Microfluidic Based Fabrication and Characterization of Highly Porous Polymeric Microspheres


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
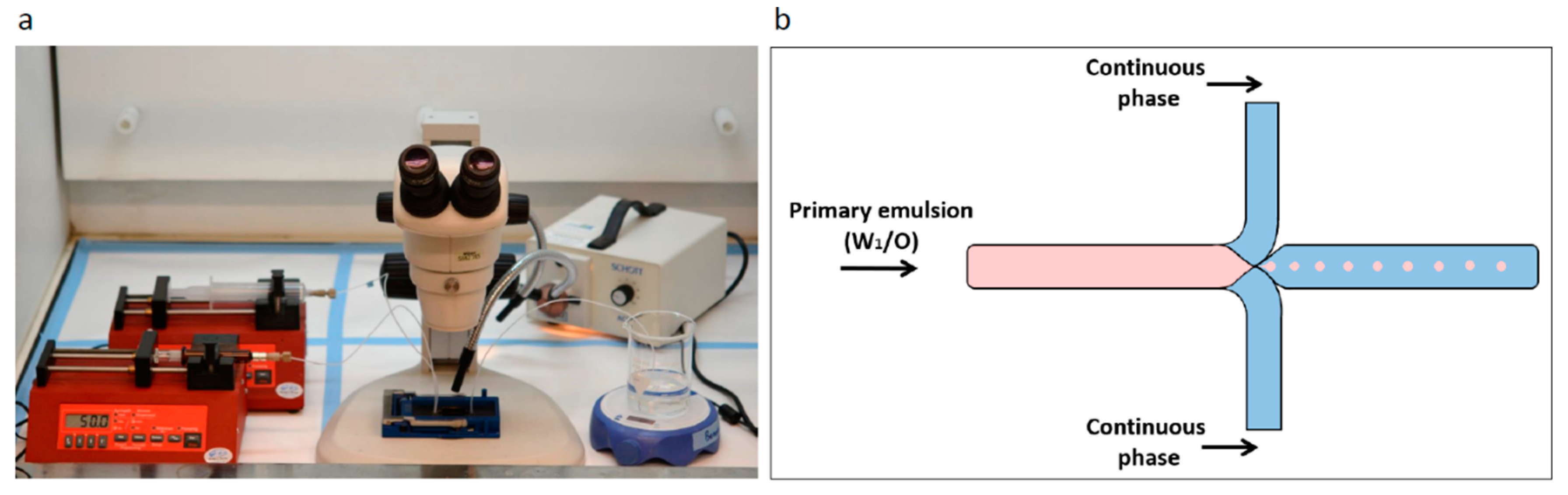
2.2. Microfluidic System and Chip
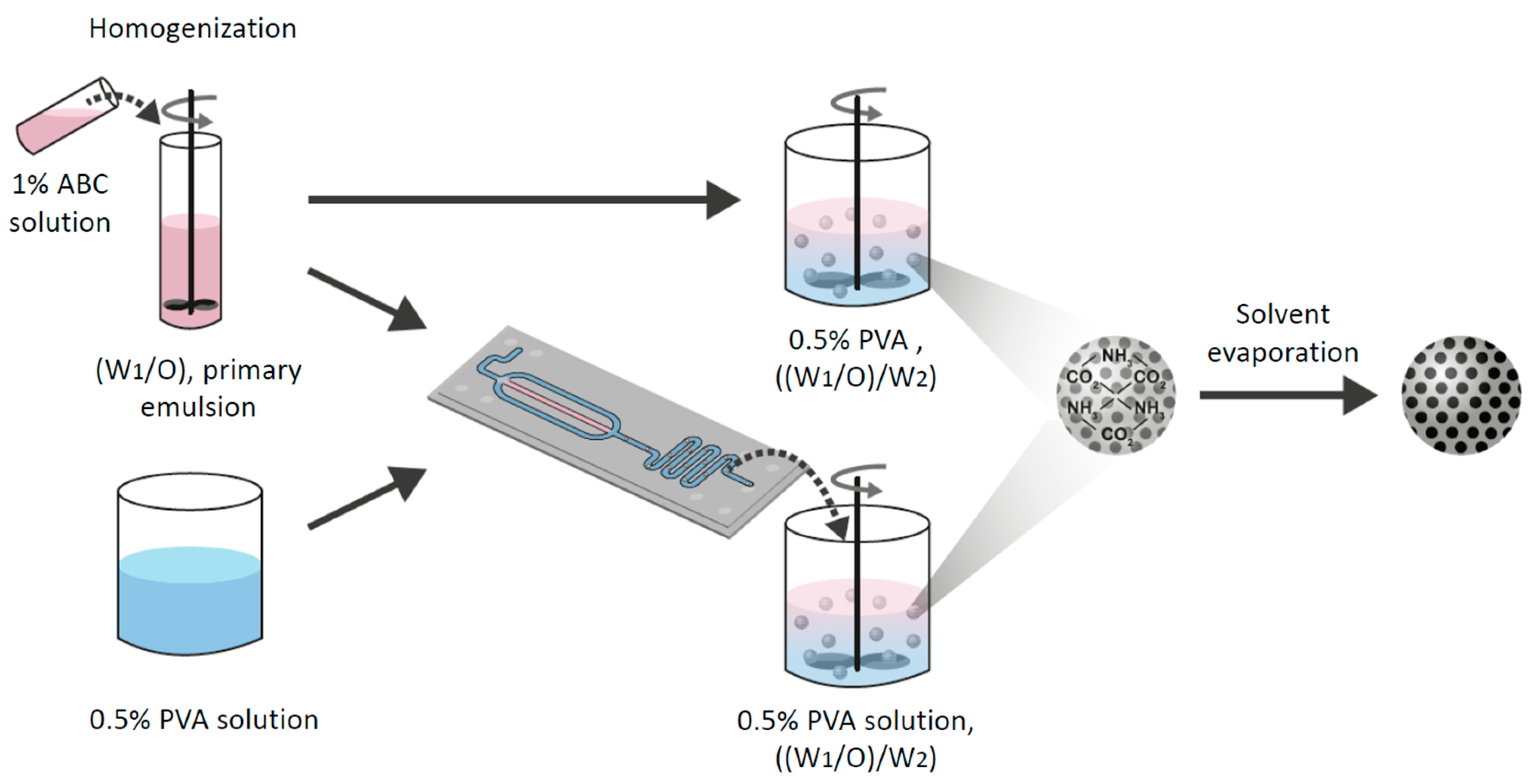
2.3. Preparation of Water-in-Oil Emulsion
2.3.1. Porous Microspheres using Microfluidics
2.3.2. Porous Microparticle Preparation using the Batch Method
2.4. Particle Characterization-Electron Microscopy
2.5. Confocal Microscopy Imaging
2.6. Porosity
2.7. In Vitro Release Kinetics Study
2.8. Statistics
3. Results and Discussion
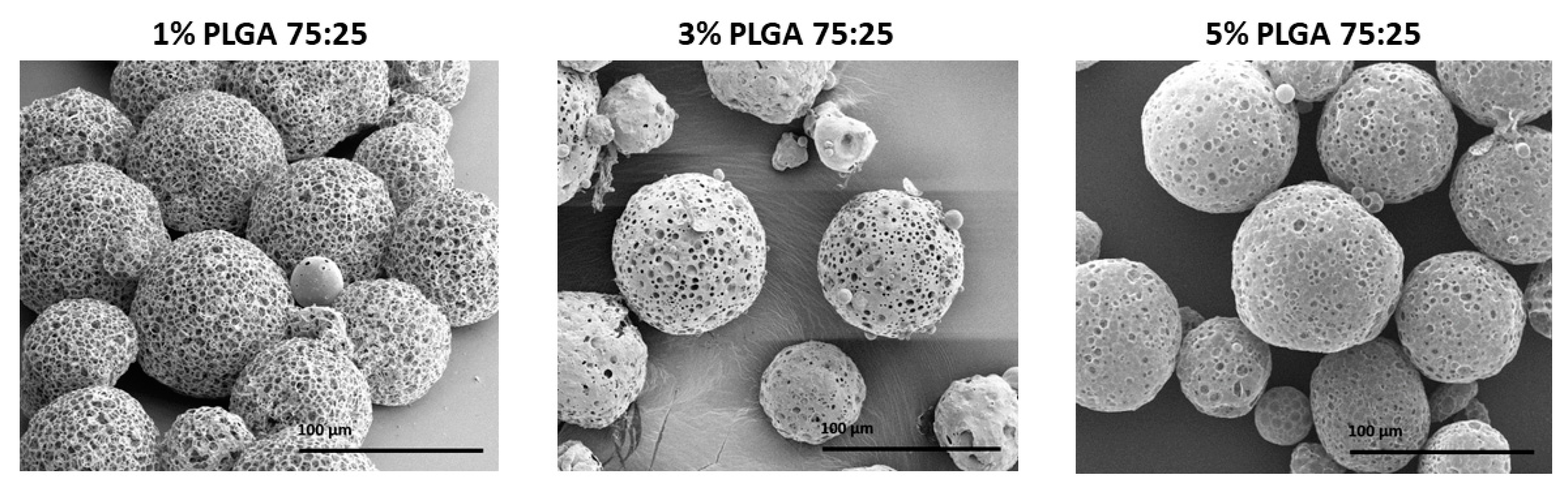
3.1. The Effect of Polymer Concentration
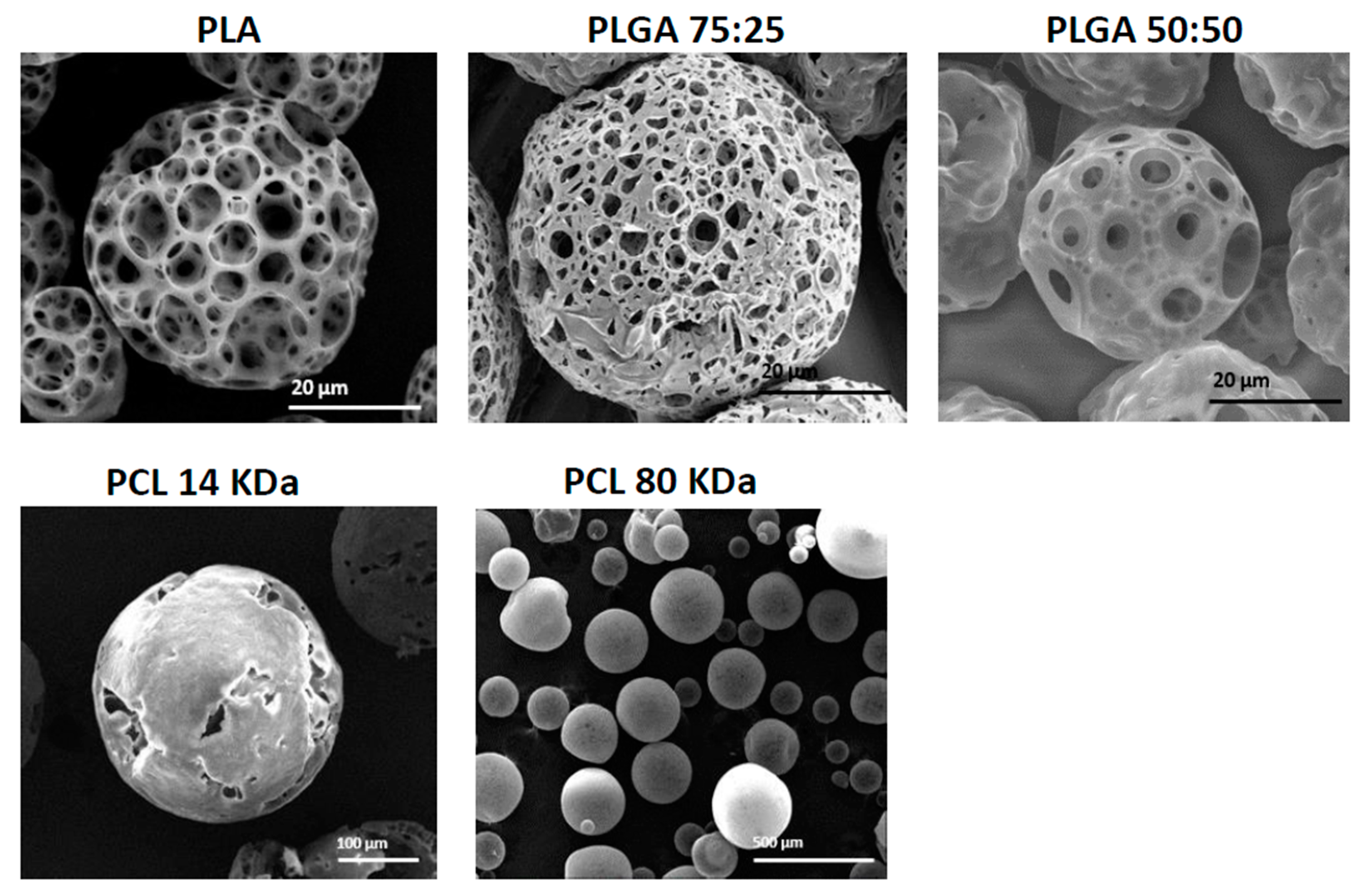
3.2. The Effect of Polymer Type and Molecular Weight
3.3. The Effect of Solvent Type
3.4. The Effect of the Synthesis Method
3.5. The Effect of NaOH Hydrolysis of the Microspheres
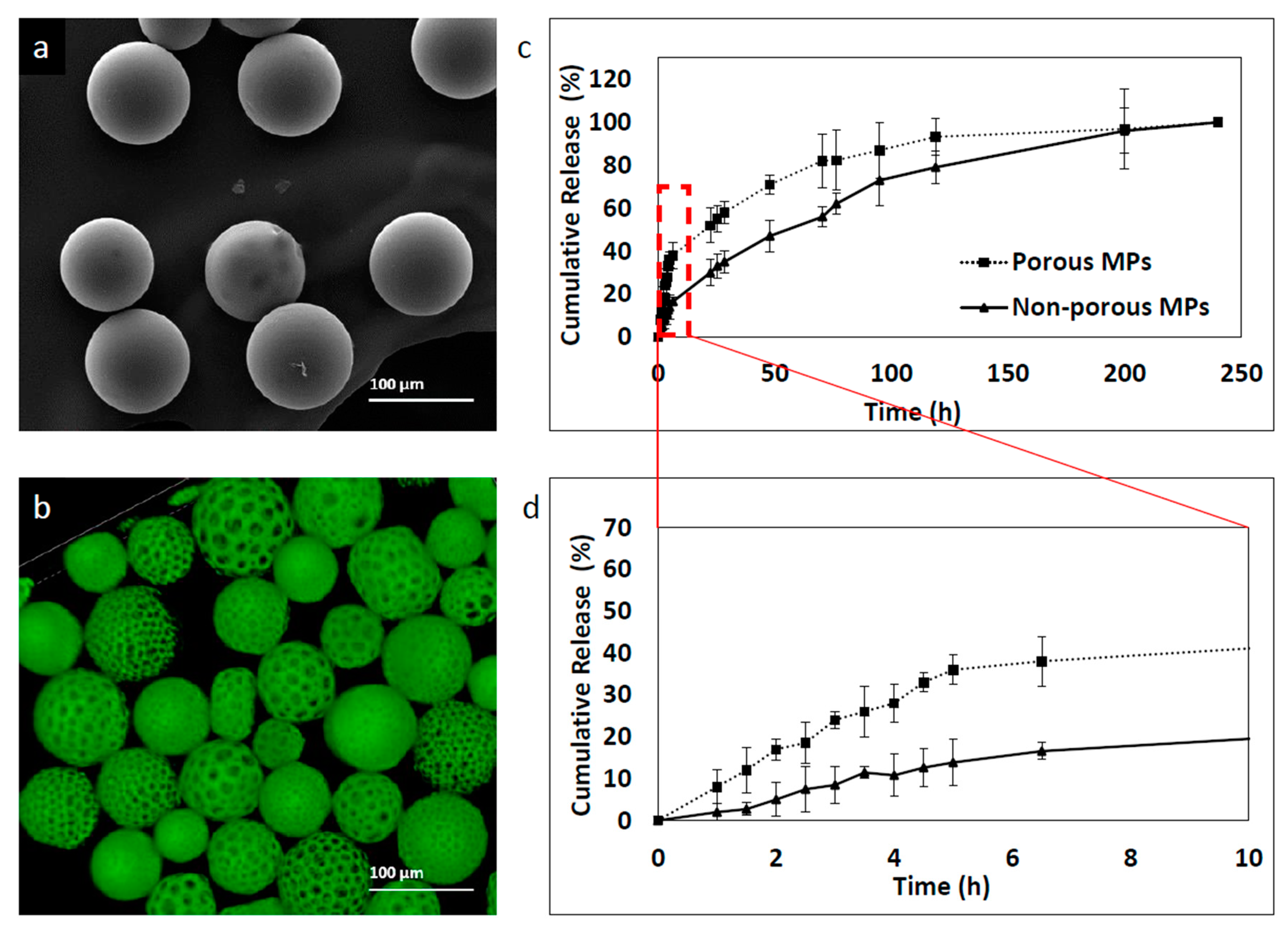
3.6. The Effect of Porosity on the Kinetics Release Profile
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cai, Y.; Chen, Y.; Hong, X.; Liu, Z.; Yuan, W. Porous microsphere and its applications. Int. J. Nanomed. 2013, 8, 1111–1120. [Google Scholar]
- Gokmen, M.T.; Du Prez, F. Progress in Polymer Science Porous polymer particles—A comprehensive guide to synthesis, characterization, functionalization and applications. Prog. Polym. Sci. 2012, 37, 365–405. [Google Scholar] [CrossRef]
