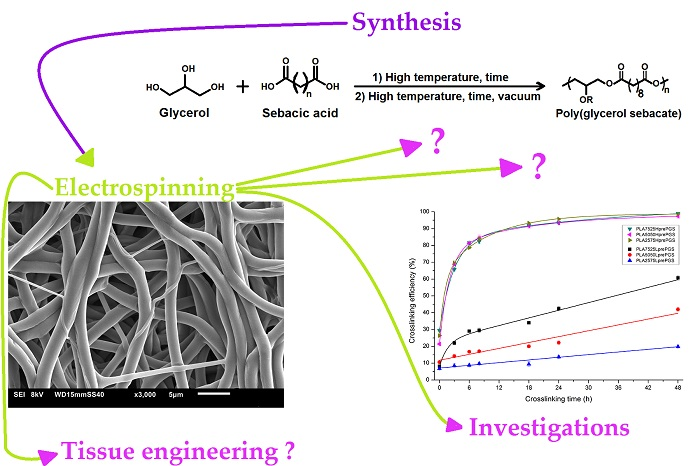
Poly(Glycerol Sebacate)–Poly(l-Lactide) Nonwovens. Towards Attractive Electrospun Material for Tissue Engineering




Abstract

1. Introduction
- Hydrolysis as a degradation mechanism and hydrolysable ester bonds;
- Adequate degree of crosslinking, providing elastomeric properties;
- Chemical bonds that are involved in crosslinking shall be hydrolysable and be identical to those present in the main chain to minimize heterogeneous degradation;
- The monomers from which the material is synthesized should be non-toxic, at least one of them should be trifunctional, and at least one of them should provide hydroxyl groups for hydrogen bonds.
PGS—Good Properties for Soft Tissues
- PGS cannot be electrospun singly, neither in pre-polymer form, nor in crosslinked form. It was highly probable that overcoming this limitation is possible by electrospinning of only slightly crosslinked