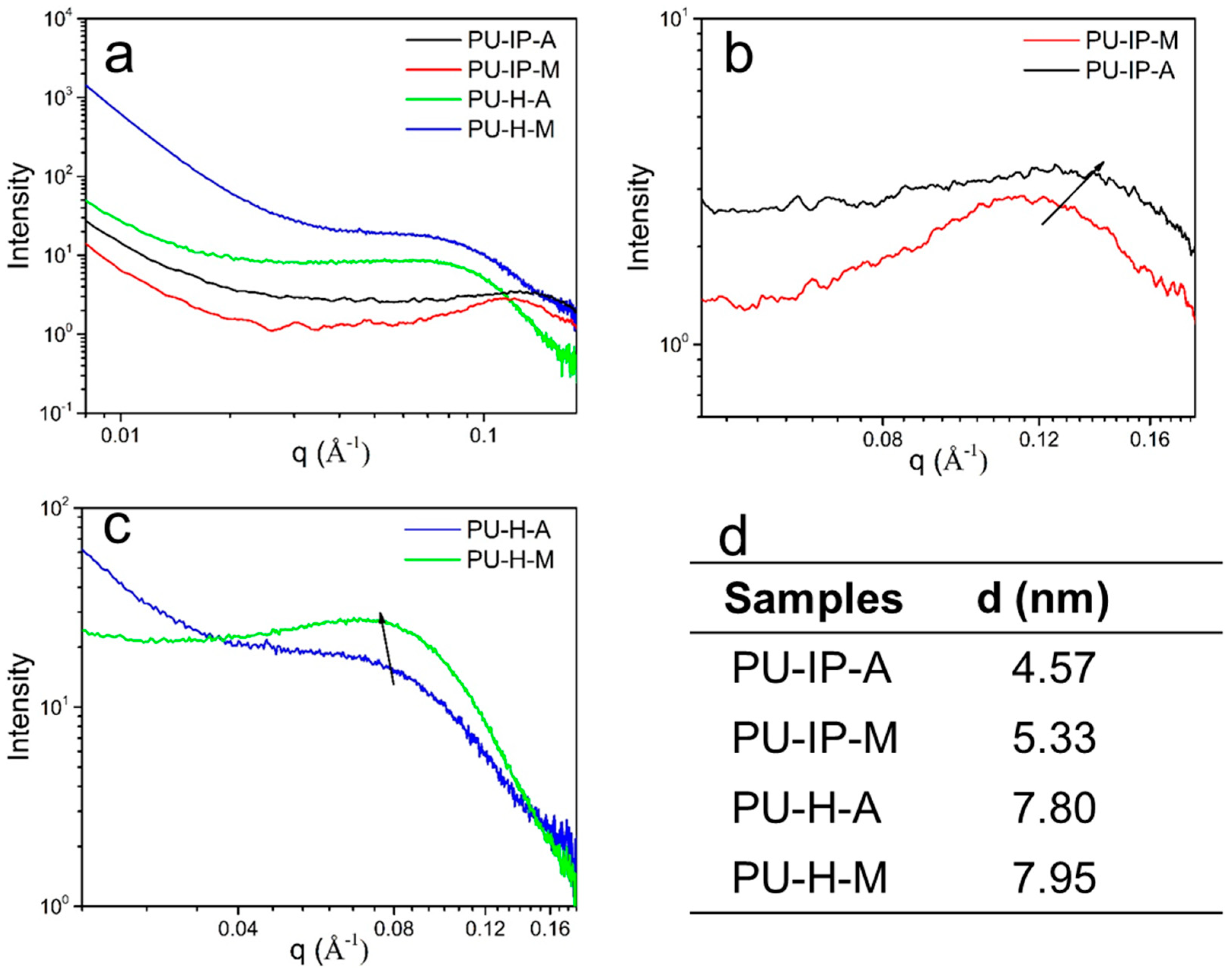
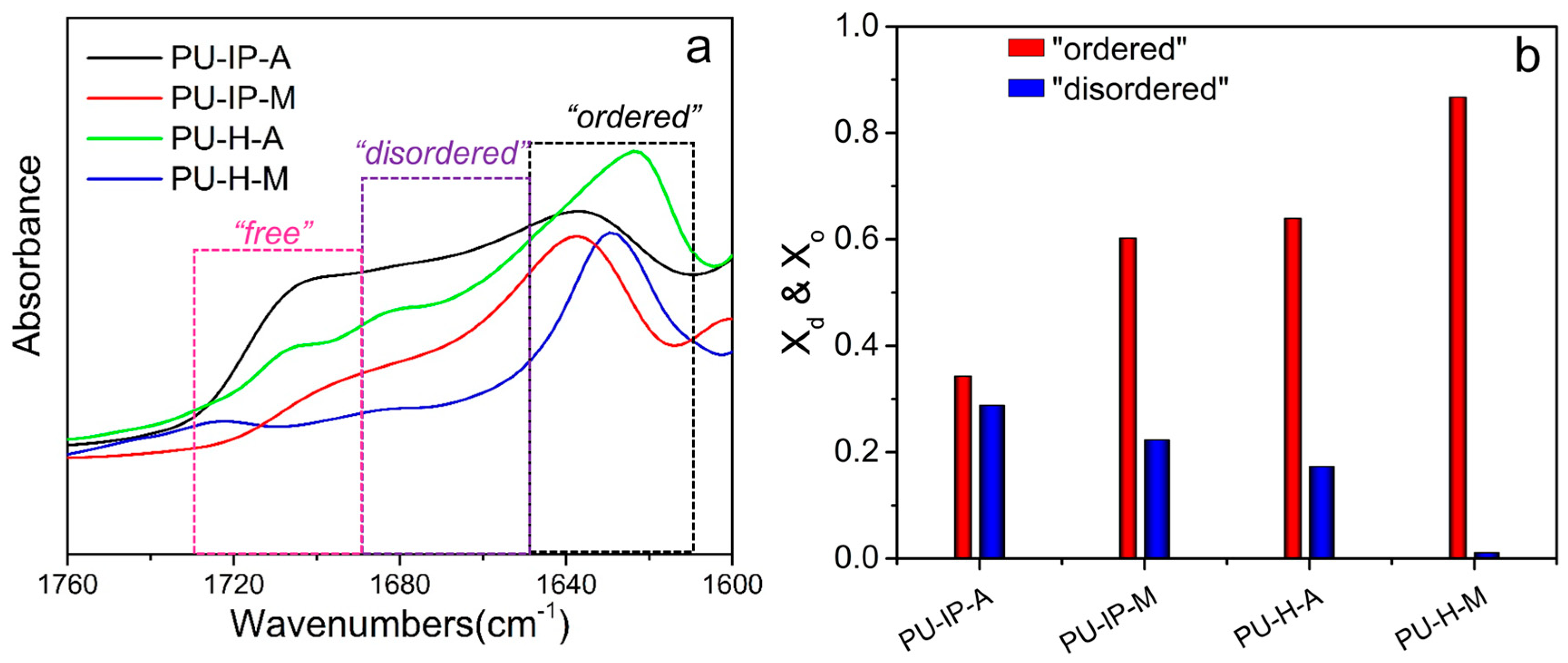
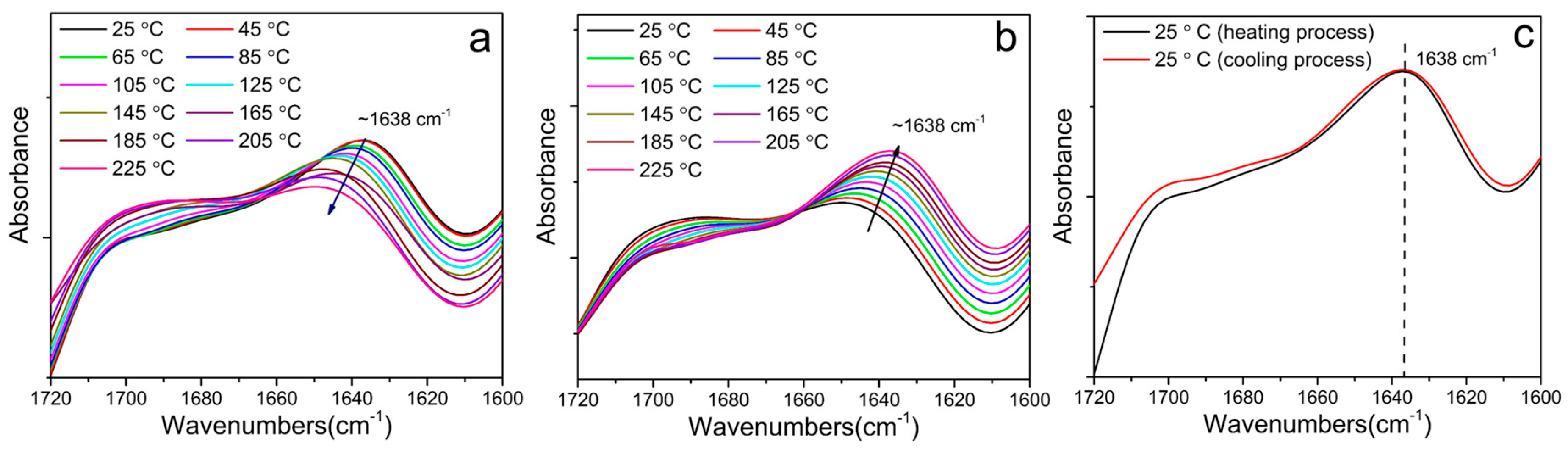
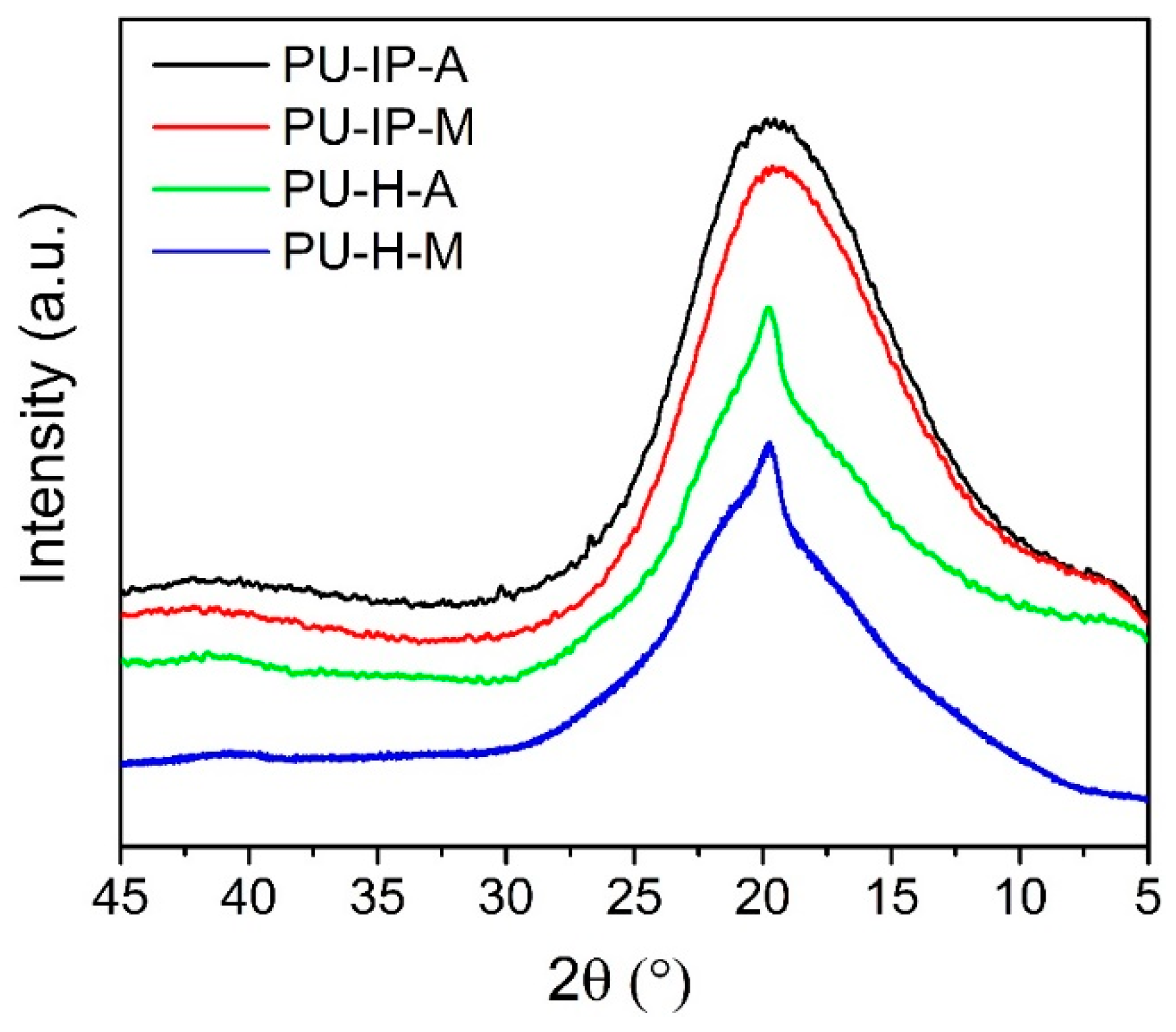
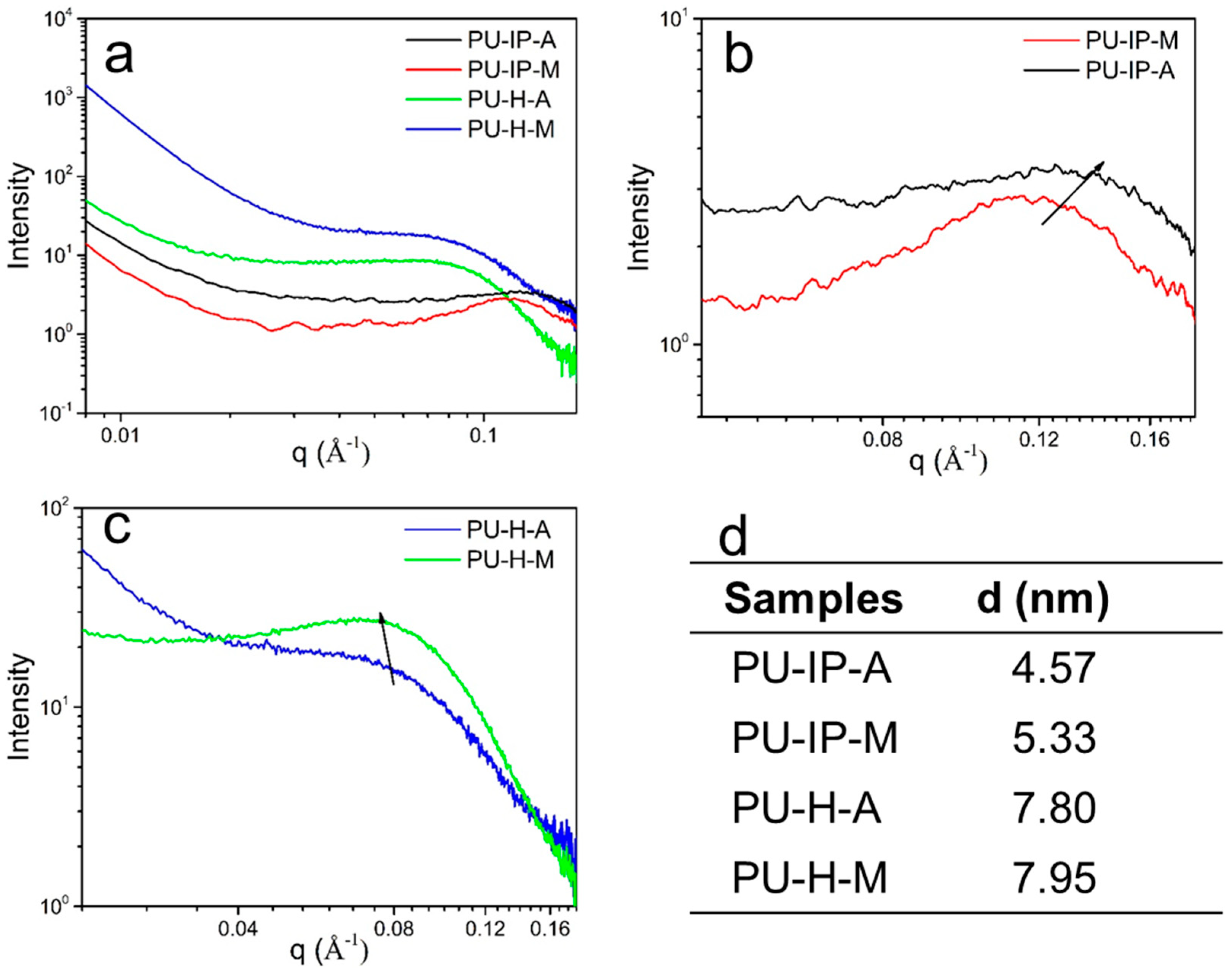
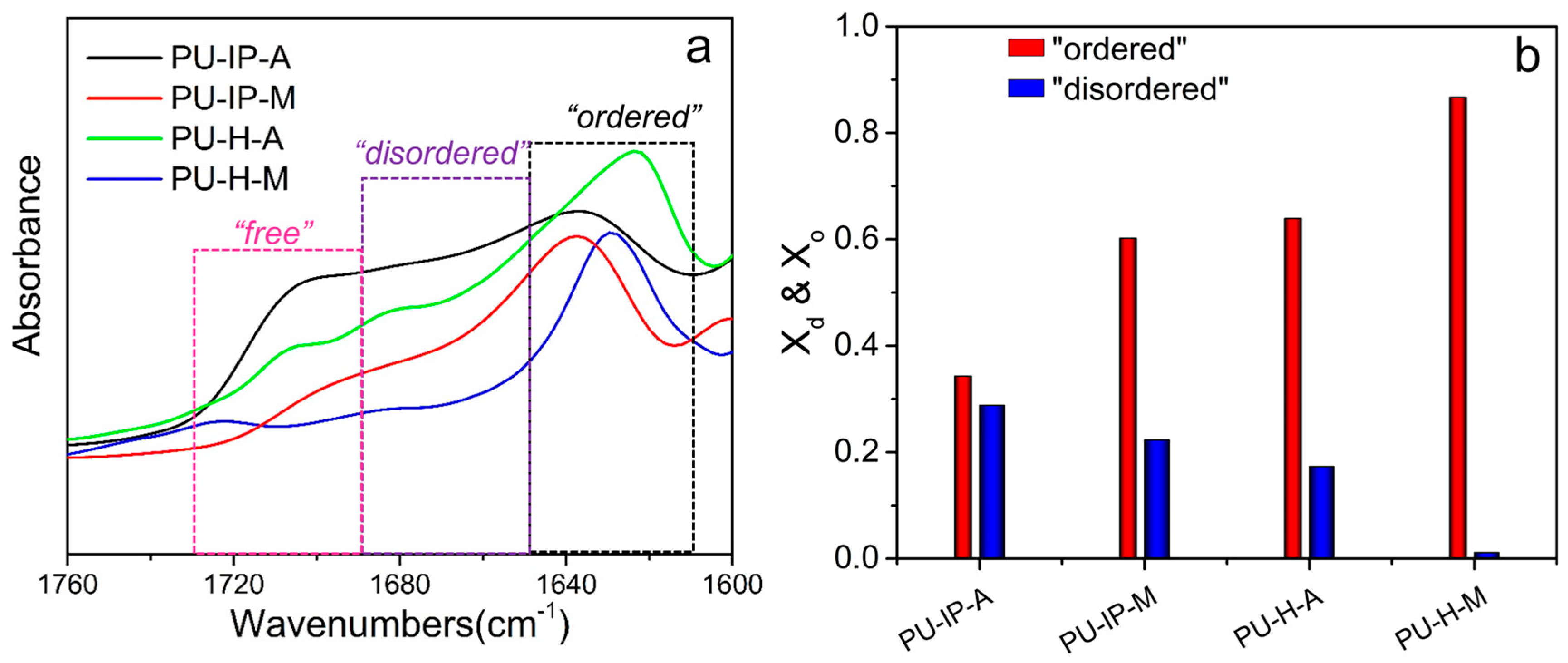
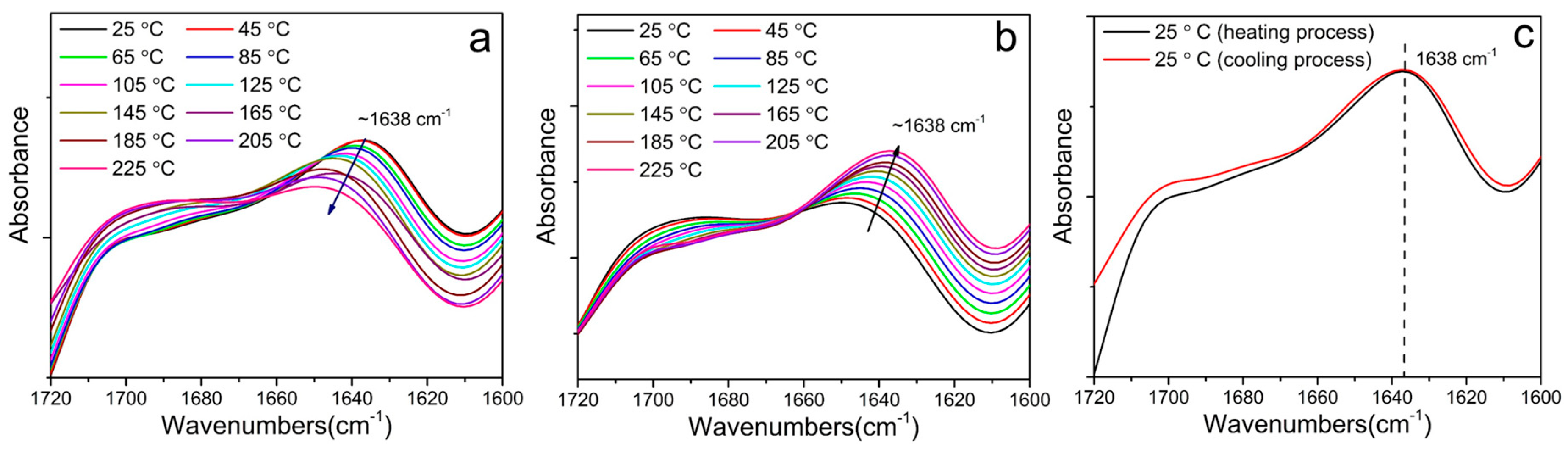
3.3. Hard Segmental Packing in All Samples
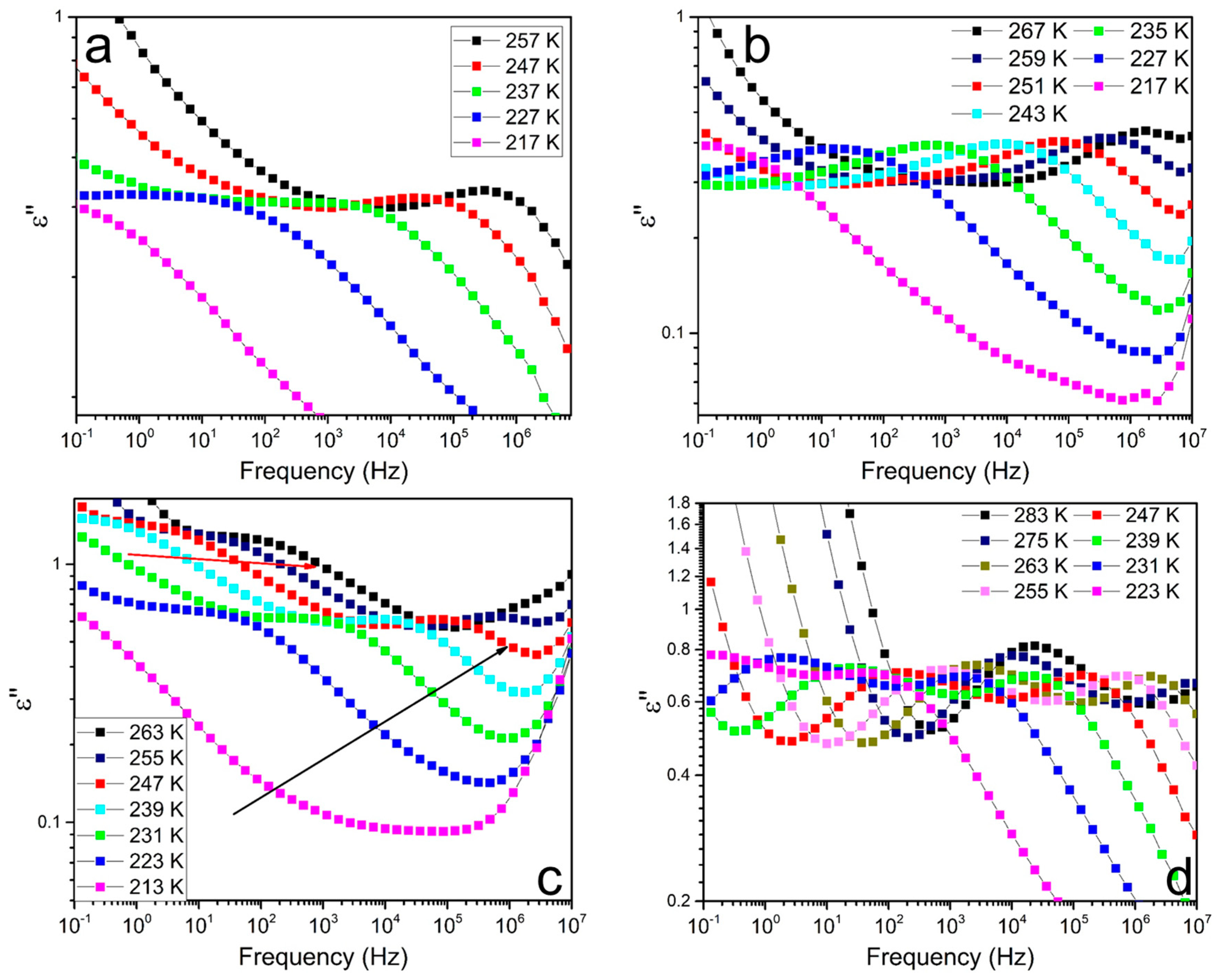
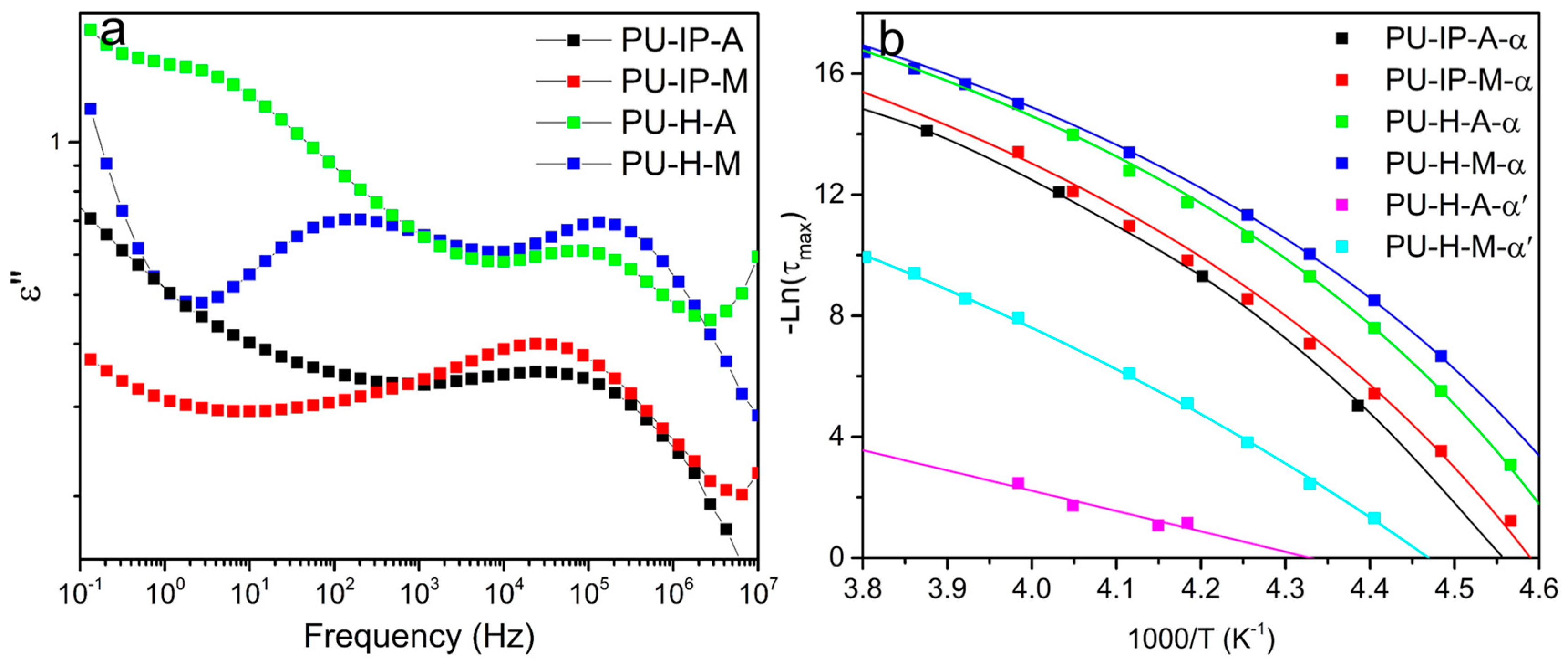
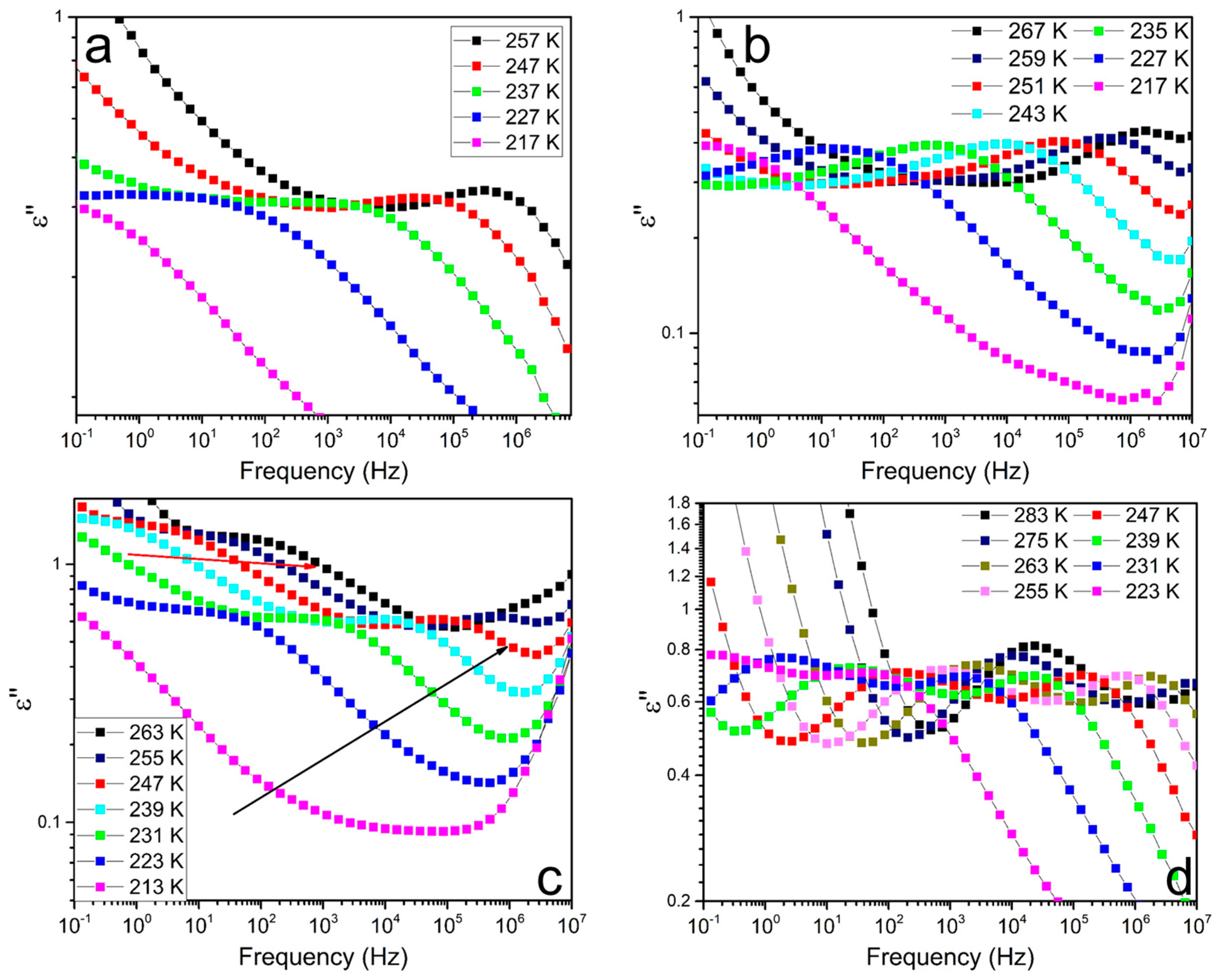
3.4. Soft Segmental Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Qian, X.; Song, L.; Tai, Q.; Hu, Y.; Yuen, R.K. Graphite oxide/polyurea and graphene/polyurea nanocomposites: A comparative investigation on properties reinforcements and mechanism. Compos. Sci. Technol. 2013, 74, 228–234. [Google Scholar] [CrossRef]
- Casalini, R.; Bogoslovov, R.; Qadri, S.; Roland, C. Nanofiller reinforcement of elastomeric polyurea. Polymer 2012, 53, 1282–1287. [Google Scholar] [CrossRef]
- Liu, C.; Xie, Q.; Ma, C.; Zhang, G. Fouling Release Property of Polydimethylsiloxane Based Polyurea with Improved Adhesion to Substrate. Ind. Eng. Chem. Res. 2016, 55, 6671–6676. [Google Scholar] [CrossRef]
- Chattopadhyay, D.K.; Raju, K.V.S.N. Structural engineering of polyurethane coatings for high performance applications. Prog. Polym. Sci. 2007, 32, 352–418. [Google Scholar] [CrossRef]
- Yang, W.J.; Tao, X.; Zhao, T.; Weng, L.; Kang, E.T.; Wang, L.H. Antifouling and Antibacterial Hydrogel Coatings with Self-healing Properties Based on Dynamic Disulfide Exchange Reaction. Polym. Chem. 2015, 6, 7027–7035. [Google Scholar] [CrossRef]
- Liu, C.; Ma, C.; Xie, Q.; Zhang, G. Self-repairing silicone coating for marine anti-biofouling. J. Mater. Chem. A 2017, 5, 15855–15861. [Google Scholar] [CrossRef]
