Influences of Hyperbranched Polyester Modification on the Crystallization Kinetics of Isotactic Polypropylene/Graphene Oxide Composites

Abstract
:
1. Introduction
2. Experimental Section
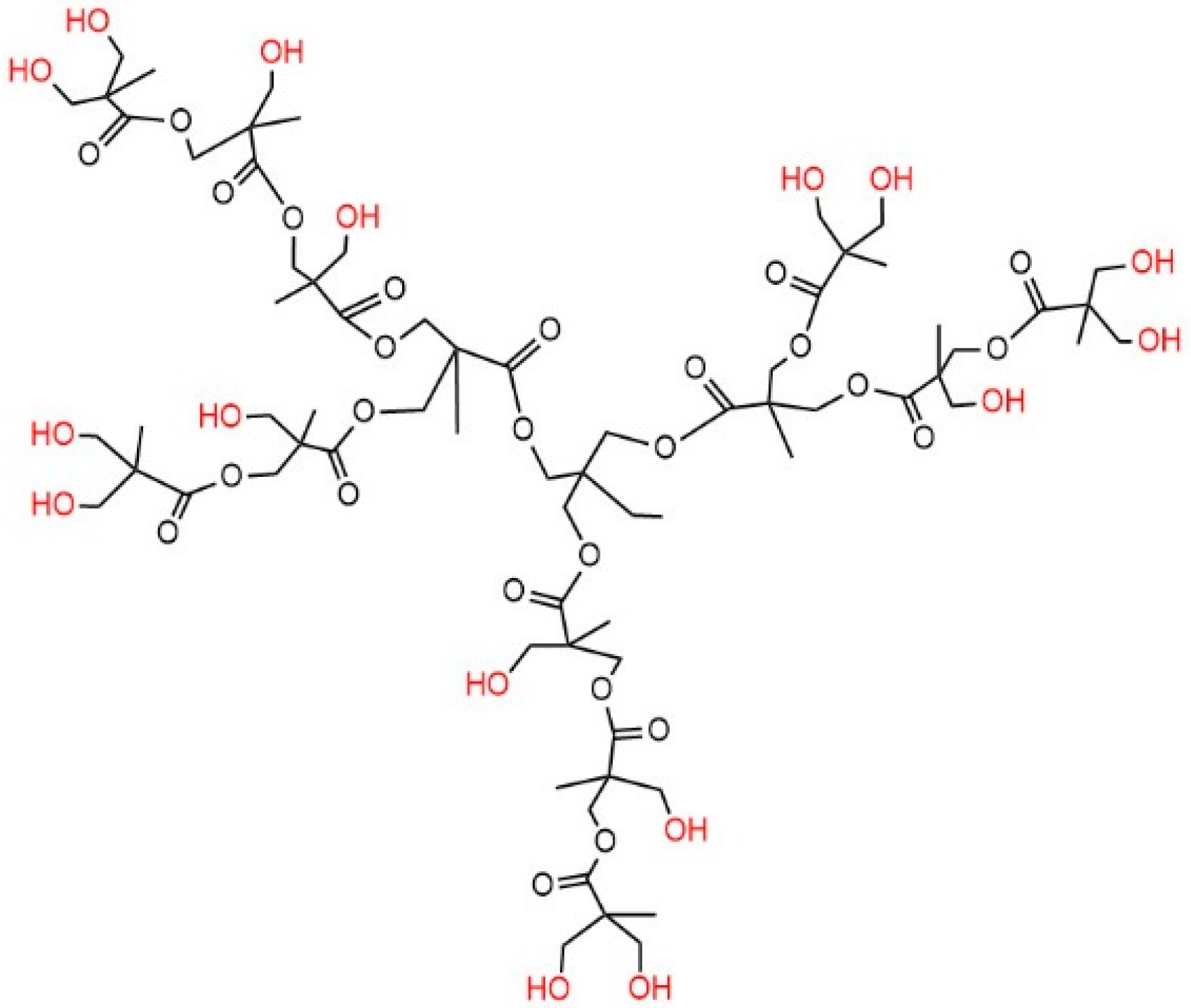
2.1. Materials
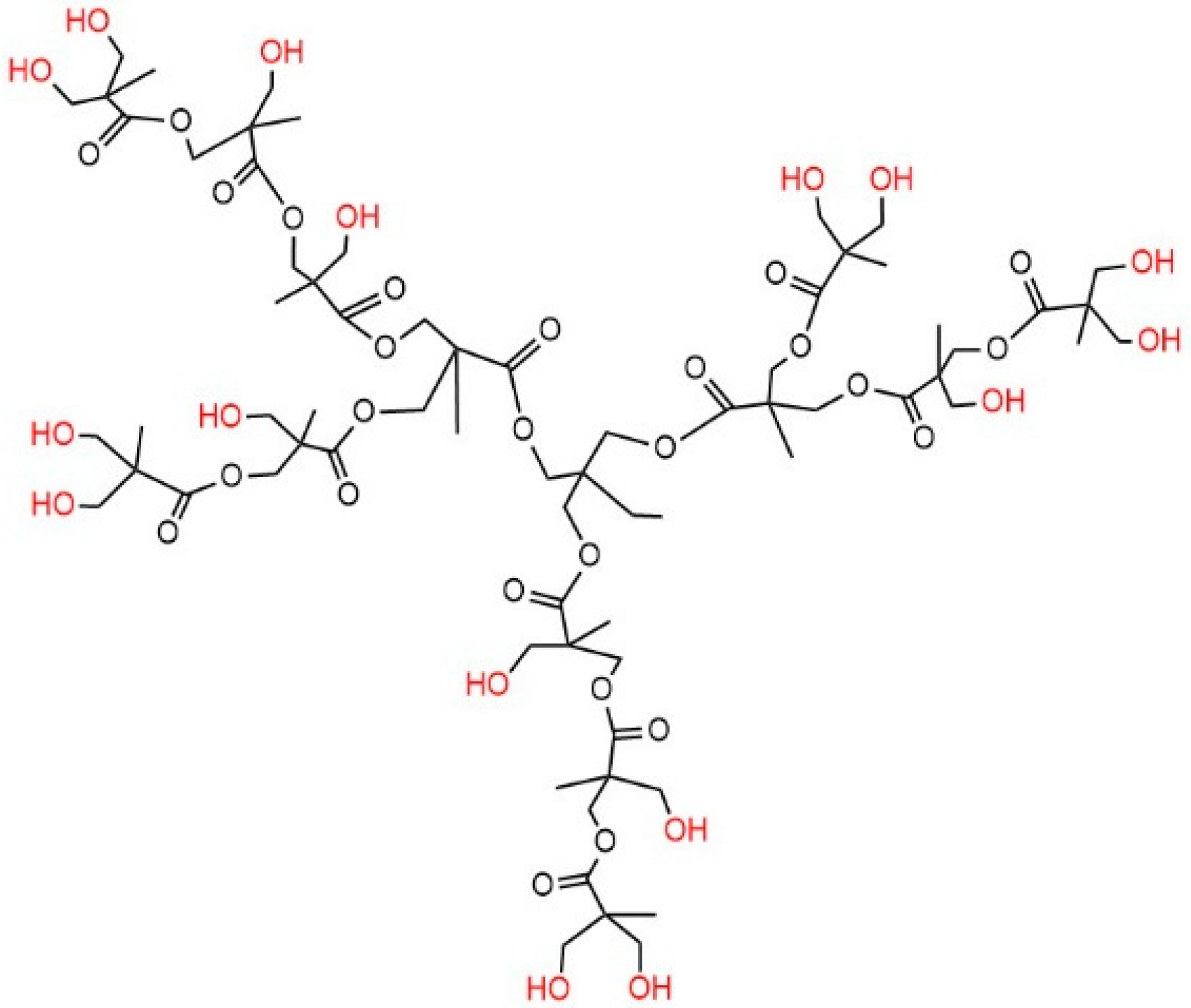
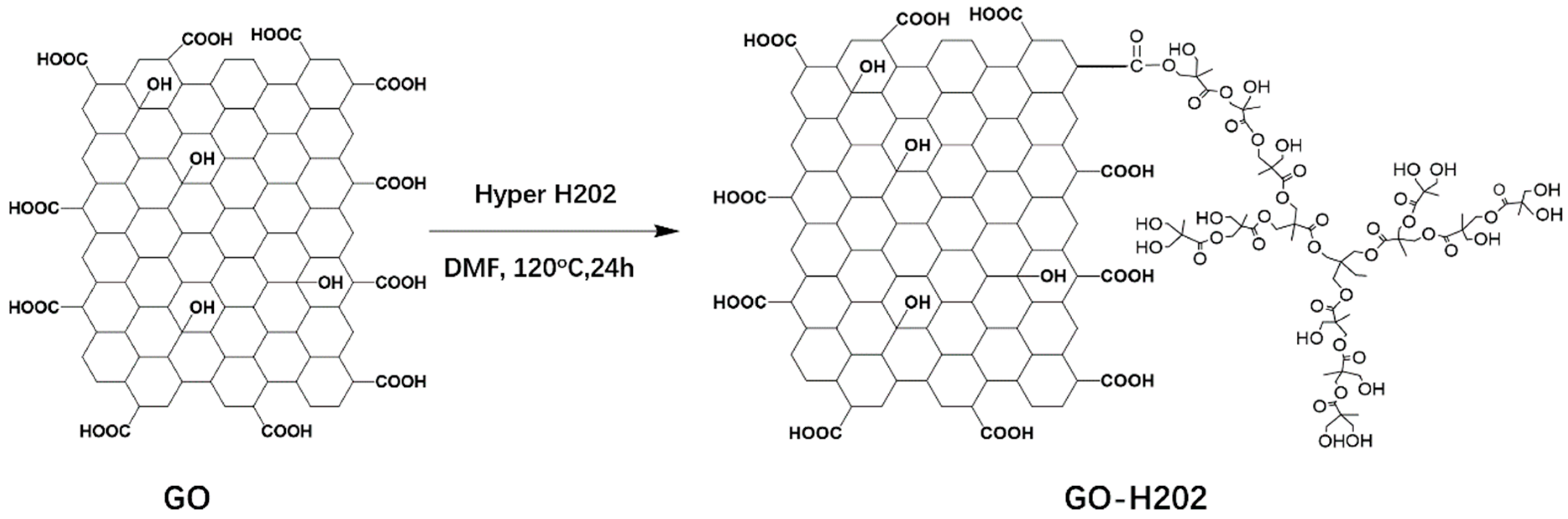
2.2. Grafting of the Graphene Oxide
2.3. Preparation of iPP/GO Composites
2.4. Characterization