- Zhou, J.; Fang, T.; Wen, J.; Shao, Z.; Dong, J. Silk coating on poly (ε-caprolactone) microspheres for the delayed release of vancomycin. J. Microencapsul. 2011, 28, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.J.; Lee, J.; Seo, J.Y.; Rhim, T.; Kim, S.H.; Yoon, H.J.; Lee, K.Y. Preparation of budesonide-loaded porous PLGA microparticles and their therapeutic efficacy in a murine asthma model. J. Control. Release 2011, 150, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Shahriari, D.; Koffler, J.Y.; Tuszynski, M.H. Hierarchically ordered porous and high-volume polycaprolactone microchannel scaffolds enhanced axon growth in transected spinal cords. Tissue Eng. Part A 2017, 23, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Ding, J. Poly (lactide-co-glycolide) porous scaffolds for tissue engineering and regenerative medicine. Interface Focus 2012, 2, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.K.; Panda, A.K.; Labhasetwar, V. Characterization of porous PLGA/PLA microparticles as a scaffold for three dimensional growth of breast cancer cells. Biomacromolecules 2005, 6, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Malikmammadov, E.; Tanir, T.E.; Kiziltay, A.; Hasirci, V.; Hasirci, N. PCL and PCL-based materials in biomedical applications. J. Biomater. Sci. Polym. Ed. 2018, 29, 863–893. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.M.; Shive, M.S. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 2012, 64, 72–82. [Google Scholar] [CrossRef]
- Hines, D.J.; Kaplan, D.L. Poly(lactic-co-glycolic) acid-controlled-release systems: Experimental and modeling insights. Crit. Rev. Ther. Drug Carr. Syst. 2013, 30, 257–276. [Google Scholar] [CrossRef]