- Chen, X.; Dam, M.A.; Ono, K.; Mal, A.; Shen, H.; Nutt, S.R.; Sheran, K.; Wudl, F. A thermally re-mendable cross-linked polymeric material. Science 2002, 295, 1698–1702. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-X.; Tournilhac, F.; Leibler, L.; Guan, Z. Making insoluble polymer networks malleable via olefin metathesis. J. Am. Chem. Soc. 2012, 134, 8424–8427. [Google Scholar] [CrossRef]
- Lei, Z.Q.; Xie, P.; Rong, M.Z.; Zhang, M.Q. Catalyst-free dynamic exchange of aromatic Schiff base bonds and its application to self-healing and remolding of crosslinked polymers. J. Mater. Chem. A 2015, 3, 19662–19668. [Google Scholar] [CrossRef]
- Zhang, Z.P.; Rong, M.Z.; Zhang, M.Q.; Yuan, C.E. Alkoxyamine with reduced homolysis temperature and its application in repeated autonomous self-healing of stiff polymers. Polym. Chem. 2013, 4, 4648–4654. [Google Scholar] [CrossRef]
- De Luzuriaga, A.R.; Martin, R.; Markaide, N.; Rekondo, A.; Cabañero, G.; Rodríguez, J.; Odriozola, I. Epoxy resin with exchangeable disulfide crosslinks to obtain reprocessable, repairable and recyclable fiber-reinforced thermoset composites. Mater. Horiz. 2016, 3, 241–247. [Google Scholar] [CrossRef]
- Li, T.; Xie, Z.; Xu, J.; Weng, Y.; Guo, B.-H. Design of a self-healing cross-linked polyurea with dynamic cross-links based on disulfide bonds and hydrogen bonding. Eur. Polym. J. 2018, 107, 249–257. [Google Scholar] [CrossRef]
- Rinaldi, R.; Boyce, M.; Weigand, S.; Londono, D.; Guise, M. Microstructure evolution during tensile loading histories of a polyurea. J. Polym. Sci. Part B Polym. Phys. 2011, 49, 1660–1671. [Google Scholar] [CrossRef]
- Castagna, A.M.; Pangon, A.; Dillon, G.P.; Runt, J. Effect of Thermal History on the Microstructure of a Poly (tetramethylene oxide)-Based Polyurea. Macromolecules 2013, 46, 6520–6527. [Google Scholar] [CrossRef]