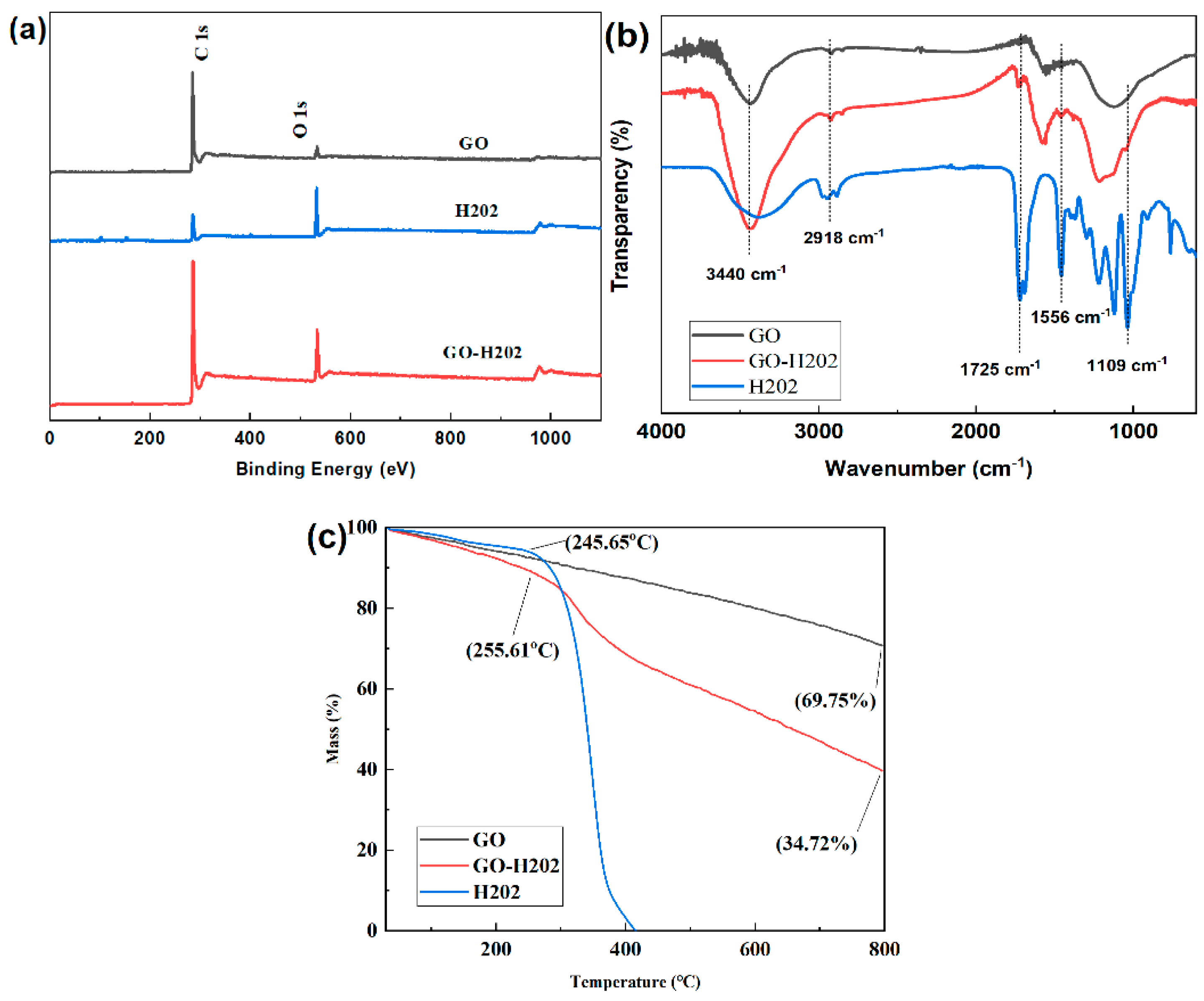
2.4.1. X-ray Photoelectron Spectra (XPS)
2.4.2. Fourier Transform Infrared (FT-IR)
2.4.3. Thermogravimetric Analysis (TGA)
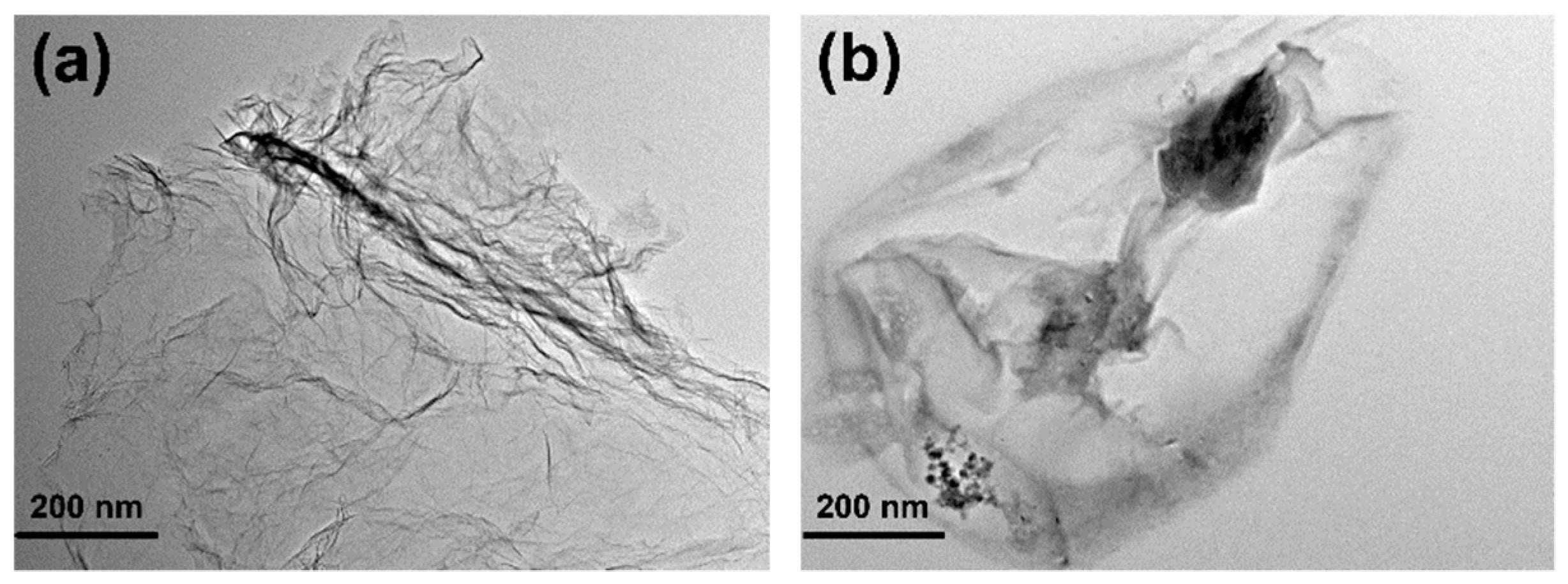
2.4.4. Transmission Electron Microscopy (TEM)
2.4.5. Differential Scanning Calorimetry (DSC)
2.4.6. Isothermal Crystallization Kinetics
- (a)
- Samples were heated from 50 to 200 °C at 10 ℃/min and kept for 5 min to erase any previous thermal history.