prepolymer with a higher molecular weight, but still soluble. However, it is already known by authors that the slightly crosslinked PGS form is highly viscous and, without any additives, non-electrospinnable.
- One of the feasible methods is to process it in core-shell geometry, with PGS prepolymer as a core. During the subsequent process—crosslinking followed by leaching—the shell polymer must be thermally stable in the crosslinking temperature range (120 °C–140 °C).
- The other method is to electrospin a blend of PGS pre-polymer with the so-called carrier polymer, (e.g., PVA), followed by curing and leaching out the carrier polymer and residuals of non-crosslinked pre-PGS; such electrospinning of blends may be more reliable and much easier to optimize than core-shell. The drawbacks are related to leaching of the carrier polymer in itself, the residuals of leached polymer, and unknown influence of interactions between PGS and the carrier polymer on PGS crosslinking.
- Electrospinning of pre-PGS as a bicomponent blend with, for example, PCL or PLA or other components, without subsequent leaching, is another promising option. It is a blend-electrospinning, with an intention that PGS prepolymer will bring novel properties, that is, hydrophilicity, with a possible effect on mechanical properties, particularly when PGS contribution is the majority. In the case of blends with PCL, PGS cannot be crosslinked, so there is an essential lack of possibility for elastomeric properties, leading to a question of whether the whole idea is worth the effort. There are other, simpler methods of increasing hydrophilicity rather than synthesis of PGS and electrospinning with addition of PCL. PLA may be a more appropriate choice—its melting point is around 160 °C–180 °C, so the PGS in such blends can be crosslinked. Such investigations