- Wool, R.P.; O’Connor, K.M. A theory crack healing in polymers. J. Appl. Phys. 1981, 52, 5953–5963. [Google Scholar] [CrossRef]
- Das, S.; Cox, D.F.; Wilkes, G.L.; Klinedinst, D.B.; Yilgor, I.; Yilgor, E.; Beyer, F. Effect of Symmetry and Hydrogen bond Strength of Hard Segments on the Structure Property Relationships of Segmented, Nonchain Extended Polyurethanes and Polyureas. J. Macromol. Sci. Part B 2007, 46, 853–875. [Google Scholar] [CrossRef]
- Kim, S.M.; Jeon, H.; Shin, S.H.; Park, S.A.; Jegal, J.; Hwang, S.Y.; Oh, D.X.; Park, J. Superior Toughness and Fast Self-Healing at Room Temperature Engineered by Transparent Elastomers. Adv. Mater. 2018, 30, 1705145. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Rekondo, A.; de Luzuriaga, A.R.; Cabañero, G.; Grande, H.J.; Odriozola, I. The processability of a poly (urea-urethane) elastomer reversibly crosslinked with aromatic disulfide bridges. J. Mater. Chem. A 2014, 2, 5710–5715. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, L.; Rowan, S.J. Trapping dynamic disulfide bonds in the hard segments of thermoplastic polyurethane elastomers. Macromol. Chem. Phys. 2017, 218, 1600320. [Google Scholar] [CrossRef]
- Sami, S.; Yildirim, E.; Yurtsever, M.; Yurtsever, E.; Yilgor, E.; Yilgor, I.; Wilkes, G.L. Understanding the influence of hydrogen bonding and diisocyanate symmetry on the morphology and properties of segmented polyurethanes and polyureas: Computational and experimental study. Polymer 2014, 55, 4563–4576. [Google Scholar] [CrossRef]
- Sheth, J.P.; Klinedinst, D.B.; Wilkes, G.L.; Yilgor, I.; Yilgor, E. Role of chain symmetry and hydrogen bonding in segmented copolymers with monodisperse hard segments. Polymer 2005, 46, 7317–7322. [Google Scholar] [CrossRef]