- (b)
- Samples were cooled down to the desired crystallization temperature TcISO at 50 °C/min.
- (c)
- Samples were isothermally kept for a certain time to complete crystallization.
2.4.7. Self-Nucleation Isothermal Crystallization Kinetics
- (a)
- (b)
- The samples were heated from 50 to 200 °C at 10 ℃/min and held for 5 min to destroy any residual nuclei.
- (c)
- After that, samples were cooled to 50 °C at 10 °C/min and held for 2 min to create a “standard” thermal history [53], and then they were heated to the self-nucleating temperature (denoted as TSN, the lowest temperature within Domain Ⅱ is preferred) and kept for 5 min.
- (d)
- Then, it was rapidly cooled to the predetermined crystallization temperature (TcSN) and held for a certain time to allow the completion of crystallization.
- (e)
- Finally, the sample was heated to 200 °C at a rate of 10 °C/min.
2.4.8. Polarized Optical Microscopy (POM)
3. Results and Discussions
3.1. Chemical Structure of GO-H202
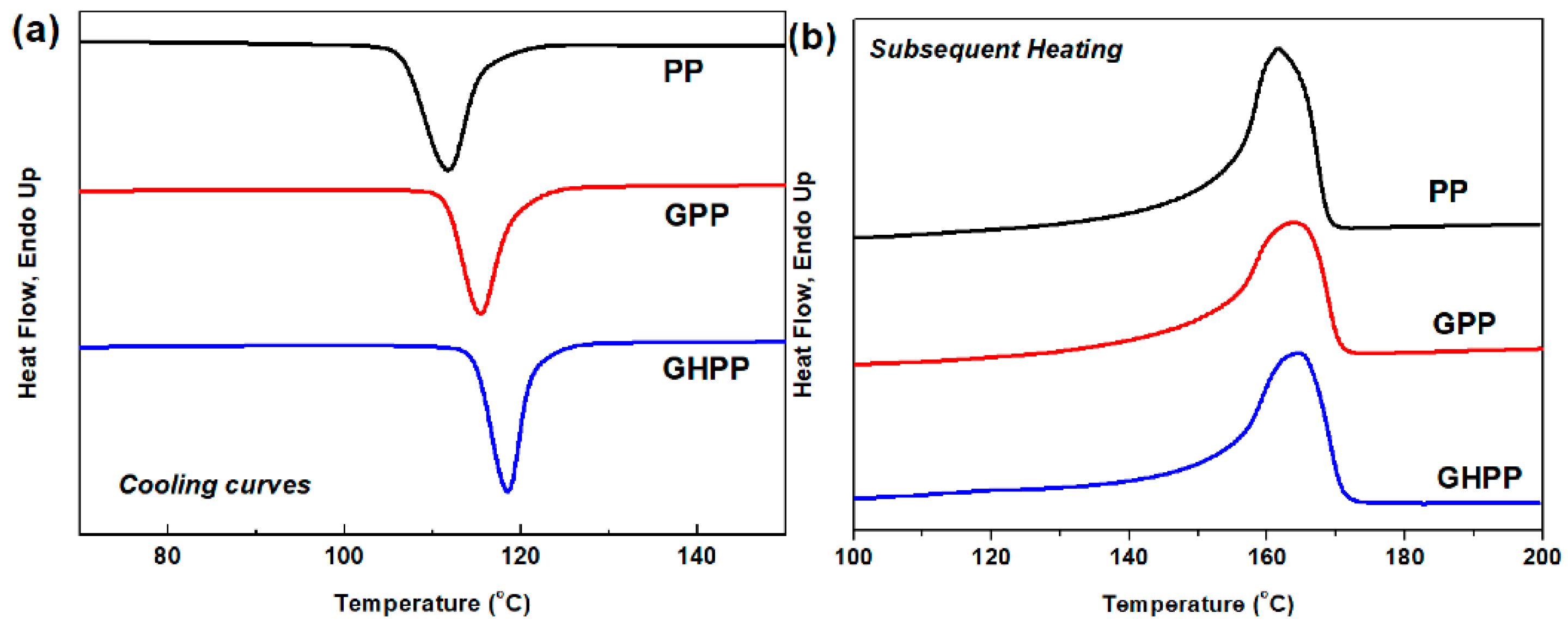
3.2. DSC Cooling and Heating Behavior of the Composites
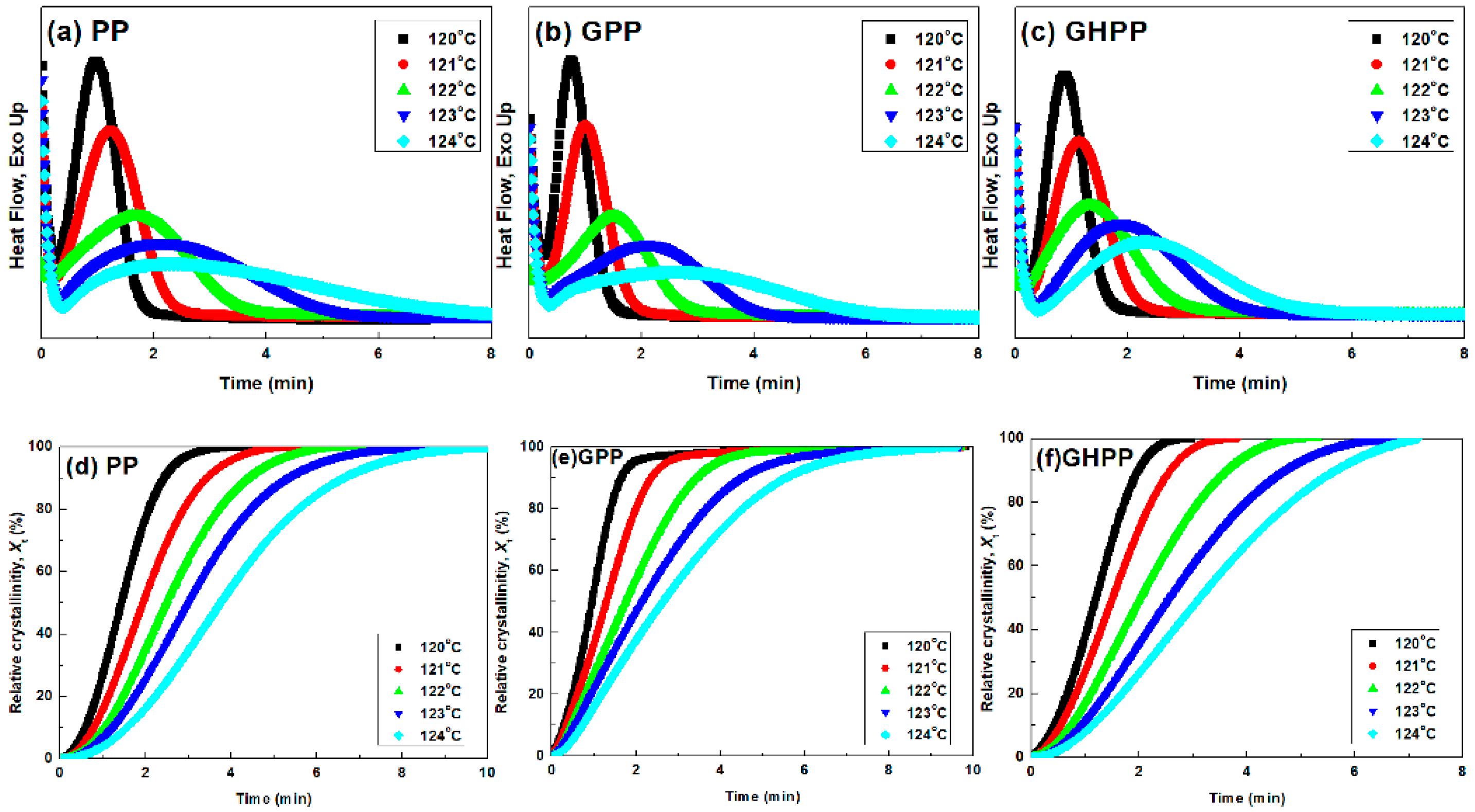
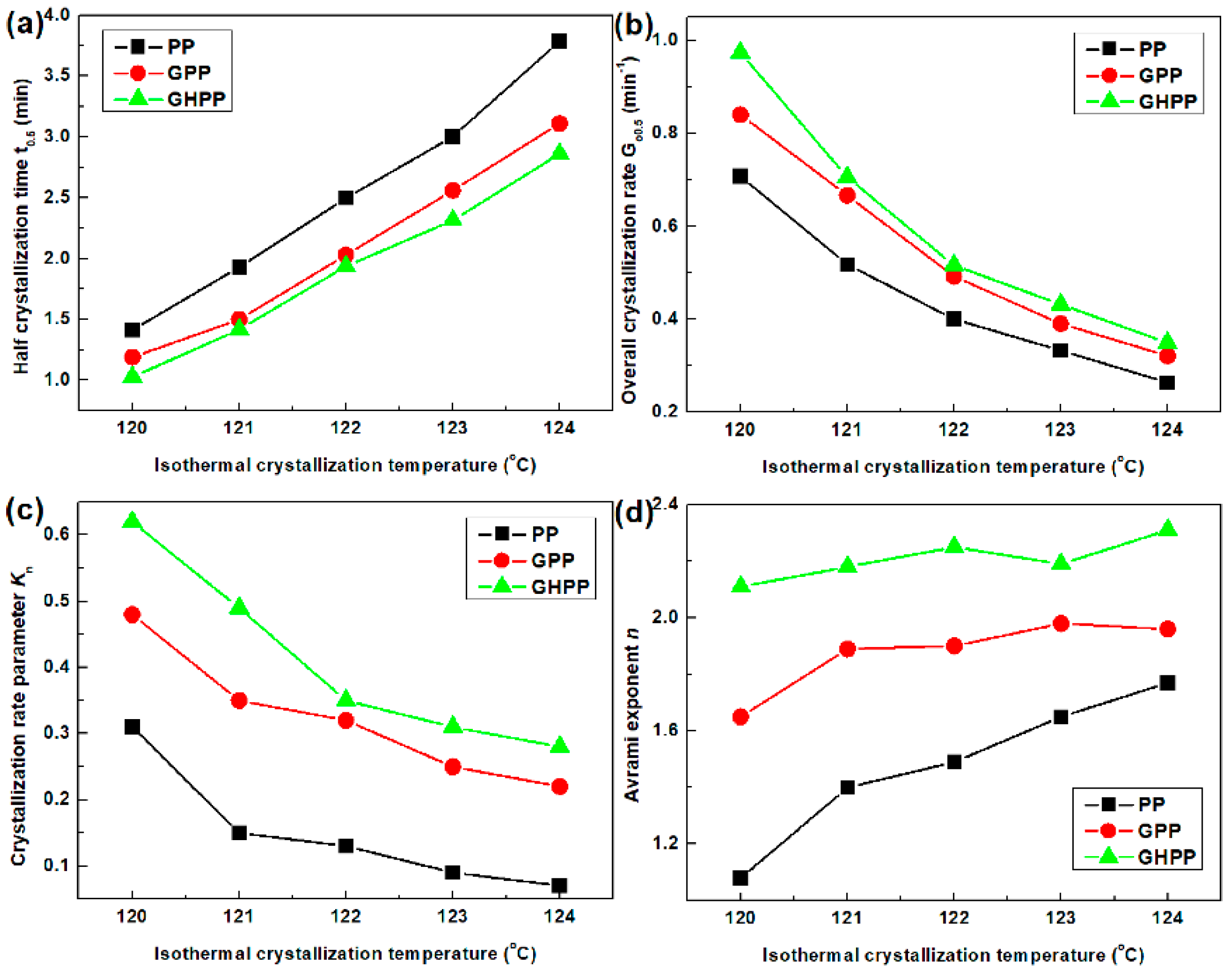
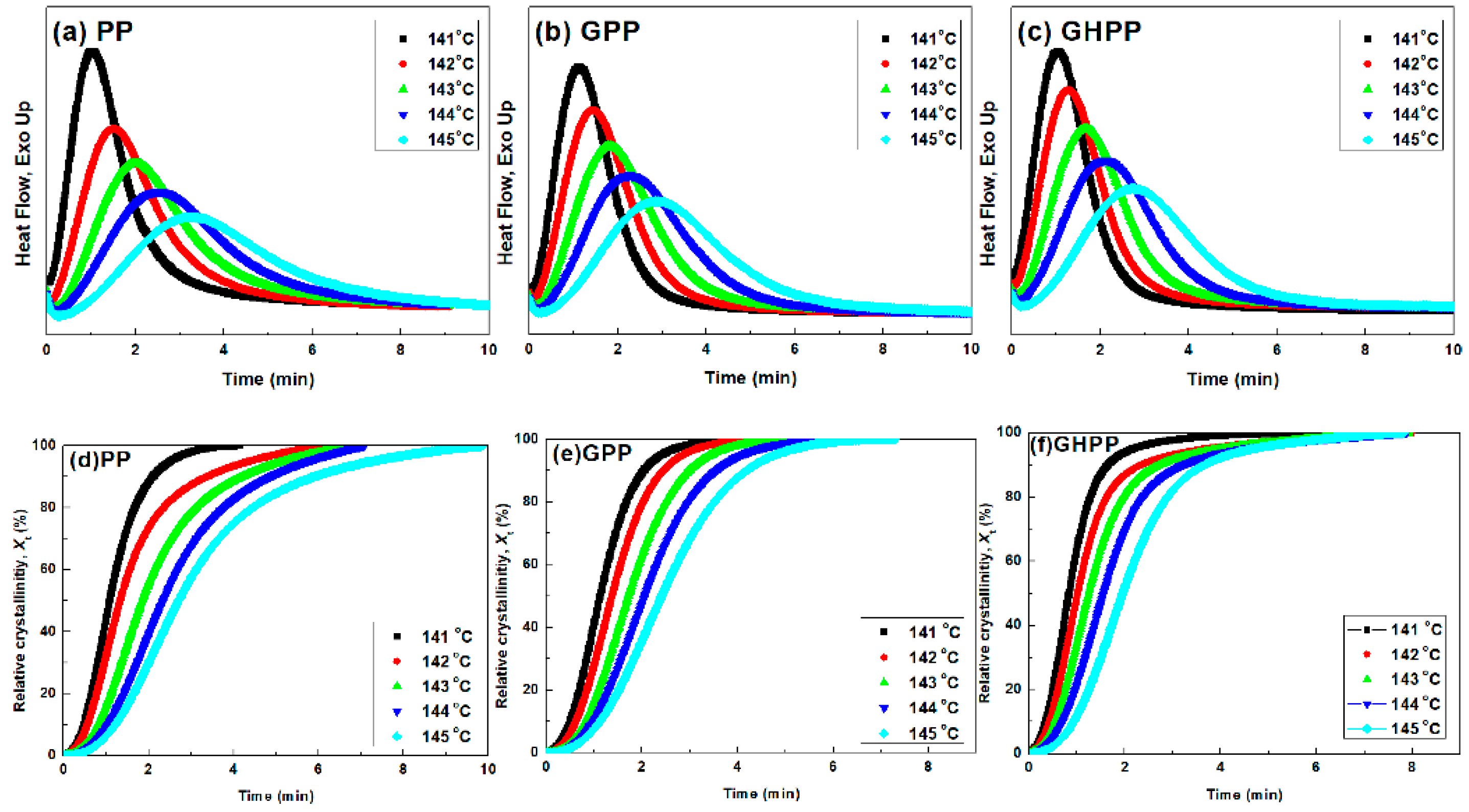
3.3. Isothermal Crystallization Kinetics of the Composites
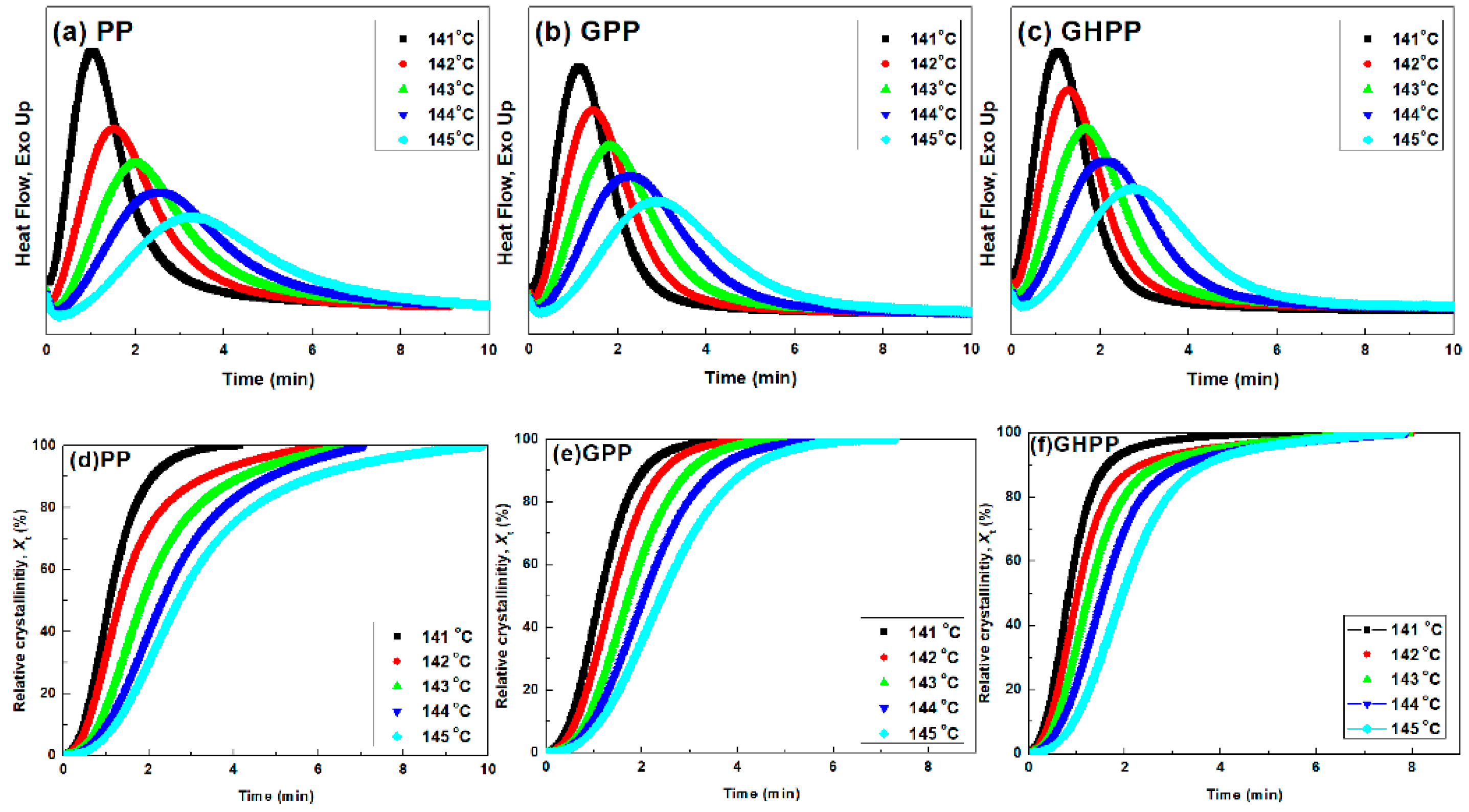
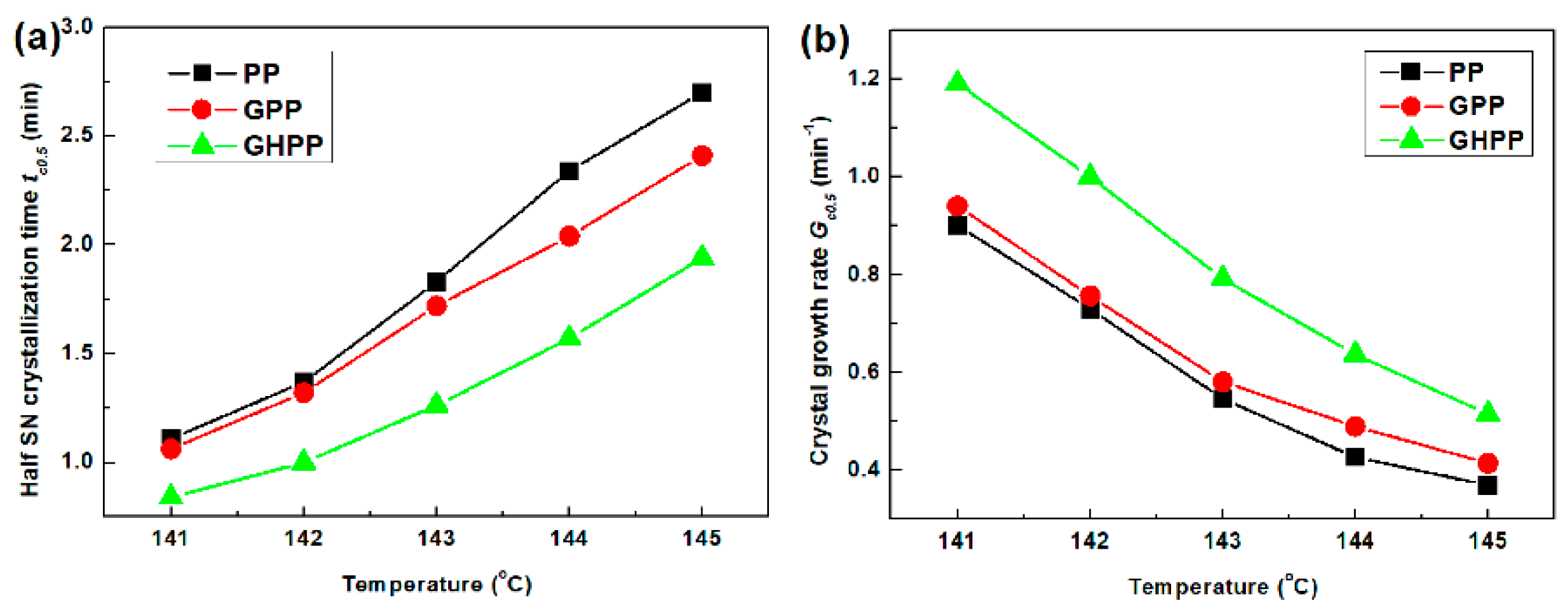
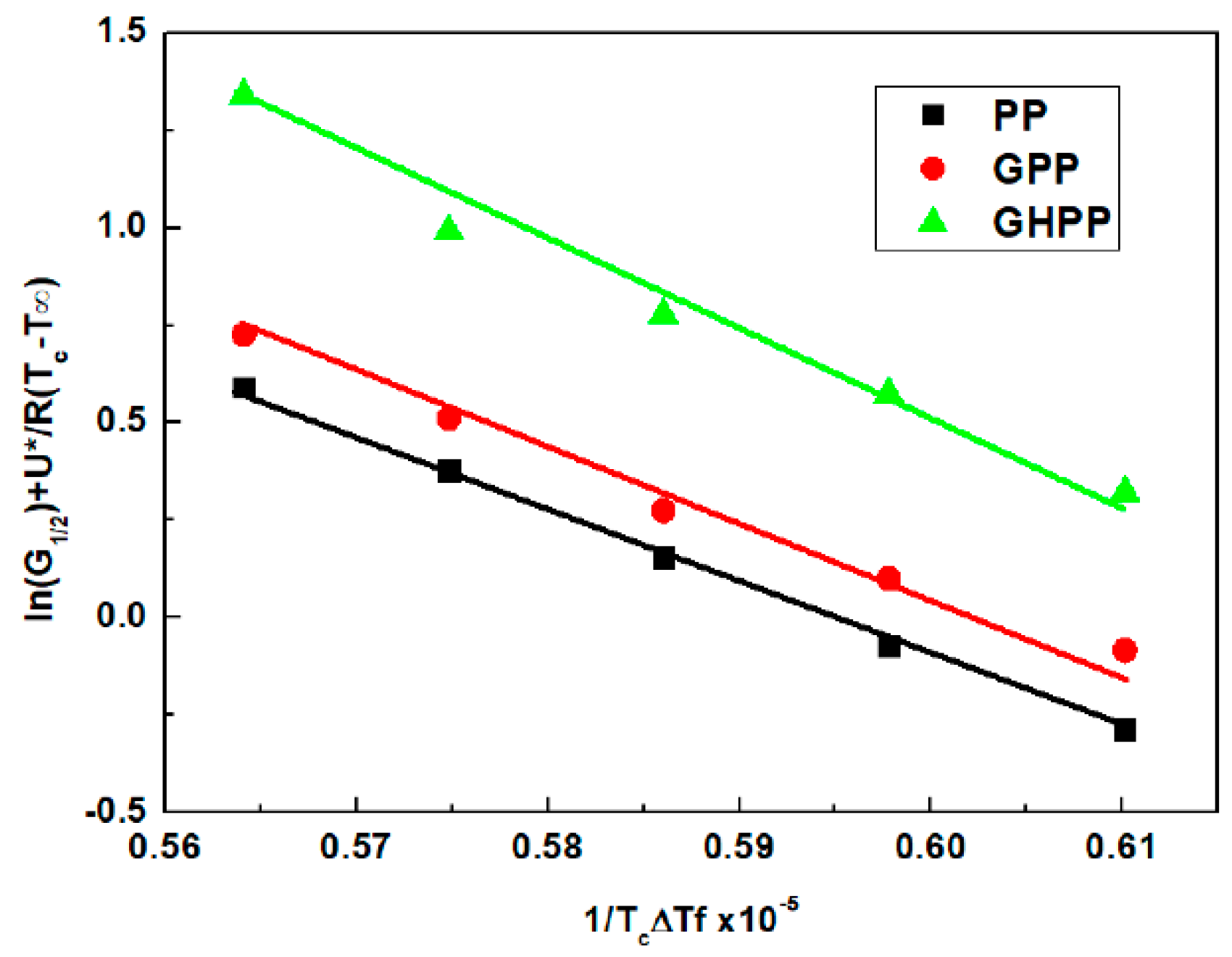
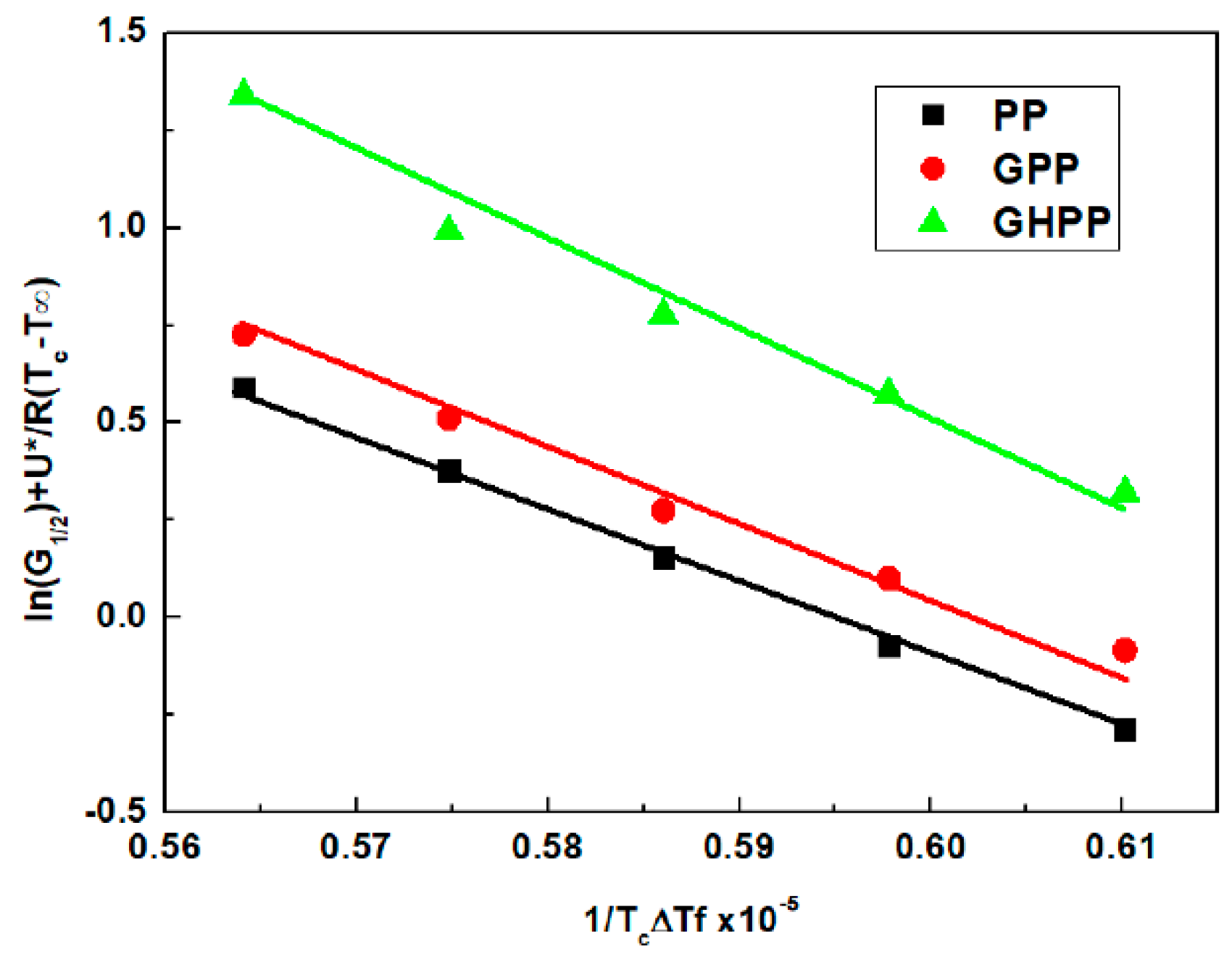
3.4. Self-Nucleation Isothermal Crystallization Kinetics
3.5. Polarized Optical Microscopy Observation
4. Conclusions
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
References
- Natta, G.; Corradini, P. Structure and properties of isotactic polypropylene. Nuovo Cim. 1960, 15, 40–51. [Google Scholar] [CrossRef]
- Natta, G.; Pino, P.; Corradini, P.; Danusso, F.; Mantica, E.; Mazzanti, G.; Moraglio, G. Crystalline high polymers of α-olefins. J. Am. Chem. Soc. 1955, 77, 1708–1710. [Google Scholar] [CrossRef]
- Zeng, F.; Chen, J.; Yang, F.; Kang, J.; Cao, Y.; Xiang, M. Effects of Polypropylene Orientation on Mechanical and Heat Seal Properties of Polymer-Aluminum-Polymer Composite Films for Pouch Lithium-Ion Batteries. Materials 2018, 11, 144. [Google Scholar] [CrossRef] [PubMed]
- Xiong, B.; Chen, R.; Zeng, F.; Kang, J.; Men, Y. Thermal shrinkage and microscopic shutdown mechanism of polypropylene separator for lithium-ion battery: In-situ ultra-small angle X-ray scattering study. J. Membr. Sci. 2018, 545, 213–220. [Google Scholar] [CrossRef]
- Yu, Y.; Xiong, B.; Zeng, F.; Xu, R.; Yang, F.; Kang, J.; Xiang, M.; Li, L.; Sheng, X.; Hao, Z. Influences of Compression on the Mechanical Behavior and Electrochemical Performances of Separators for Lithium Ion Batteries. Ind. Eng. Chem. Res. 2018, 57, 17142–17151. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, X.; Zheng, G.; Dai, K.; Liu, C.; Shen, C.; Yin, R.; Guo, Z. Mechanical enhancement of melt-stretched β-nucleated isotactic polypropylene: The role of lamellar branching of β-crystal. Polym. Test. 2017, 58, 227–235. [Google Scholar] [CrossRef]