are being undertaken by the authors of this publication, using various contributions of PGS in blends with PLA, ranging between 25% and 75%.
2. Materials and Methods
2.1. Synthesis of Two Types of PGS
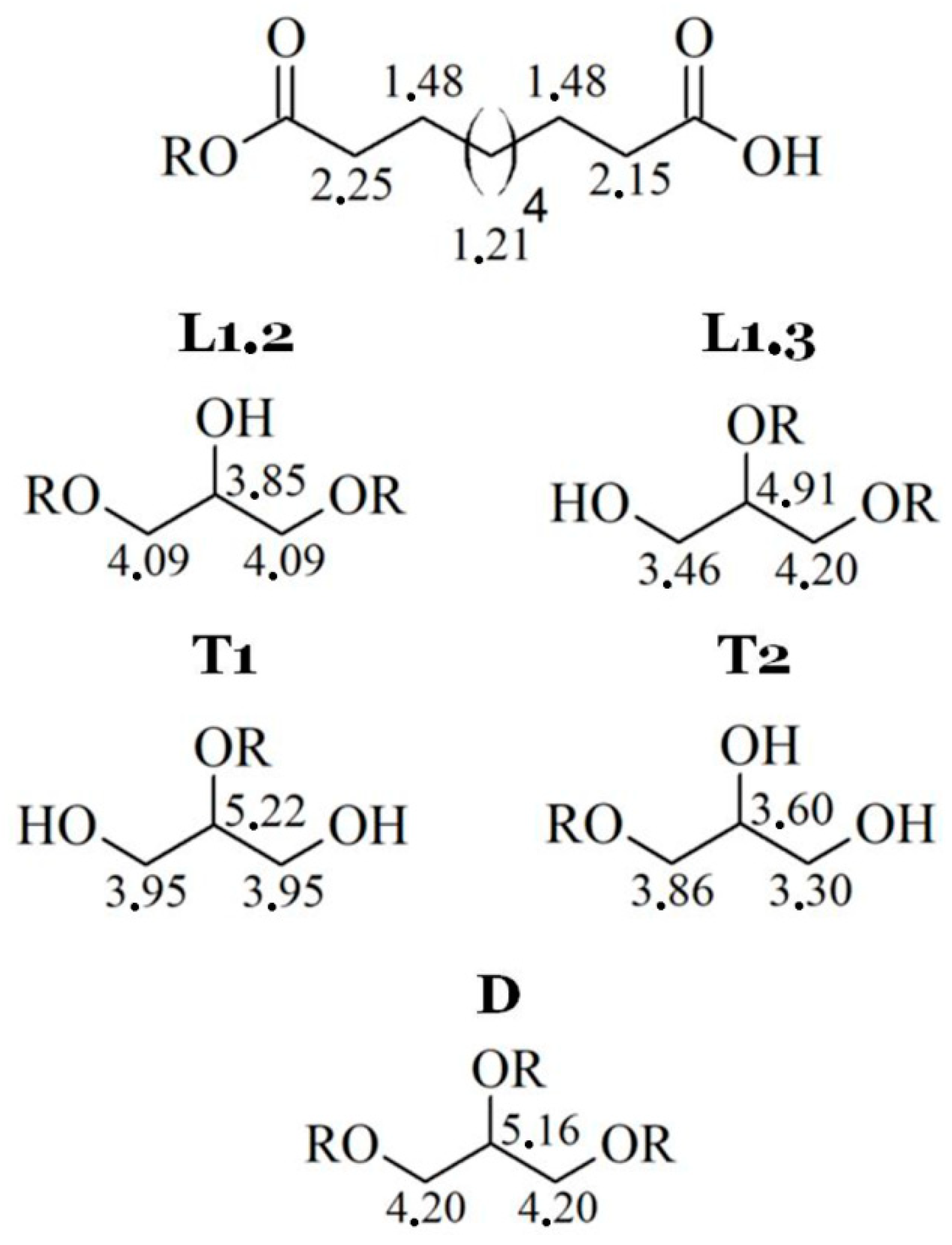
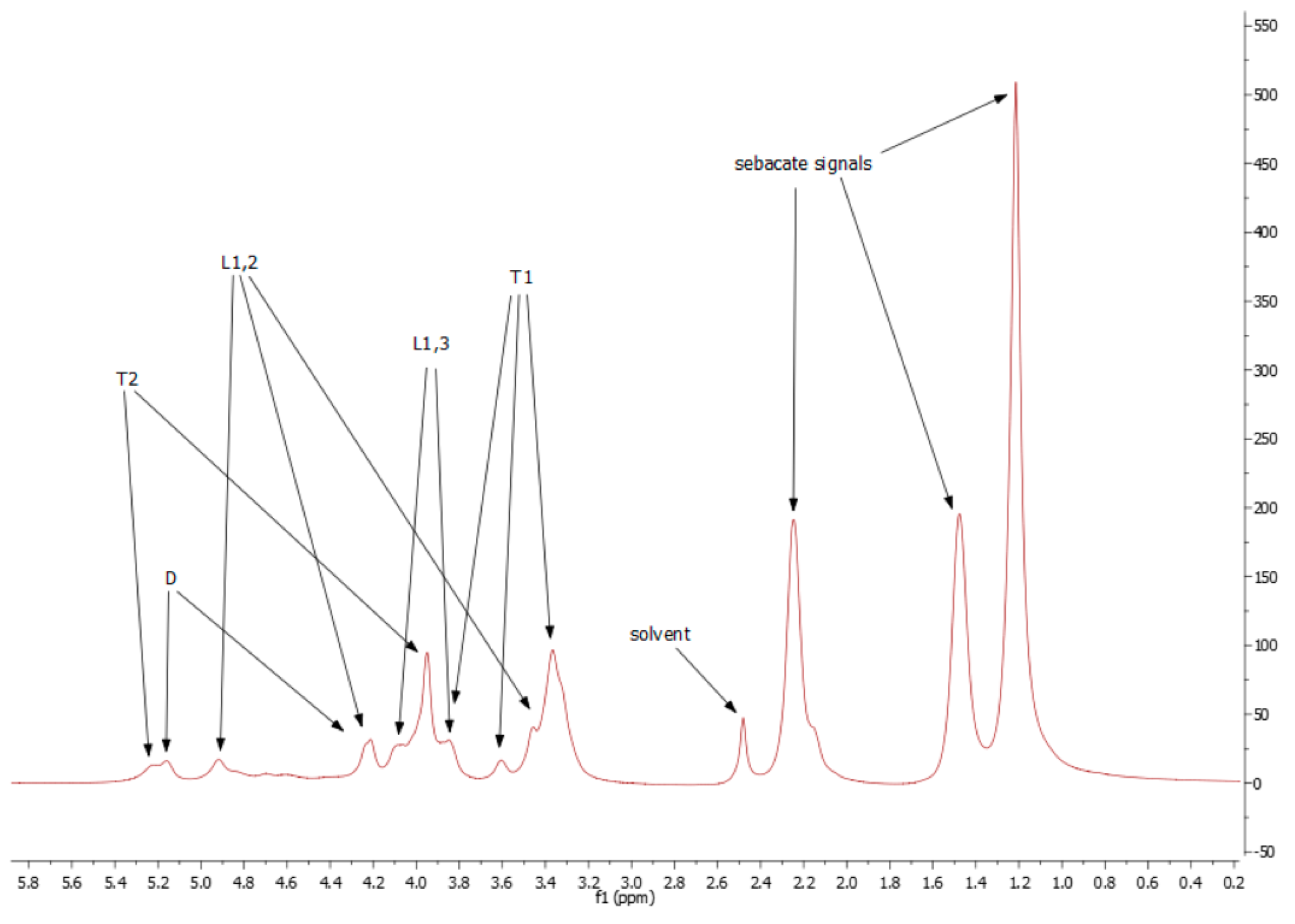
2.1.1. Nuclear Magnetic Resonance (NMR) Spectroscopy
2.1.2. Esterification Degree
2.1.3. Molecular Weight
2.2. Electrospinning of PLA–PGS Blends
2.3. Crosslinking of Nonwovens
2.4. Scanning Electron Microscopy (SEM)
2.5. Wettability
2.6. Differential Scanning Calorimetry (DSC) Analysis
2.7. FTIR Analysis
3. Results and Discussion
3.1. PGS Characterization
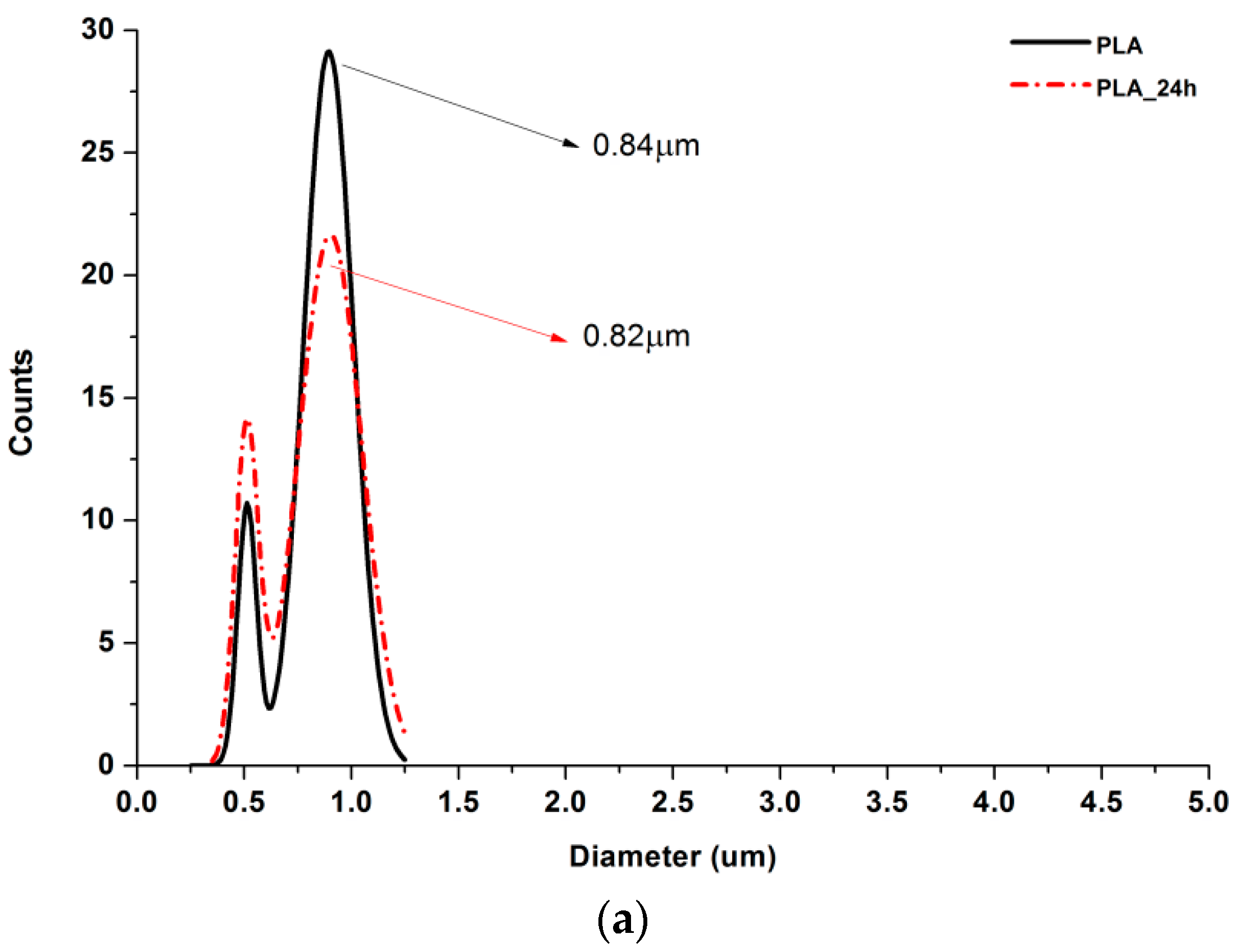
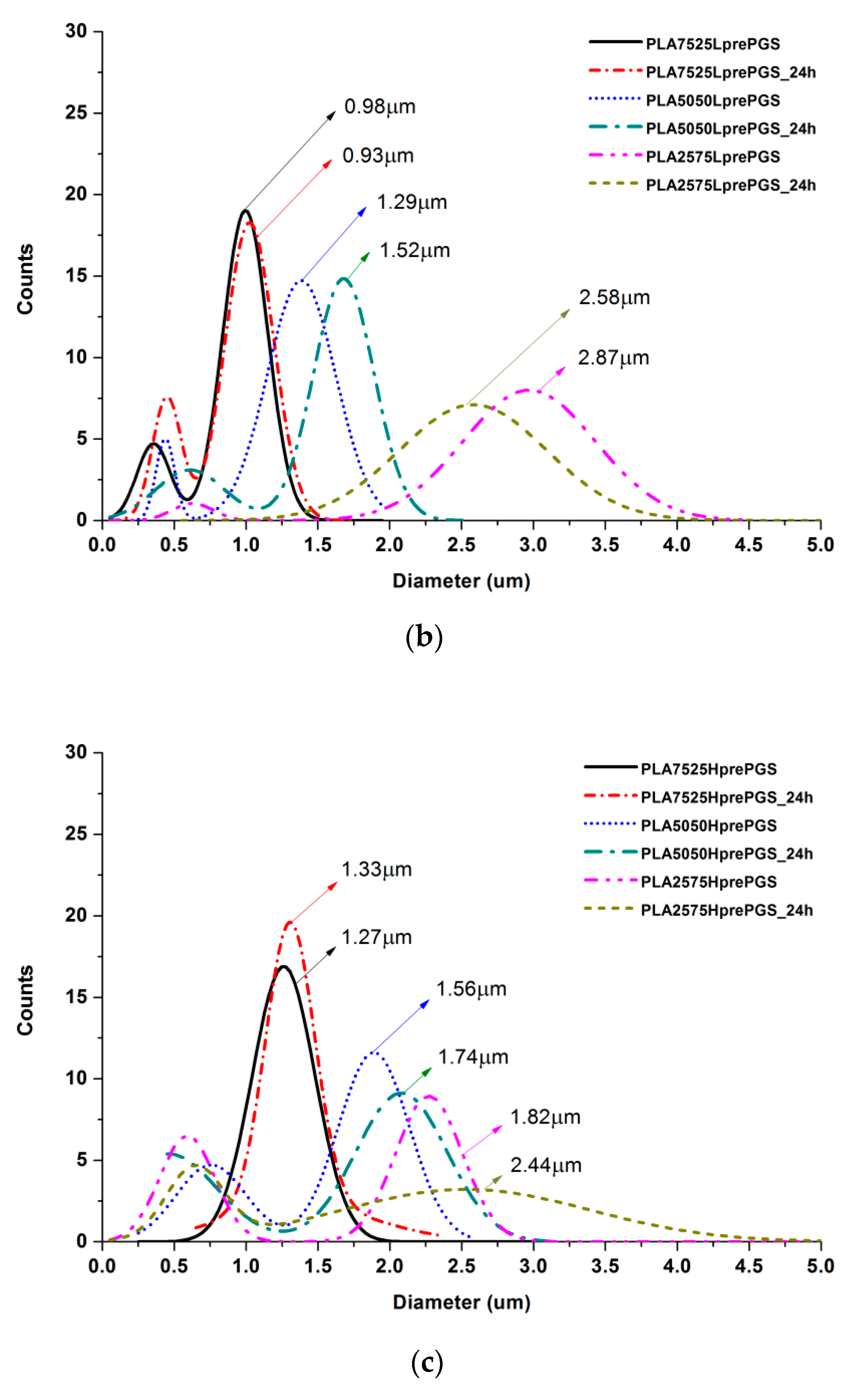
3.2. SEM Nonwovens Morphology Analysis
3.3. Crosslinking Efficiency
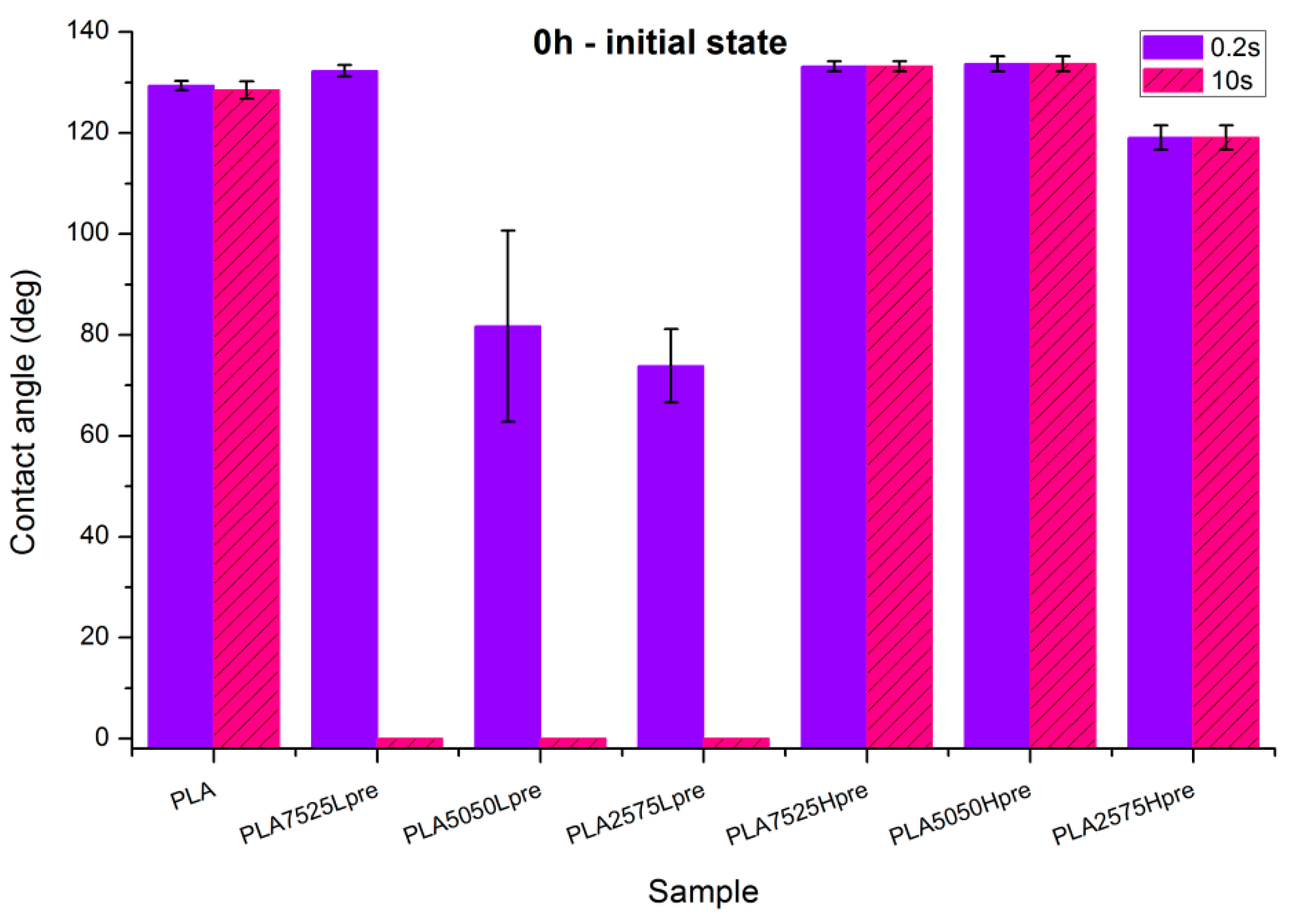
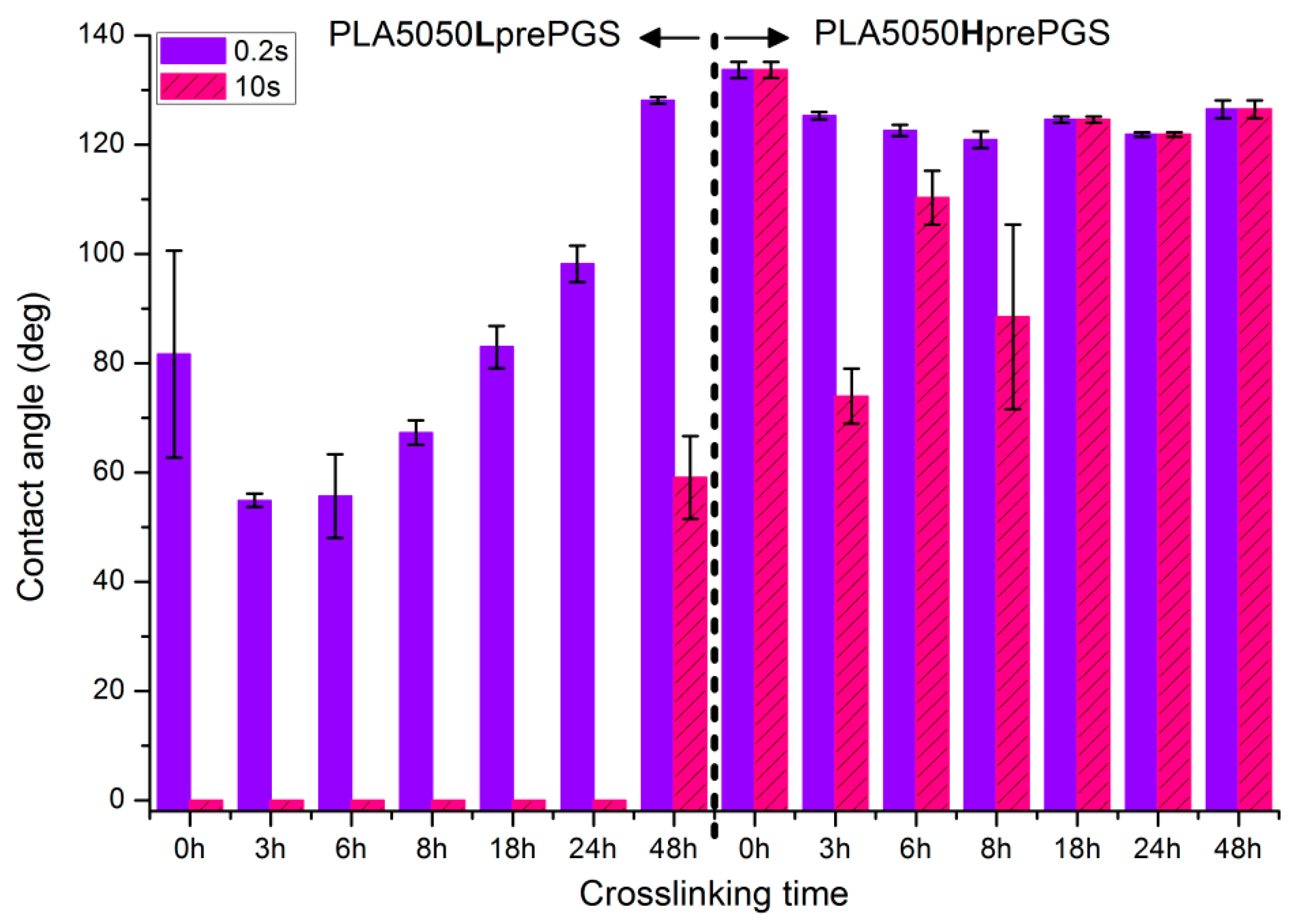
3.4. Wettability