- Chen, X.; Wudl, F.; Mal, A.K.; Shen, H.; Nutt, S.R. New thermally remendable highly cross-linked polymeric materials. Macromolecules 2003, 36, 1802–1807. [Google Scholar] [CrossRef]
- Xu, W.M.; Rong, M.Z.; Zhang, M.Q. Sunlight driven self-healing, reshaping and recycling of robust, transparent and yellowing-resistant polymer. J. Mater. Chem. A 2016, 4, 10683–10690. [Google Scholar] [CrossRef]
- Burattini, S.; Colquhoun, H.M.; Fox, J.D.; Friedmann, D.; Greenland, B.W.; Harris, P.J.; Hayes, W.; Mackay, M.E.; Rowan, S.J. A self-repairing, supramolecular polymer system: Healability as a consequence of donor-acceptor pi-pi stacking interactions. Chem. Commun. 2009, 44, 6717. [Google Scholar] [CrossRef] [PubMed]
- Ono, T.; Fujii, S.; Nobori, T.; Lehn, J.M. Soft-to-hard transformation of the mechanical properties of dynamic covalent polymers through component incorporation. Chem. Commun. 2007, 43, 46–48. [Google Scholar] [CrossRef] [PubMed]
- Hentschel, J.; Kushner, A.M.; Ziller, J.; Guan, Z. Self-healing supramolecular block copolymers. Angew. Chem. Int. Ed. 2012, 51, 10561–10565. [Google Scholar] [CrossRef]
- Varley, R.J.; Sybrand, V.D.Z. Autonomous damage initiated healing in a thermo-responsive ionomer. Polym. Int. 2010, 59, 1031–1038. [Google Scholar] [CrossRef]
- Zhang, Y.; Dai, Z.; Han, J.; Li, T.; Xu, J.; Guo, B.H. Interplay between crystallization and Diels-Alder reaction in biobased multiblock copolyesters possessing dynamic covalent bond. Polym. Chem. 2017, 8, 4280–4289. [Google Scholar] [CrossRef]
- Zhang, D.D.; Ruan, Y.B.; Zhang, B.Q.; Qiao, X.; Deng, G.; Chen, Y.; Liu, C.Y. A self-healing PDMS elastomer based on acylhydrazone groups and the role of hydrogen bonds. Polymer 2017, 120, 189–196. [Google Scholar] [CrossRef]