- Xiong, J.K.B.; Chen, R.; Men, Y. Initiation of cavitation upon drawing of pre-oriented polypropylene film: In situ SAXS and WAXD studies. Polymer 2017, 128, 57–64. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, B.; Yang, J.; Ding, D.; Yan, X.; Zheng, G.; Dai, K.; Liu, C.; Guo, Z. Synergies among the self-assembled β-nucleating agent and the sheared isotactic polypropylene matrix. Polymer 2015, 60, 40–49. [Google Scholar] [CrossRef]
- Kang, J.H.J.; Chen, Z.; Yu, H.; Chen, J.; Yang, F.; Cao, Y.; Xiang, M. Investigation on the crystallization behavior and polymorphic composition of isotactic polypropylene / multi-walled carbon nanotubes composites nucleated with β-nucleating agent—The role of melt structures. J. Therm. Anal. Calorim. 2015, 119, 1769–1780. [Google Scholar] [CrossRef]
- Bruckner, S.; Meille, S.V.; Petraccone, V.; Pirozzi, B. Polymorphism in isotactic polypropylene. Prog. Polym. Sci. 1991, 16, 361–404. [Google Scholar] [CrossRef]
- Kang, J.; Chen, J.; Cao, Y.; Li, H. Effects of ultrasound on the conformation and crystallization behavior of isotactic polypropylene and [beta]-isotactic polypropylene. Polymer 2010, 51, 249–256. [Google Scholar] [CrossRef]
- Kang, J.; Weng, G.; Chen, Z.; Chen, J.; Cao, Y.; Yang, F.; Xiang, M. New understanding in the influence of melt structure and β-nucleating agents on the polymorphic behavior of isotactic polypropylene. Rsc Adv. 2014, 56, 29514–29526. [Google Scholar] [CrossRef]
- Yansong, Y.; Fangxinyu, Z.; Jinyao, C.; Jian, K.; Feng, Y.; Ya, C.; Ming, X. Isothermal Crystallization Kinetics and Subsequent Melting Behavior of β-Nucleated Isotactic Polypropylene / Graphene Oxide Composites with Different Ordered Structure. Polym. Int. 2018. [Google Scholar] [CrossRef]
- Brückner, S.; Phillips, P.J.; Mezghani, K.; Meille, S.V. On the crystallization of γ-isotactic polypropylene: A high pressure study. Macromol. Rapid Commun. 1997, 18, 1–7. [Google Scholar] [CrossRef]
- Krache, R.; Benavente, R.; López-Majada, J.M.; Pereña, J.M.; Cerrada, M.L.; Pérez, E. Competition between α, β, and γ Polymorphs in a β-Nucleated Metallocenic Isotactic Polypropylene. Macromolecules 2007, 40, 6871–6878. [Google Scholar] [CrossRef]
- Horvath, Z.; Sajó, I.E.; Stoll, K.; Menyhárd, A.; Varga, J. The effect of molecular mass on the polymorphism and crystalline structure of isotactic polypropylene. Express Polym. Lett. 2010, 4, 101–114. [Google Scholar] [CrossRef]
- Varga, J. β-Modification of Isotactic Polypropylene: Preparation, Structure, Processing, Properties, and Application. J. Macromol. Sci. Part B 2002, 41, 1121–1171. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, J.; Ji, F.; Zhang, X.; Zheng, G.; Shen, C. Effects of melt structure on shear-induced β-cylindrites of isotactic polypropylene. Polymer 2012, 53, 1791–1800. [Google Scholar] [CrossRef]
- Kang, J.; He, J.; Chen, Z.; Yang, F.; Chen, J.; Cao, Y.; Xiang, M. Effects of β-nucleating agent and crystallization conditions on the crystallization behavior and polymorphic composition of isotactic polypropylene/multi-walled carbon nanotubes composites. Polym. Adv. Technol. 2015, 26, 32–40. [Google Scholar] [CrossRef]