- PLA is a polyester with contact angle values around 70°–80° for film samples, and contact angle values increase in nonwoven form, which is a known phenomenon [35]
- Nonwovens consisting of PLA and LprePGS are hydrophilic owing to LprePGS, which is a hydrophilic polymer thanks to the –OH groups. The more LprePGS, the more apparent the hydrophilic nature. For all samples with LprePGS, after 10 s, a whole water droplet is adsorbed by the materials
- HprePGS, which is supposed to have much less –OH groups owing to the higher degree of conversion of sebacic acid and glycerol, higher crosslinking degree, and higher esterification degree after the second step of polycondensation, compared with LprePGS, does not bring hydrophilicity to nonwovens. A slightly lower contact angle for the PLA2575Hpre sample (but still in the hydrophobic range) may be because of the higher content of HprePGS, as well as (or even more likely) because of the different morphology of nonwoven compared with samples with a lower HprePGS content.
- Nonwoven of pure PLA became slightly less hydrophobic, for 10 s, being not much above the hydrophilic boundary value of 90°. After the crosslinking process, shrinking appears; fibers are closer to each other; and the morphology of the nonwoven is closer to casted film, which is characterized by lower values of contact angle. The difference between the contact angle values for 0.2 s and 10 s also indicates that the water droplet is being adsorbed by the material, slowly but constantly. Such behavior indicates that the material is rather hydrophilic than hydrophobic, even if the contact angle values at the beginning are above 90 degrees.
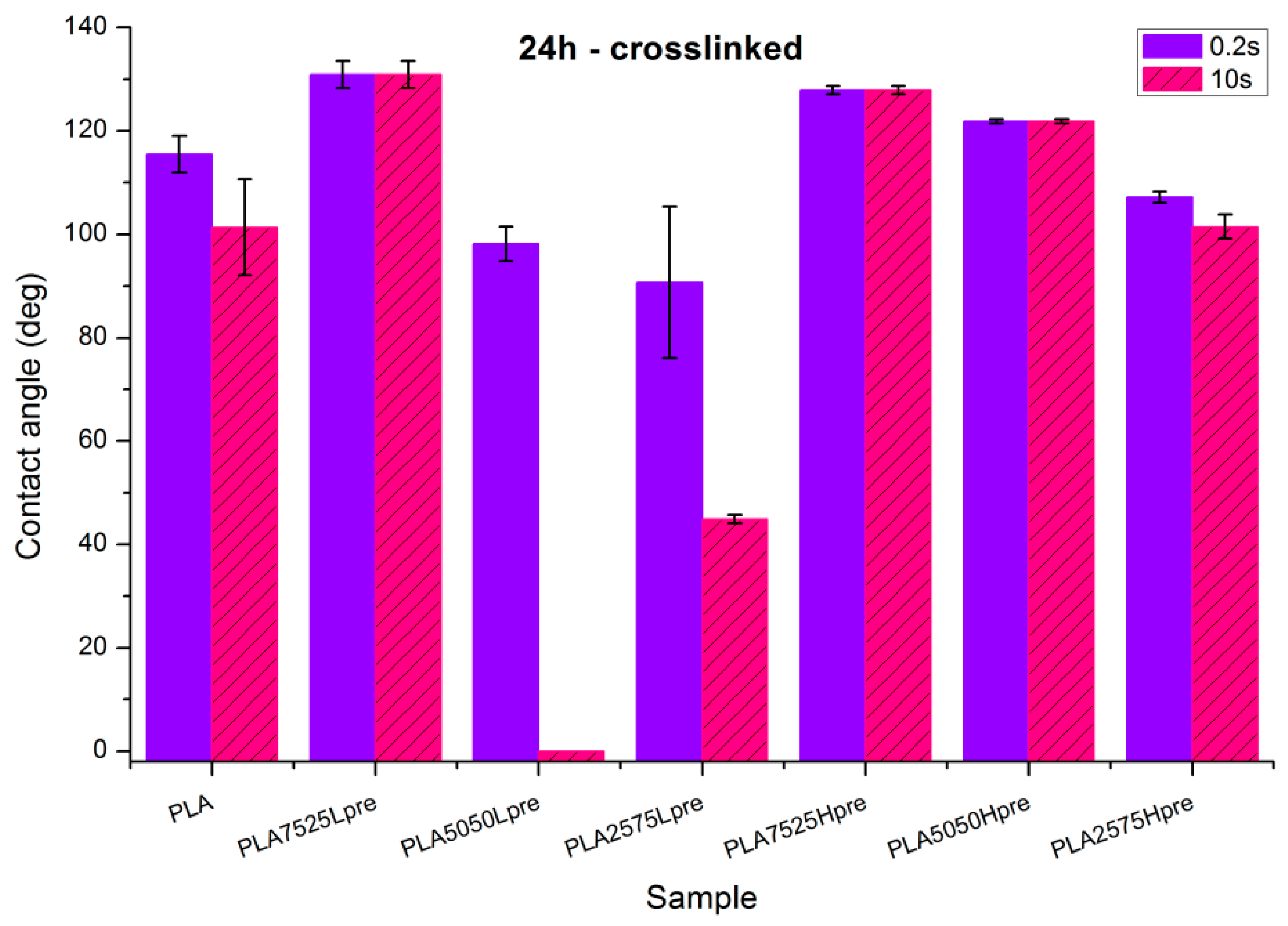
- Nonwovens with LprePGS, after 24 h of crosslinking, changed as expected. The blend with 25% addition of LprePGS, with the highest crosslinking degree, became