- Wang, J.; Li, Y.; Wang, X.; Wang, J.; Tian, H.; Zhao, P.; Tian, Y.; Gu, Y.; Wang, L.; Wang, C. Droplet microfluidics for the production of microparticles and nanoparticles. Micromachines 2017, 8, 22. [Google Scholar] [CrossRef]
- Marre, S.; Jensen, K.F. Synthesis of micro and nanostructures in microfluidic systems. Chem. Soc. Rev. 2010, 39, 1183–1202. [Google Scholar] [CrossRef] [PubMed]
- Whitesides, G.M. The origins and the future of microfluidics. Nature 2006, 442, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Valencia, P.M.; Farokhzad, O.C.; Karnik, R.; Langer, R. Microfluidic technologies for accelerating the clinical translation of nanoparticles. Nat. Nanotechnol. 2012, 7, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Riahi, R.; Tamayol, A.; Shaegh, S.A.M.; Ghaemmaghami, A.M.; Dokmeci, M.R.; Khademshosseini, A. Microfluidics for advanced drug delivery systems. Curr. Opin. Chem. Eng. 2015, 7, 101–112. [Google Scholar] [CrossRef]
- Damiati, S.; Kompella, U.B.; Damiati, S.A.; Kodzius, R. Microfluidic devices for drug delivery systems and drug screening. Genes 2018, 9, 103. [Google Scholar] [CrossRef] [PubMed]
- Amoyav, B.; Benny, O. Controlled and tunable polymer particles’ production using a single microfluidic device. Appl. Nanosci. 2018, 8, 905–914. [Google Scholar] [CrossRef]
- Taek, K.K.; Jun, J.Y.; Doo, S.L.; Park, T.G. Gas foamed open porous biodegradable polymeric microspheres. Biomaterials 2006, 27, 152–159. [Google Scholar]