- Pawlak, A.; Piorkowska, E. Crystallization of isotactic polypropylene in a temperature gradient. Colloid Polym. Sci. 2001, 279, 939–946. [Google Scholar] [CrossRef]
- Qiyan, Z.; Hongmei, P.; Jian, K.; Ya, C.; Ming, X. Effects of melt structure on non-isothermal crystallization behavior of isotactic polypropylene nucleated with α/β compounded nucleating agents. Polym. Eng. Sci. 2017, 57, 989–997. [Google Scholar]
- Kang, Z.C.J.; Yang, F.; Chen, J.; Cao, Y.; Weng, G.; Xiang, M. Understanding the effects of nucleating agent concentration on the polymorphic behavior of β-nucleated isotactic polypropylene with different melt structures. Colloid Polym. Sci. 2015, 293, 2061–2073. [Google Scholar] [CrossRef]
- Davies, A.; Yu, A. Material advancements in supercapacitors: From activated carbon to carbon nanotube and graphene. Can. J. Chem. Eng. 2011, 89, 1342–1357. [Google Scholar] [CrossRef]
- Dong, X.; Wang, X.; Wang, J.; Song, H.; Li, X.; Wang, L.; Chan-Park, M.B.; Li, C.M.; Chen, P. Synthesis of a MnO2–graphene foam hybrid with controlled MnO2 particle shape and its use as a supercapacitor electrode. Carbon 2012, 50, 4865–4870. [Google Scholar] [CrossRef]
- Choi, H.-J.; Jung, S.-M.; Seo, J.-M.; Chang, D.W.; Dai, L.; Baek, J.-B. Graphene for energy conversion and storage in fuel cells and supercapacitors. Nano Energy 2012, 1, 534–551. [Google Scholar] [CrossRef]
- Yang, X.; Zhu, J.; Qiu, L.; Li, D. Bioinspired effective prevention of restacking in multilayered graphene films: Towards the next generation of high-performance supercapacitors. Adv. Mater. 2011, 23, 2833–2838. [Google Scholar] [CrossRef] [PubMed]
- Jaidev, S. Ramaprabhu, Poly(p-phenylenediamine)/graphene nanocomposites for supercapacitor applications. J. Mater. Chem. 2012, 22, 18775. [Google Scholar] [CrossRef]
- Chae, H.-R.; Lee, J.; Lee, C.-H.; Kim, I.-C.; Park, P.-K. Graphene oxide-embedded thin-film composite reverse osmosis membrane with high flux, anti-biofouling, and chlorine resistance. J. Membr. Sci. 2015, 483, 128–135. [Google Scholar] [CrossRef]
- Bao, R.-Y.; Cao, J.; Liu, Z.-Y.; Yang, W.; Xie, B.-H.; Yang, M.-B. Towards balanced strength and toughness improvement of isotactic polypropylene nanocomposites by surface functionalized graphene oxide. J. Mater. Chem. A 2014, 2, 3190–3199. [Google Scholar] [CrossRef]
- Yu, Y.; Zeng, F.; Chen, J.; Kang, J.; Yang, F.; Cao, Y.; Xiang, M. Regulating polycrystalline behavior of the β-nucleated isotactic polypropylene/graphene oxide composites by melt memory effect. Polym. Compos. 2018. [Google Scholar] [CrossRef]
- Hao, Z.; Li, L.; Liao, X.; Sheng, X.; Zhang, Y. Preparation and toughening performance investigation of epoxy resins containing carbon nanotubes modified with hyperbranched polyester. Polym. Bull. 2018, 75, 1013–1026. [Google Scholar] [CrossRef]
- Zhang, J.; Hu, C.P. Synthesis, characterization and mechanical properties of polyester-based aliphatic polyurethane elastomers containing hyperbranched polyester segments. Eur. Polym. J. 2008, 44, 3708–3714. [Google Scholar] [CrossRef]