- Zheng, N.; Fang, Z.; Zou, W.; Zhao, Q.; Xie, T. Thermoset Shape-Memory Polyurethane with Intrinsic Plasticity Enabled by Transcarbamoylation. Angew. Chem. Int. Ed. 2016, 55, 11421–11425. [Google Scholar] [CrossRef]
- Yuan, C.E.; Rong, M.Z.; Zhang, M.Q.; Zhang, Z.P.; Yuan, Y.C. Self-Healing of Polymers via Synchronous Covalent Bond Fission/Radical Recombination. Chem. Mater. 2011, 23, 5076–5081. [Google Scholar] [CrossRef]
- Imato, K.; Kanehara, T.; Nojima, S.; Ohishi, T.; Higaki, Y.; Takahara, A.; Otsuka, H. Repeatable mechanochemical activation of dynamic covalent bonds in thermoplastic elastomers. Chem. Commun. 2016, 52, 10482–10485. [Google Scholar] [CrossRef] [PubMed]
- Ohishi, T.; Iki, Y.; Imato, K.; Higaki, Y.; Takahara, A.; Otsuka, H. Insertion Metathesis Depolymerization of Aromatic Disulfide-containing Dynamic Covalent Polymers under Weak Intensity Photoirradiation. Chem. Lett. 2013, 42, 1346–1348. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, D. A novel self-healing polyurethane based on disulfide bonds. Macromol. Chem. Phys. 2016, 10, 1191–1196. [Google Scholar] [CrossRef]
- Grande, A.M.; Martin, R.; Odriozola, I.; Zwaag, S.V.D.; Garcia, S.J. Effect of the polymer structure on the viscoelastic and interfacial healing behaviour of poly (urea-urethane) networks containing aromatic disulphides. Eur. Polym. J. 2017, 97, 120–128. [Google Scholar] [CrossRef]
- Havriliak, S.; Negami, S. A complex plane representation of dielectric and mechanical relaxation processes in some polymers. Polymer 1967, 8, 161–210. [Google Scholar] [CrossRef]
- Yu, W.; Du, M.; Zhang, D.; Lin, Y.; Zheng, Q. Influence of dangling chains on molecular dynamics of polyurethanes. Macromolecules 2013, 46, 7341–7351. [Google Scholar] [CrossRef]