- Choi, S.W.; Yeh, Y.C.; Zhang, Y.; Sung, H.W.; Xia, Y. Uniform beads with controllable pore sizes for biomedical applications. Small 2010, 6, 1492–1498. [Google Scholar] [CrossRef] [PubMed]
- Choi, N.W.; Cabodi, M.; Held, B.; Gleghorn, J.P.; Bonassar, L.J.; Stroock, A.D. Microfluidic scaffolds for tissue engineering. Nat. Mater. 2007, 6, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Ping, Q.; Gao, Y. Effects of formulation factors on encapsulation efficiency and release behaviour in vitro of huperzine A-PLGA microspheres. J. Microencapsul. 2005, 22, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Freed, K.F.; Edwards, S.F. Polymer viscosity in concentrated solutions. J. Chem. Phys. 1974, 61, 3626–3633. [Google Scholar] [CrossRef]
- Davies, O.R.; Marlow, M.; Stolnik, S. Macroporous surface modified microparticles. Soft Matter 2008, 4, 1597–1601. [Google Scholar] [CrossRef]
- Wischke, C.; Schwendeman, S.P. Principles of encapsulating hydrophobic drugs in PLA/PLGA microparticles. Int. J. Pharm. 2008, 364, 298–327. [Google Scholar] [CrossRef] [PubMed]
- Rosca, I.D.; Watari, F.; Uo, M. Microparticle formation and its mechanism in single and double emulsion solvent evaporation. J. Control. Release 2004, 99, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Sander, E.A.; Alb, A.M.; Nauman, E.A.; Reed, W.F.; Dee, K.C. Solvent effects on the microstructure and properties of 75/25 poly(d,l-lactide-co-glycolide) tissue scaffolds. J. Biomed. Mater. Res. Part A 2004, 70, 506–513. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Li, X.; Zhu, T.; Shan, M.; Zhu, L.; Si, T.; Wang, H.; Sun, Y. Controlling the internal structures of polymeric microspheres via the introduction of a water-soluble organic solvent. Polymers 2018, 10, 789. [Google Scholar] [CrossRef]
- He, Y.; Battat, S.; Fan, J.; Abbaspourrad, A.; Weitz, D.A. Preparation of microparticles through co-flowing of partially miscible liquids. Chem. Eng. J. 2017, 320, 144–150. [Google Scholar] [CrossRef]
- Capretto, L.; Cheng, W.; Hill, M.; Zhang, X. Micromixing Within Microfluidic Devices. In Microfluidics; Springer: Berlin/Heidelberg, Germany, 2011; pp. 27–68. [Google Scholar]
- Joseph, M.F.; Bradford, A.B. Shear stress measurement in microfluidic systems: Liquid crystal technique. In Volume 10: Heat Transfer, Fluid Flows, and Thermal Systems, Parts A, B, and C, Proceedings of the ASME 2008 International Mechanical Engineering Congress and Exposition, Boston, MA, USA, 31 October–6 November 2008; Bruno Union College: Schenectady, NY, USA, 2008; pp. 2009–2018. [Google Scholar]
- Tsao, C.W.; Cheng, Y.C.; Cheng, J.H. Fluid flow shear stress stimulation on a multiplex microfluidic device for rat bone marrow stromal cell differentiation enhancement. Micromachines 2015, 6, 1996–2009. [Google Scholar] [CrossRef]
- Keles, H.; Naylor, A.; Clegg, F.; Sammon, C. Investigation of factors influencing the hydrolytic degradation of single PLGA microparticles. Polym. Degrad. Stable 2015, 119, 228–241. [Google Scholar] [CrossRef]