- Han, W.; Liao, X.; Yang, Q.; Li, G.; He, B.; Zhu, W.; Hao, Z. Crystallization and morphological transition of poly(l-lactide)–poly(ε-caprolactone) diblock copolymers with different block length ratios. Rsc Adv. 2017, 7, 22515–22523. [Google Scholar] [CrossRef]
- Hao, Z.; Tan, Y.; Zhang, X.; Zhang, F. Epithermal ageing mechanism of gussasphalt. J. Wuhan Univ. Technol. Mater. Sci. Ed. 2009, 24, 466–470. [Google Scholar] [CrossRef]
- Lu, L.; Xia, L.; Zengheng, H.; Xingyue, S.; Yi, Z.; Pan, L. Investigation on cure kinetics of epoxy resin containing carbon nanotubes modified with hyper-branched polyester. Rsc Adv. 2018, 8, 29830–29839. [Google Scholar] [CrossRef]
- Yu, Y.; Zeng, F.; Chen, J.; Kang, J.; Yang, F.; Cao, Y.; Xiang, M. Effects of ordered structure on non-isothermal crystallization kinetics and subsequent melting behavior of β-nucleated isotactic polypropylene/graphene oxide composites. J. Therm. Anal. Calorim. 2018, 67, 1212–1220. [Google Scholar] [CrossRef]
- Xu, R.; Xu, G.; Wang, J.; Chen, J.; Yang, F.; Kang, J.; Xiang, M. Influence of l-lysine on the permeation and antifouling performance of polyamide thin film composite reverse osmosis membranes. Rsc Adv. 2018, 8, 25236–25247. [Google Scholar] [CrossRef]
- Xu, R.; Wang, J.; Chen, D.; Yang, F.; Kang, J.; Xiang, M.; Li, L.; Sheng, X. Preparation of pH-responsive asymmetric polysulfone ultrafiltration membranes with enhanced anti-fouling properties and performance by incorporating poly(2-ethyl-2-oxazoline) additive. Rsc Adv. 2018, 8, 41270–41279. [Google Scholar] [CrossRef]
- Wang, J.; Xu, R.; Yang, F.; Kang, J.; Cao, Y.; Xiang, M. Probing influences of support layer on the morphology of polyamide selective layer of thin film composite membrane. J. Membr. Sci. 2018, 556, 374–383. [Google Scholar] [CrossRef]
- Qin, Y.; Xu, Y.; Zhang, L.; Zheng, G.; Yan, X.; Dai, K.; Liu, C.; Shen, C.; Guo, Z. Interfacial interaction enhancement by shear-induced β-cylindrite in isotactic polypropylene/glass fiber composites. Polymer 2016, 100, 111–118. [Google Scholar] [CrossRef]
- Kang, J.; Yang, F.; Wu, T.; Li, H.; Cao, Y.; Xiang, M. Polymerization control and fast characterization of the stereo-defect distribution of heterogeneous Ziegler–Natta isotactic polypropylene. Eur. Polym. J. 2012, 48, 425–434. [Google Scholar] [CrossRef]
- Zhang, L.; Qin, Y.; Zheng, G.; Dai, K.; Liu, C.; Yan, X.; Guo, J.; Shen, C.; Guo, Z. Interfacial crystallization and mechanical property of isotactic polypropylene based single-polymer composites. Polymer 2016, 90, 18–25. [Google Scholar] [CrossRef]
- Sun, S.; Zhu, L.; Liu, X.; Wu, L.; Dai, K.; Liu, C.; Shen, C.; Guo, X.; Zheng, G.; Guo, Z. Superhydrophobic Shish-kebab Membrane with Self-Cleaning and Oil/Water Separation Properties. Acs Sustain. Chem. Eng. 2018, 6, 9866–9875. [Google Scholar] [CrossRef]
- Kang, J.; Chen, Z.; Chen, J.; Yang, F.; Weng, G.; Cao, Y.; Xiang, M. Crystallization and melting behaviors of the ß-nucleated isotactic polypropylene with different melt structures—The role of molecular weight. Thermochim. Acta 2015, 599, 42–51. [Google Scholar] [CrossRef]
- Kang, J.Z.J.; Chen, Z.; Yang, F.; Chen, J.; Cao, Y.; Xiang, M. Isothermal crystallization behavior of β-nucleated isotactic polypropylene with different melt structures. J. Polym. Res. 2014. [Google Scholar] [CrossRef]