- Lei, Z.Q.; Xiang, H.P.; Yuan, Y.J.; Rong, M.Z.; Zhang, M.Q. Room-Temperature Self-Healable and Remoldable Cross-linked Polymer Based on the Dynamic Exchange of Disulfide Bonds. Chem. Mater. 2014, 26, 2038–2046. [Google Scholar] [CrossRef]
- Montarnal, D.; Capelot, M.; Tournilhac, F.; Leibler, L. Silica-like malleable materials from permanent organic networks. Science 2011, 334, 965–968. [Google Scholar] [CrossRef]
- Lai, Y.; Kuang, X.; Zhu, P.; Huang, M.; Dong, X.; Wang, D. Colorless, Transparent, Robust, and Fast Scratch-Self-Healing Elastomers via a Phase-Locked Dynamic Bonds Design. Adv. Mater. 2018, 30, 1802556. [Google Scholar] [CrossRef]
- Kwon, Y.J.; Hong, S.M.; Koo, C.M. Viscoelastic properties of decrosslinked irradiation-crosslinked polyethylenes in supercritical methanol. J. Polym. Sci. Part B Polym. Phys. 2010, 48, 1265–1270. [Google Scholar] [CrossRef]
- Wang, C.B.; Cooper, S.L. Morphology and properties of segmented polyether polyurethaneureas. Macromolecules 1983, 16, 775–786. [Google Scholar] [CrossRef]
- Versteegen, R.M.; Kleppinger, R.; Sijbesma, R.P.; Meijer, E.W. Properties and Morphology of Segmented Copoly(ether urea)s with Uniform Hard Segments. Macromolecules 2005, 39, 772–783. [Google Scholar] [CrossRef]
- Kautz, H.; Beek, D.J.M.V.; Sijbesma, R.P.; Meijer, E.W. Cooperative End-to-End and Lateral Hydrogen-Bonding Motifs in Supramolecular Thermoplastic Elastomers. Macromolecules 2006, 39, 4265–4267. [Google Scholar] [CrossRef]
- Li, T.; Zhang, C.; Xie, Z.; Xu, J.; Guo, B.-H. A multi-scale investigation on effects of hydrogen bonding on micro-structure and macro-properties in a polyurea. Polymer 2018, 145, 261–271. [Google Scholar] [CrossRef]
- And, J.M.; Painter, P. A Comparison of Hydrogen Bonding and Order in a Polyurethane and Poly(urethane−urea) and Their Blends with Poly(ethylene glycol). Macromolecules 2007, 40, 1546–1554. [Google Scholar]