- Holy, C.E.; Dang, S.M.; Davies, J.E.; Shoichet, M.S. In vitro degradation of a novel poly(lactide-co-glycolide) 75/25 foam. Biomaterials 1999, 20, 1177–1185. [Google Scholar] [CrossRef]
- Zolnik, B.S.; Burgess, D.J. Effect of acidic pH on PLGA microsphere degradation and release. J. Control. Release 2007, 122, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Vysloužil, J.; Doležel, P.; Kejdušová, M.; Mašková, E.; Mašek, J.; Lukáč, R.; Košťál, V.; Vetchý, D.; Dvořáčková, K. Influence of different formulations and process parameters during the preparation of drug-loaded PLGA microspheres evaluated by multivariate data analysis. Acta Pharm. 2014, 64, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Klose, D.; Siepmann, F.; Elkharraz, K.; Krenzlin, S.; Siepmann, J. How porosity and size affect the drug release mechanisms from PLGA-based microparticles. Int. J. Pharm. 2006, 314, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Chew, S.A.; Arriaga, M.A.; Hinojosa, V.A. Effects of surface area to volume ratio of PLGA scaffolds with different architectures on scaffold degradation characteristics and drug release kinetics. J. Biomed. Mater. Res. Part A 2016, 104, 1202–1211. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor | PLGA 75.25 Concentration (% w/v) | ||
|---|---|---|---|
| 1% | 3% | 5% | |
| Average diameter (μm) | 91 ± 1 | 86 ± 2 | 87 ± 3 |
| Average surface pore diameter (μm) | 11 ± 3 | 10 ± 4 | 8 ± 3 |
| Porosity (%) | 83.7 | 71.3 | 68.4 |
| Factor | Type of Solvent | ||
|---|---|---|---|
| DCM | CF | EA | |
| Average diameter (μm) | 150 ± 6 | 41 ± 4 | - |