- Kang, J.; Cao, Y.; Li, H.; Li, J.; Chen, S.; Yang, F.; Xiang, M. Influence of the stereo-defect distribution on the crystallization behavior of Ziegler-Natta isotactic polypropylene. J. Polym. Res. 2012, 19, 1–11. [Google Scholar] [CrossRef]
- Kang, J.; Li, J.; Chen, S.; Zhu, S.; Li, H.; Cao, Y.; Yang, F.; Xiang, M. Hydrogenated petroleum resin effect on the crystallization of isotactic polypropylene. J. Appl. Polym. Sci. 2013, 130, 25–38. [Google Scholar] [CrossRef]
- Lorenzo, A.T.; Müller, A.J. Estimation of the nucleation and crystal growth contributions to the overall crystallization energy barrier. J. Polym. Sci. Part B Polym. Phys. 2008, 46, 1478–1487. [Google Scholar] [CrossRef]
- Müller, A.J.; Arnal, M.L. Thermal fractionation of polymers. Prog. Polym. Sci. 2005, 30, 559–603. [Google Scholar] [CrossRef]
- Fillon, B.; Wittmann, J.; Lotz, B.; Thierry, A. Self-nucleation and recrystallization of isotactic polypropylene (α phase) investigated by differential scanning calorimetry. J. Polym. Sci. Part B Polym. Phys. 1993, 31, 1383–1393. [Google Scholar] [CrossRef]
- Fillon, B.; Thierry, A.; Wittmann, J.; Lotz, B. Self-nucleation and recrystallization of polymers. Isotactic polypropylene, β phase: β-α conversion and β-α growth transitions. J. Polym. Sci. Part B Polym. Phys. 1993, 31, 1407–1424. [Google Scholar] [CrossRef]
- Fillon, B.; Lotz, B.; Thierry, A.; Wittmann, J. Self-nucleation and enhanced nucleation of polymers. Definition of a convenient calorimetric “efficiency scale” and evaluation of nucleating additives in isotactic polypropylene (α phase). J. Polym. Sci. Part B Polym. Phys. 1993, 31, 1395–1405. [Google Scholar] [CrossRef]
- Wang, B.; Chen, Z.; Kang, J.; Yang, F.; Chen, J.; Cao, Y.; Xiang, M. Influence of melt structure on the crystallization behavior and polymorphic composition of polypropylene random copolymer. Thermochim. Acta 2015, 604, 67–76. [Google Scholar] [CrossRef]
- Kang, J.; Chen, Z.; Zhou, T.; Yang, F.; Chen, J.; Cao, Y.; Xiang, M. Dynamic crystallization and melting behavior of β-nucleated isotactic polypropylene with different melt structures. J. Polym. Res. 2014. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, B.; Kang, J.; Peng, H.; Chen, J.; Yang, F.; Cao, Y.; Li, H.; Xiang, M. Crystallization behavior and morphology of β-nucleated isotactic polypropylene with different stereo-defect distribution. Polym. Adv. Technol. 2014, 25, 353–363. [Google Scholar] [CrossRef]
- Kang, J.; Li, J.; Chen, S.; Peng, H.; Wang, B.; Cao, Y.; Li, H.; Chen, J.; Gai, J.; Yang, F.; et al. Investigation of the crystallization behavior of isotactic polypropylene polymerized with different Ziegler-Natta catalysts. J. Appl. Polym. Sci. 2013, 129, 2663–2670. [Google Scholar] [CrossRef]
- Kang, G.W.J.; Chen, J.; Yang, F.; Cao, Y.; Xiang, M. Influences of pre-ordered melt structures on the crystallization behavior and polymorphic composition of β-nucleated isotactic polypropylene with different stereo-defect distribution. J. Appl. Polym. Sci. 2015, 132, 42632. [Google Scholar] [CrossRef]