- Coleman, M.M.; Lee, K.H.; Skrovanek, D.J.; Painter, P.C. Hydrogen bonding in polymers. 4. Infrared temperature studies of a simple polyurethane. Macromolecules 1986, 19, 2149–2157. [Google Scholar] [CrossRef]
- Han, S.L.; Ying, K.W.; Hsu, S.L. Spectroscopic analysis of phase separation behavior of model polyurethanes. Macromolecules 1988, 21, 98–99. [Google Scholar]
- Teo, L.S.; Chuhyung Chen, A.; Kuo, J.F. Fourier Transform Infrared Spectroscopy Study on Effects of Temperature on Hydrogen Bonding in Amine-Containing Polyurethanes and Poly(urethane−urea)s. Macromolecules 1997, 30, 1793–1799. [Google Scholar] [CrossRef]
- Srichatrapimuk, V.; Cooper, S. Infrared thermal analysis of polyurethane block polymers. J. Macromol. Sci. Part B 2012, 15, 267–311. [Google Scholar] [CrossRef]
- Fragiadakis, D.; Gamache, R.; Bogoslovov, R.; Roland, C. Segmental dynamics of polyurea: Effect of stoichiometry. Polymer 2010, 51, 178–184. [Google Scholar] [CrossRef]
- Raftopoulos, K.; Pandis, C.; Apekis, L.; Pissis, P.; Janowski, B.; Pielichowski, K.; Jaczewska, J. Polyurethane–POSS hybrids: Molecular dynamics studies. Polymer 2010, 51, 709–718. [Google Scholar] [CrossRef]
- Castagna, A.M.; Fragiadakis, D.; Lee, H.; Choi, T.; Runt, J. The role of hard segment content on the molecular dynamics of poly (tetramethylene oxide)-based polyurethane copolymers. Macromolecules 2011, 44, 7831–7836. [Google Scholar] [CrossRef]
- Luo, M.C.; Zhang, X.K.; Zeng, J.; Gao, X.X.; Huang, G.S. Enhanced relaxation behavior below glass transition temperature in diene elastomer with heterogeneous physical network. Polymer 2016, 91, 81–88. [Google Scholar] [CrossRef]
- Castagna, A.M.; Wang, W.; Winey, K.I.; Runt, J. Structure and dynamics of zinc-neutralized sulfonated polystyrene ionomers. Macromolecules 2011, 44, 2791–2798. [Google Scholar] [CrossRef]