| Average surface pore diameter (μm) | 6 ± 2 | 8 ± 6 | - |
| Porosity (%) | 78.4 | 93.3 | - |
| Factor | Type of Polymer (Flow Rate in mL/min) | |||
|---|---|---|---|---|
| PLGA 75:25 (0.3) | PLGA 75:25 (0.6) | PLA (0.3) | PLA (0.6) | |
| Average diameter (μm) | 77 ± 4 | 43 ± 5 | 112 ± 4 | 53 ± 7 |
| Average surface pore diameter (μm) | 4 ± 7 | 3 ± 8 | 23 ± 8 | 12 ± 5 |
| Porosity (%) | 71.3 | 78.3 | 81.5 | 86.6 |
| Factor | NaOH 0.2% Solution Immersing Time (min) | ||||
|---|---|---|---|---|---|
| 0 | 2 | 4 | 6 | 8 | |
| Average diameter (μm) | 298 ± 11 | 307 ± 6 | 312 ± 19 | 238 ± 7 | 225 ± 14 |
| Average surface pore diameter (μm) | 8 ± 3 | 14 ± 5 | 32 ± 12 | 31 ± 9 | 53 ± 13 |
| Porosity (%) | 68.3 | 79.3 | 84.7 | 91.8 | 98.6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amoyav, B.; Benny, O. Microfluidic Based Fabrication and Characterization of Highly Porous Polymeric Microspheres. Polymers 2019, 11, 419. https://doi.org/10.3390/polym11030419
Amoyav B, Benny O. Microfluidic Based Fabrication and Characterization of Highly Porous Polymeric Microspheres. Polymers. 2019; 11(3):419. https://doi.org/10.3390/polym11030419
Chicago/Turabian StyleAmoyav, Benzion, and Ofra Benny. 2019. "Microfluidic Based Fabrication and Characterization of Highly Porous Polymeric Microspheres" Polymers 11, no. 3: 419. https://doi.org/10.3390/polym11030419
APA StyleAmoyav, B., & Benny, O. (2019). Microfluidic Based Fabrication and Characterization of Highly Porous Polymeric Microspheres. Polymers, 11(3), 419. https://doi.org/10.3390/polym11030419