- Peng, H.; Wang, B.; Gai, J.; Chen, J.; Yang, F.; Cao, Y.; Li, H.; Kang, J.; Xiang, M. Morphology and mechanical behavior of isotactic polypropylene with different stereo-defect distribution in injection molding. Polym. Adv. Technol. 2014, 25, 1464–1470. [Google Scholar] [CrossRef]
- Müller, A.J.; Michell, R.M.; Pérez, R.A.; Lorenzo, A.T. Successive Self-nucleation and Annealing (SSA): Correct design of thermal protocol and applications. Eur. Polym. J. 2015, 65, 132–154. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Miller, R.L. Kinetic of crystallization from the melt and chain folding in polyethylene fractions revisited: Theory and experiment. Polymer 1997, 38, 3151–3212. [Google Scholar] [CrossRef]
- Clark, E.J.; Hoffman, J.D. Regime III crystallization in polypropylene. Macromolecules 1984, 17, 878–885. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Miller, R.L. Response to criticism of nucleation theory as applied to crystallization of lamellar polymers. Macromolecules 1989, 22, 3502–3505. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Miller, R.L. Test of the reptation concept: Crystal growth rate as a function of molecular weight in polyethylene crystallized from the melt. Macromolecules 1988, 21, 3038–3051. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | O 1s (%) | C 1s (%) | n(O)/n(C) |
|---|---|---|---|
| H202 | 60.5 | 39.5 | 1.53 |
| GO | 16.2 | 83.8 | 0.19 |
| GO-H202 | 27.7 | 72.3 | 0.38 |
| Sample | Cooling Scan | Subsequent Heating Scan | ||||||
|---|---|---|---|---|---|---|---|---|
| Tc (°C) | Tconset (°C) | Tcendset (°C) | Width (°C) | Tm (°C) | Tmonset (°C) | Tmendset (°C) | Xc (%) | |
| PP | 111.7 | 118.5 | 106.5 | 12.0 | 161.5 | 153.1 | 166.0 | 48.6 |
| GPP | 115.4 | 121.0 | 110.7 | 10.3 | 163.6 | 153.9 | 167.2 | 49.5 |
| GHPP | 118.5 | 122.6 | 114.4 | 8.2 | 164.8 | 155.2 | 168.7 | 50.9 |
| Sample | Kg × 10−4 (K2) | σe (erg cm−2) |
|---|---|---|
| PP | 23.4 | 96.6 |
| GPP | 20.2 | 83.1 |
| GHPP | 17.9 | 76.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, Z.; Li, L.; Yang, B.; Sheng, X.; Liao, X.; He, L.; Liu, P. Influences of Hyperbranched Polyester Modification on the Crystallization Kinetics of Isotactic Polypropylene/Graphene Oxide Composites. Polymers 2019, 11, 433. https://doi.org/10.3390/polym11030433
Hao Z, Li L, Yang B, Sheng X, Liao X, He L, Liu P. Influences of Hyperbranched Polyester Modification on the Crystallization Kinetics of Isotactic Polypropylene/Graphene Oxide Composites. Polymers. 2019; 11(3):433. https://doi.org/10.3390/polym11030433
Chicago/Turabian StyleHao, Zengheng, Lu Li, Bo Yang, Xingyue Sheng, Xia Liao, Leilei He, and Pan Liu. 2019. "Influences of Hyperbranched Polyester Modification on the Crystallization Kinetics of Isotactic Polypropylene/Graphene Oxide Composites" Polymers 11, no. 3: 433. https://doi.org/10.3390/polym11030433
APA StyleHao, Z., Li, L., Yang, B., Sheng, X., Liao, X., He, L., & Liu, P. (2019). Influences of Hyperbranched Polyester Modification on the Crystallization Kinetics of Isotactic Polypropylene/Graphene Oxide Composites. Polymers, 11(3), 433. https://doi.org/10.3390/polym11030433