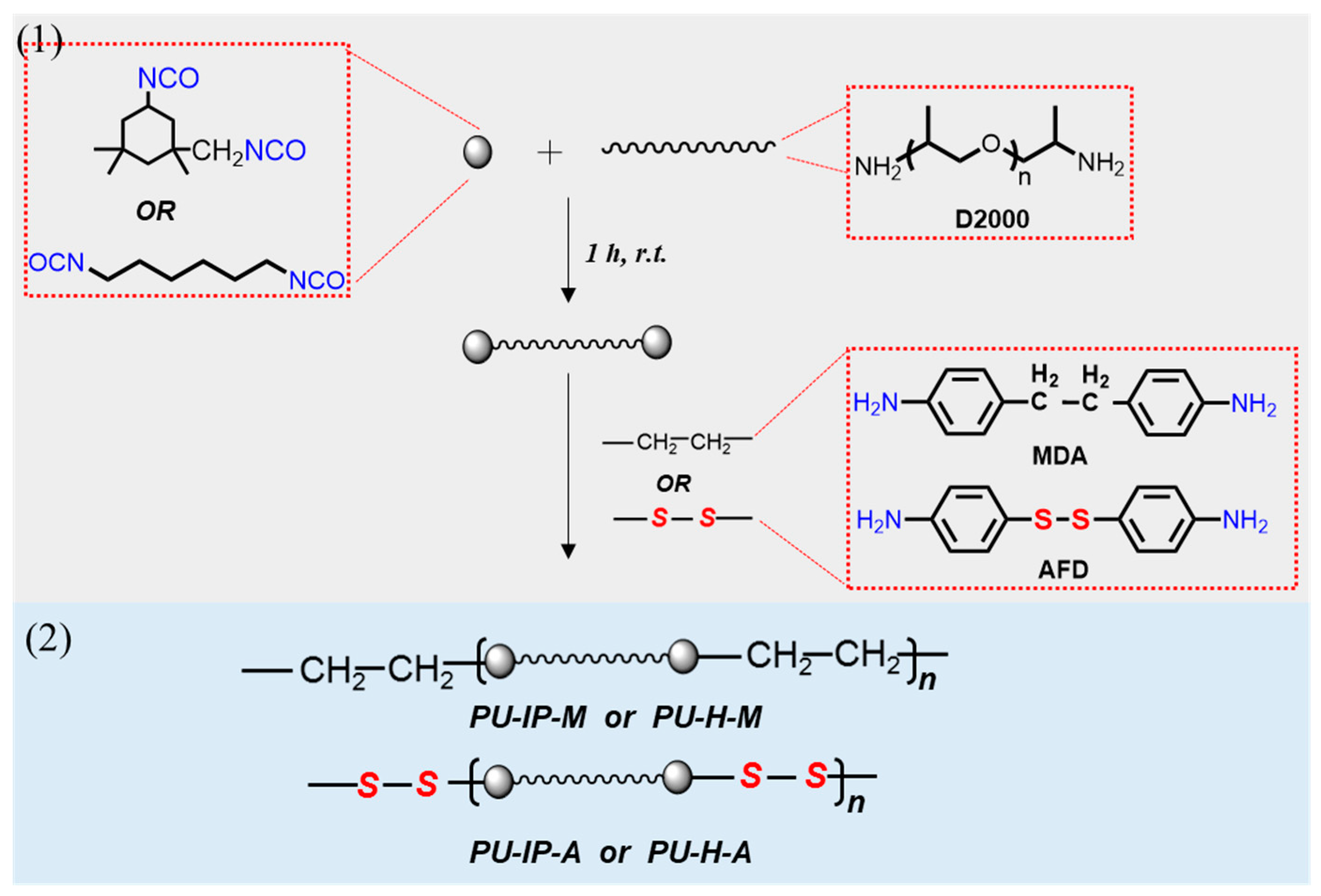

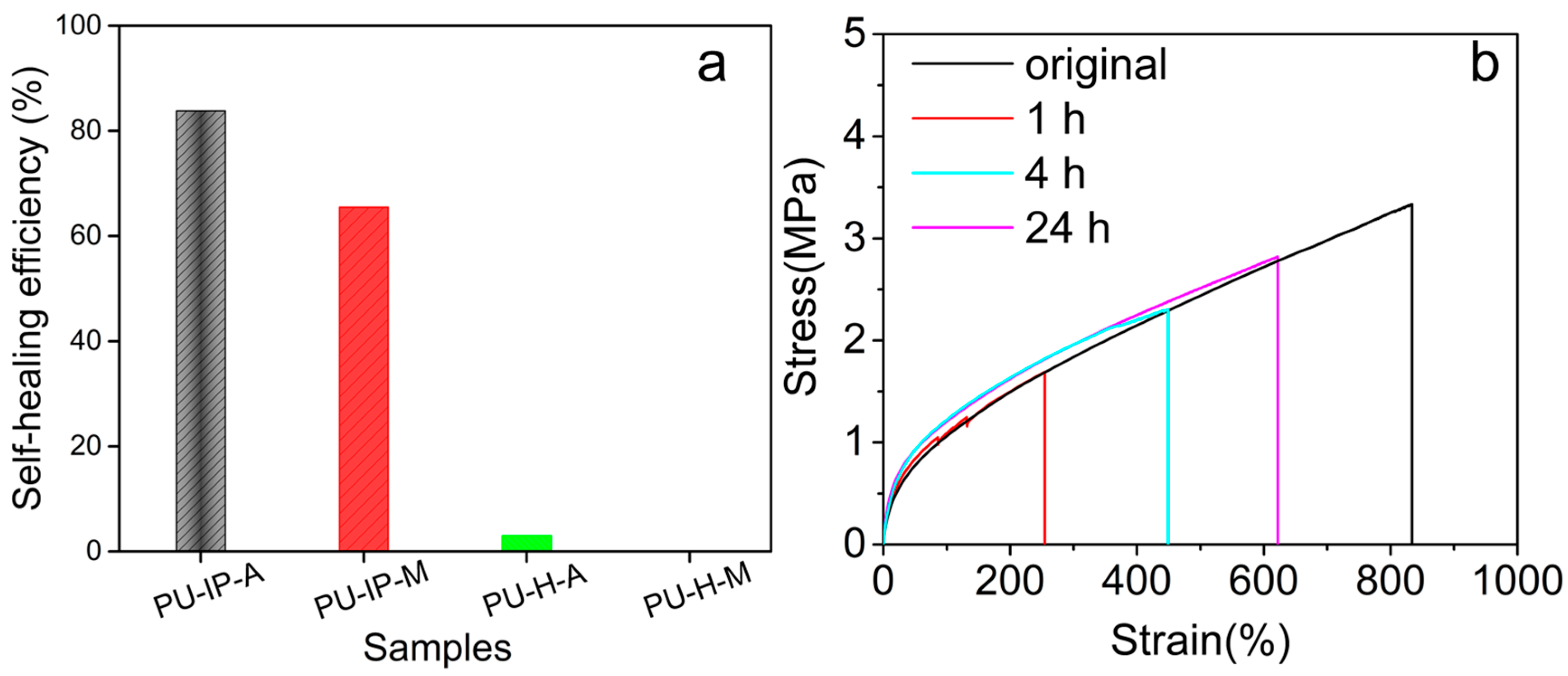
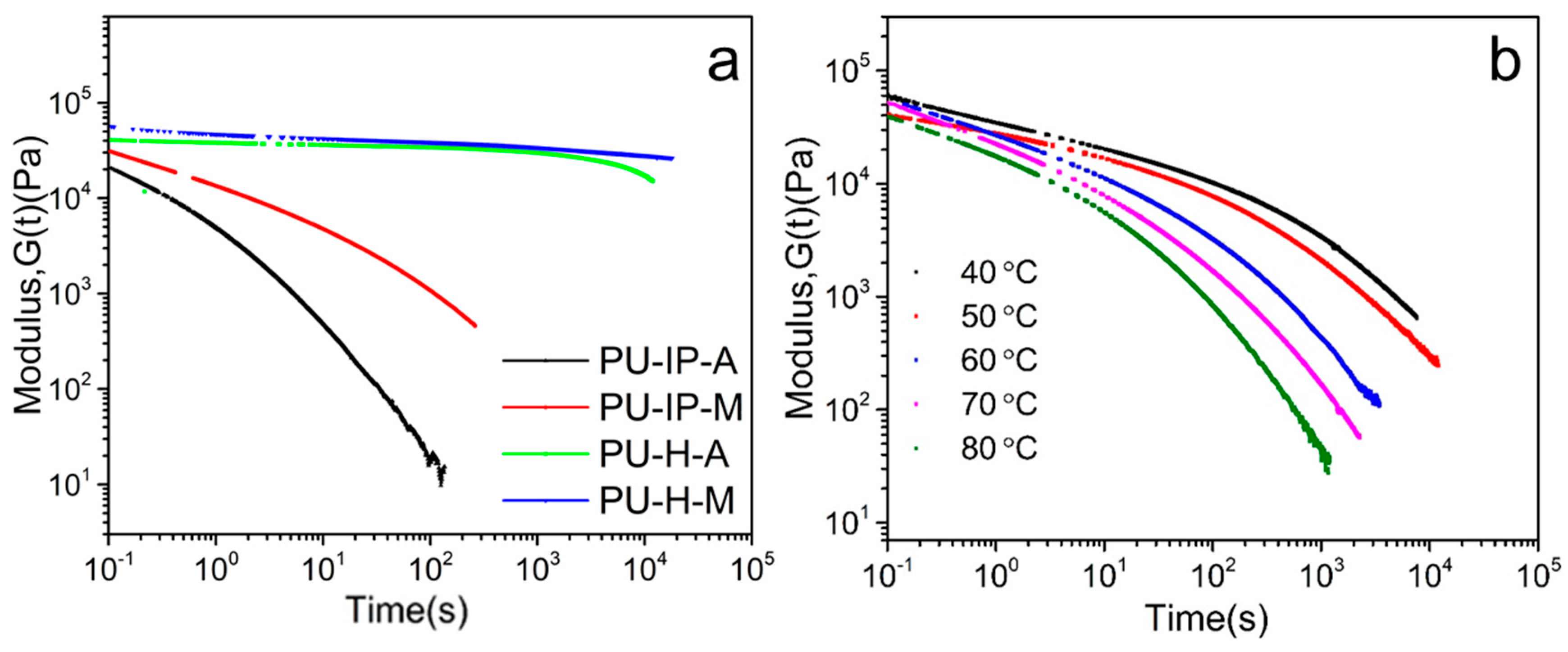


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
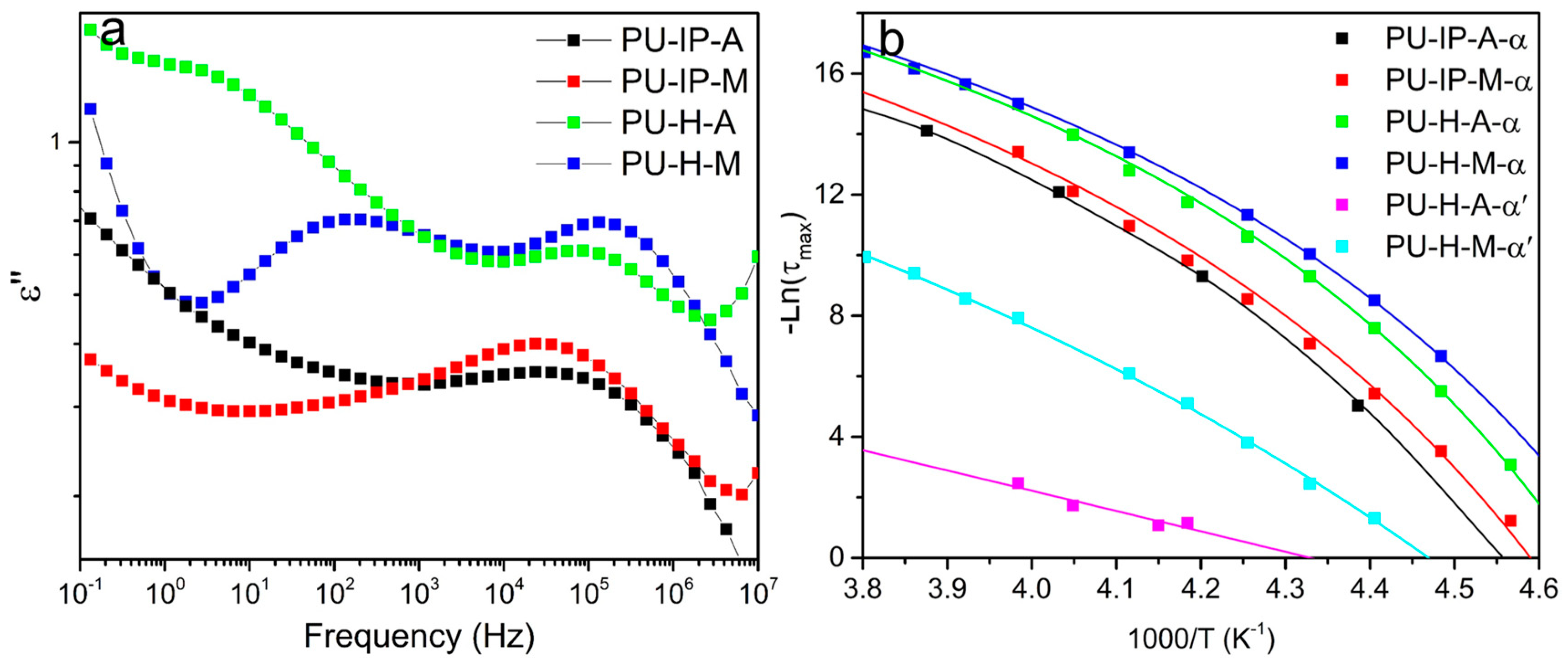{kind=link}
| Samples | PU-IP-A | PU-IP-M | PU-H-A | PU-H-M |
|---|---|---|---|---|
| Tg,DMA (°C) | −47.7 | −51.1 | −55.2 | −54.2 |
| Tg,DSC (°C) | −47.3 | −52.9 | −56.2 | −58.2 |
| Tg,DRS (°C) | −54.6 | −58.6 | −60.2 | −60.3 |
| Tg,DRS,α’ (°C) | -- | -- | −52.3 | −54.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.; Zheng, T.; Han, J.; Liu, Z.; Guo, Z.-X.; Zhuang, Z.; Xu, J.; Guo, B.-H. Effects of Diisocyanate Structure and Disulfide Chain Extender on Hard Segmental Packing and Self-Healing Property of Polyurea Elastomers. Polymers 2019, 11, 838. https://doi.org/10.3390/polym11050838
Li T, Zheng T, Han J, Liu Z, Guo Z-X, Zhuang Z, Xu J, Guo B-H. Effects of Diisocyanate Structure and Disulfide Chain Extender on Hard Segmental Packing and Self-Healing Property of Polyurea Elastomers. Polymers. 2019; 11(5):838. https://doi.org/10.3390/polym11050838
Chicago/Turabian StyleLi, Ting, Tianze Zheng, Jiarui Han, Zhanli Liu, Zhao-Xia Guo, Zhuo Zhuang, Jun Xu, and Bao-Hua Guo. 2019. "Effects of Diisocyanate Structure and Disulfide Chain Extender on Hard Segmental Packing and Self-Healing Property of Polyurea Elastomers" Polymers 11, no. 5: 838. https://doi.org/10.3390/polym11050838
APA StyleLi, T., Zheng, T., Han, J., Liu, Z., Guo, Z.-X., Zhuang, Z., Xu, J., & Guo, B.-H. (2019). Effects of Diisocyanate Structure and Disulfide Chain Extender on Hard Segmental Packing and Self-Healing Property of Polyurea Elastomers. Polymers, 11(5), 838. https://doi.org/10.3390/polym11050838