hydrophobic, and the hydrophilicity of other two blends with a higher content of LprePGS was altered toward higher contact angle values, owing to the diminishing of free –OH groups’ content.
- For nonwovens with HprePGS, the contact angle values slightly decreased, but remained in the hydrophobic range. Observations may indicate that, when it comes to the hydrophobic materials analyzed here, the change in the morphology of nonwoven after heating (shrinking) may lead it to less hydrophobic properties, just as was observed for pure PLA nonwoven.
- For nonwovens with LprePGS, with the shortest time of crosslinking, during which LprePGS is crosslinked just slightly, the major effect of increasing hydrophilicity probably comes from the change in morphology of nonwoven—shrinking and reduced spaces between fibers. Longer times of crosslinking lead to increasing of the contact angle registered at 0.2 s. Nonwovens, however, remain hydrophilic even after 48 h of crosslinking
- For nonwovens with HprePGS, similar effects are observed, but within the range of much higher values of the contact angle; after 3 h of crosslinking, a decrease in the contact angle is observed compared with non-crosslinked nonwoven—because of the change of nonwoven morphology, which overcomes influence of increasing the crosslinking degree. After longer times of crosslinking, however, the contact angle values increase, especially those registered after 10 s—nonwovens become more and more hydrophobic up to 18 h of crosslinking. After that, with the crosslinking degree of SC PGS at the level of 90% and above, the contact angle stays above 120 degrees, more or less within the error range.
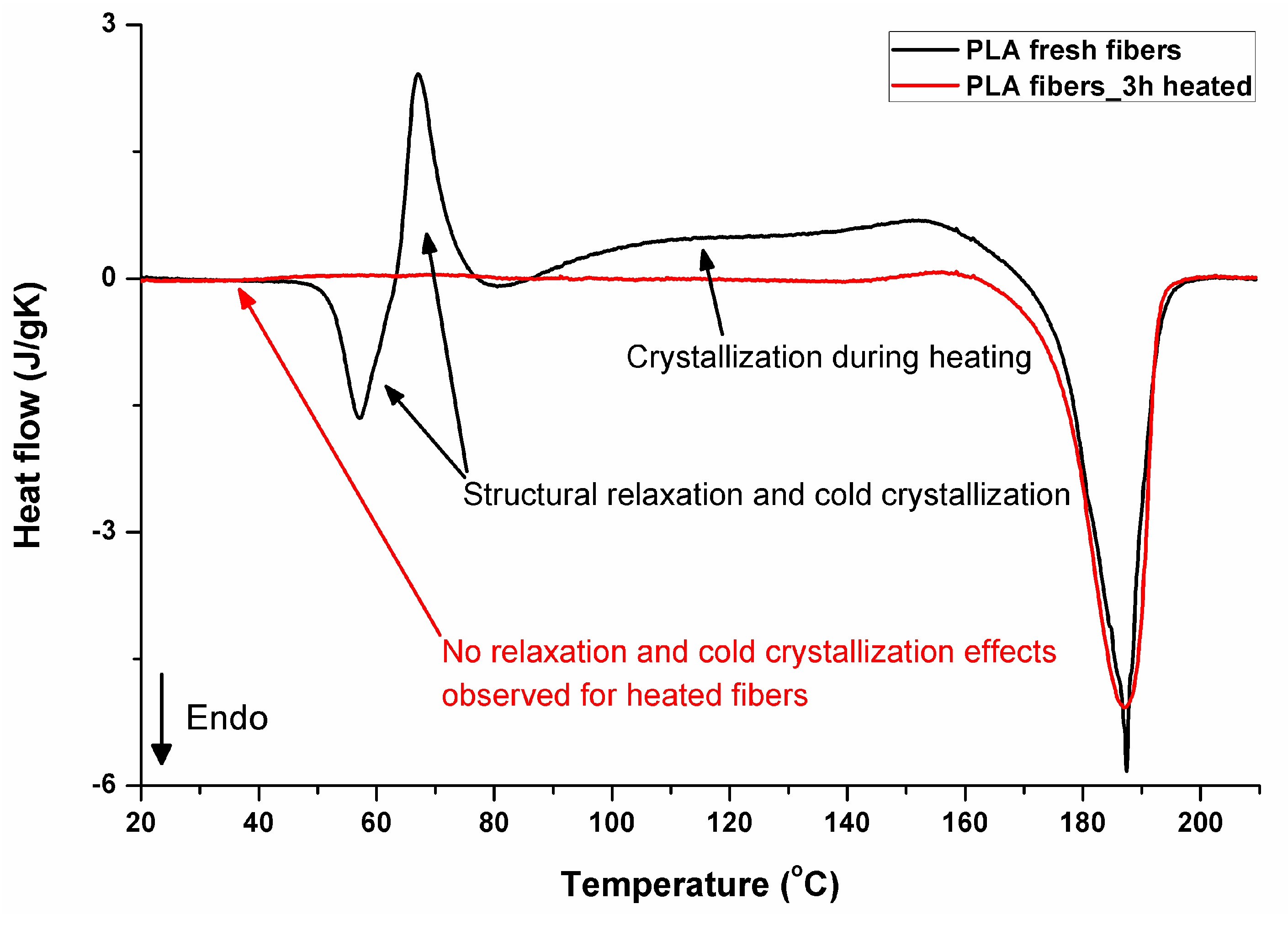
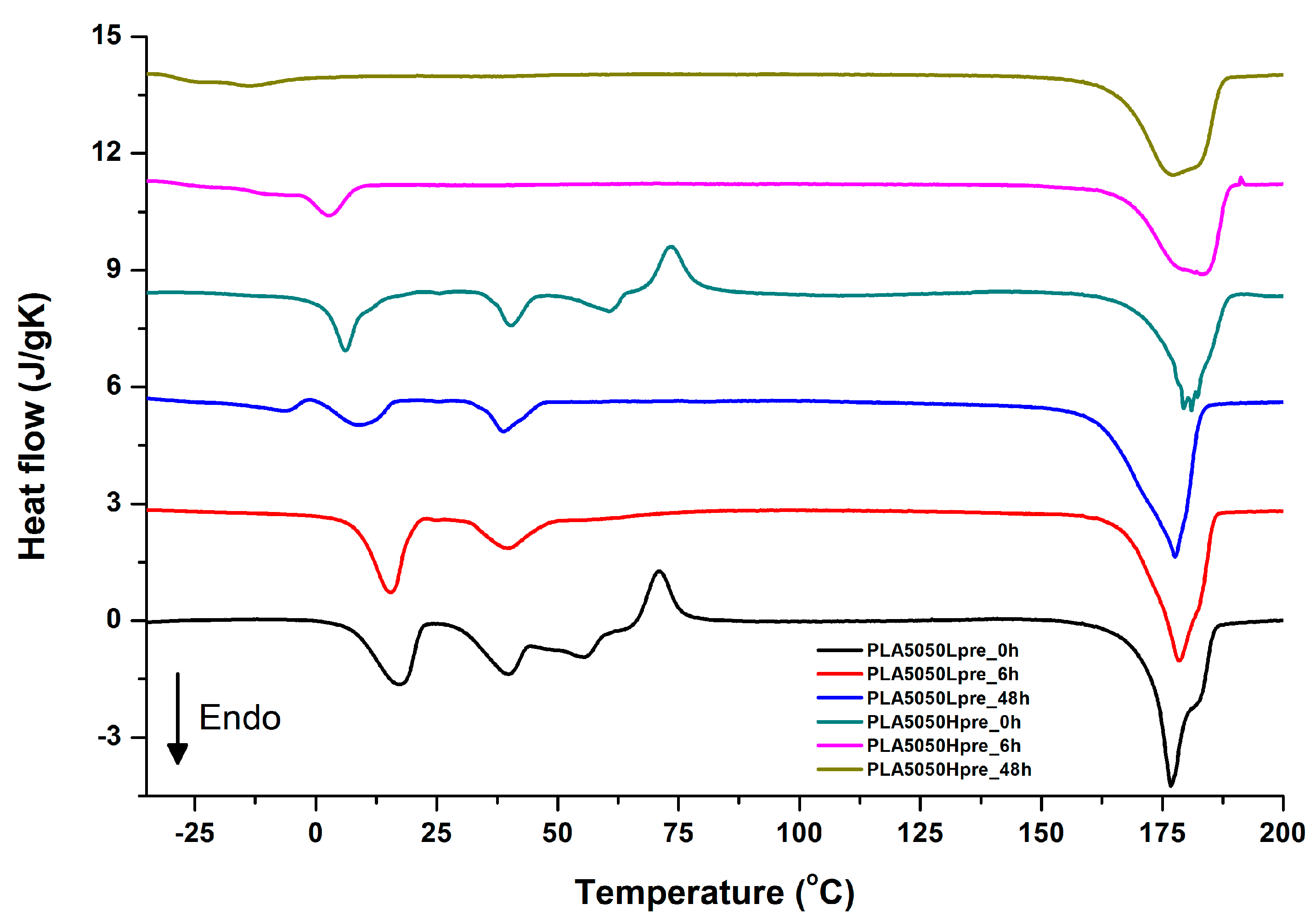
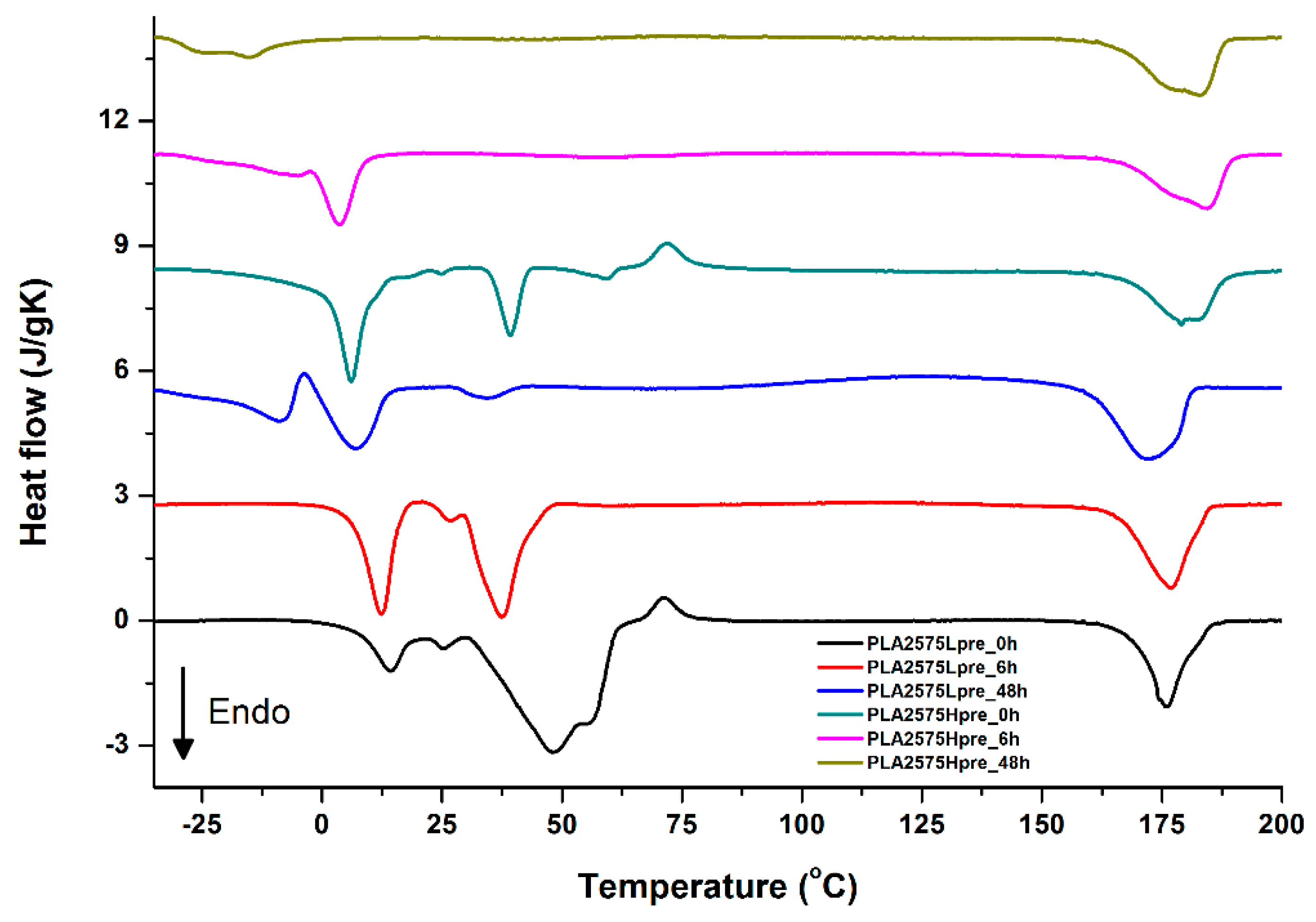
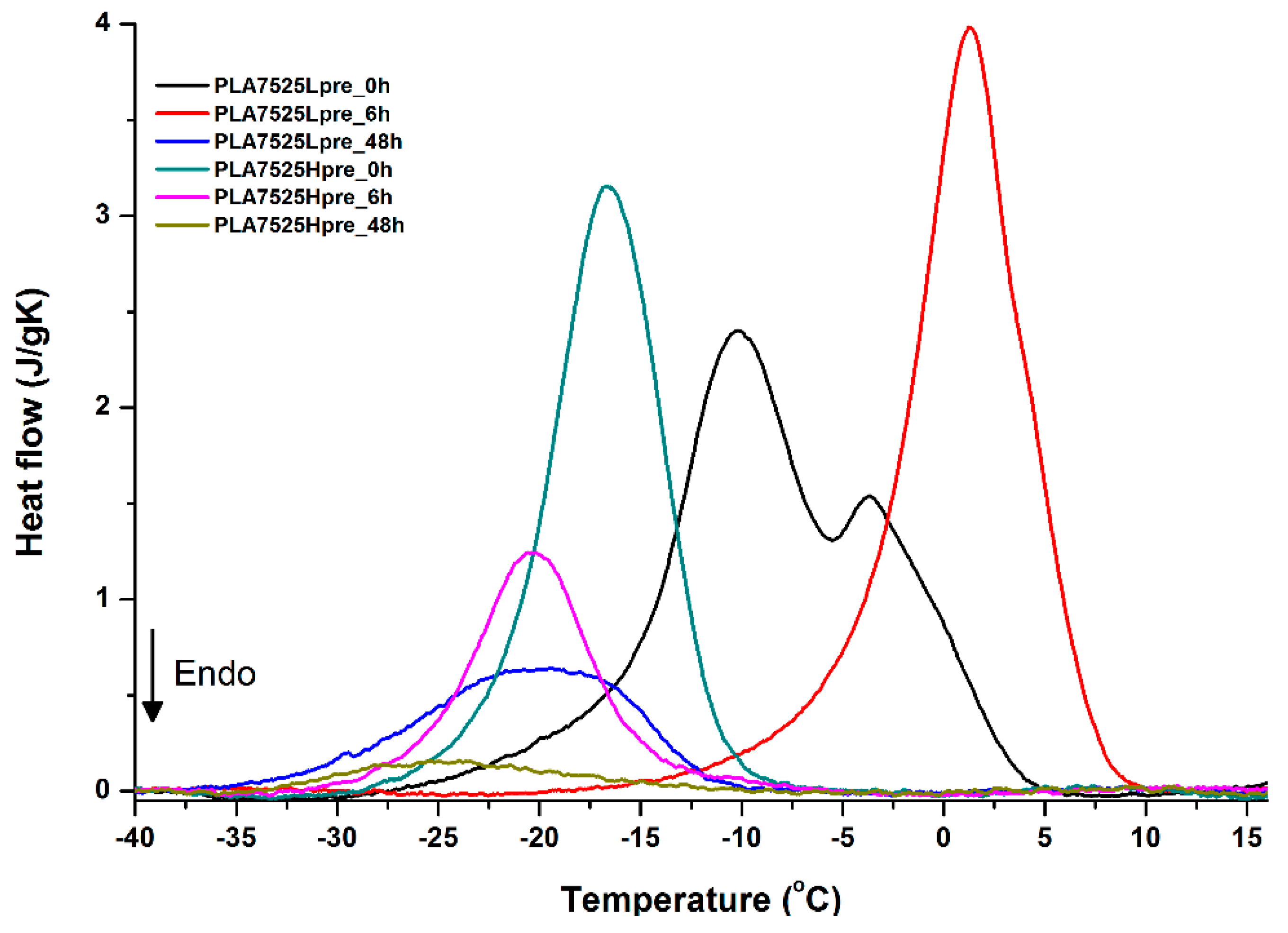
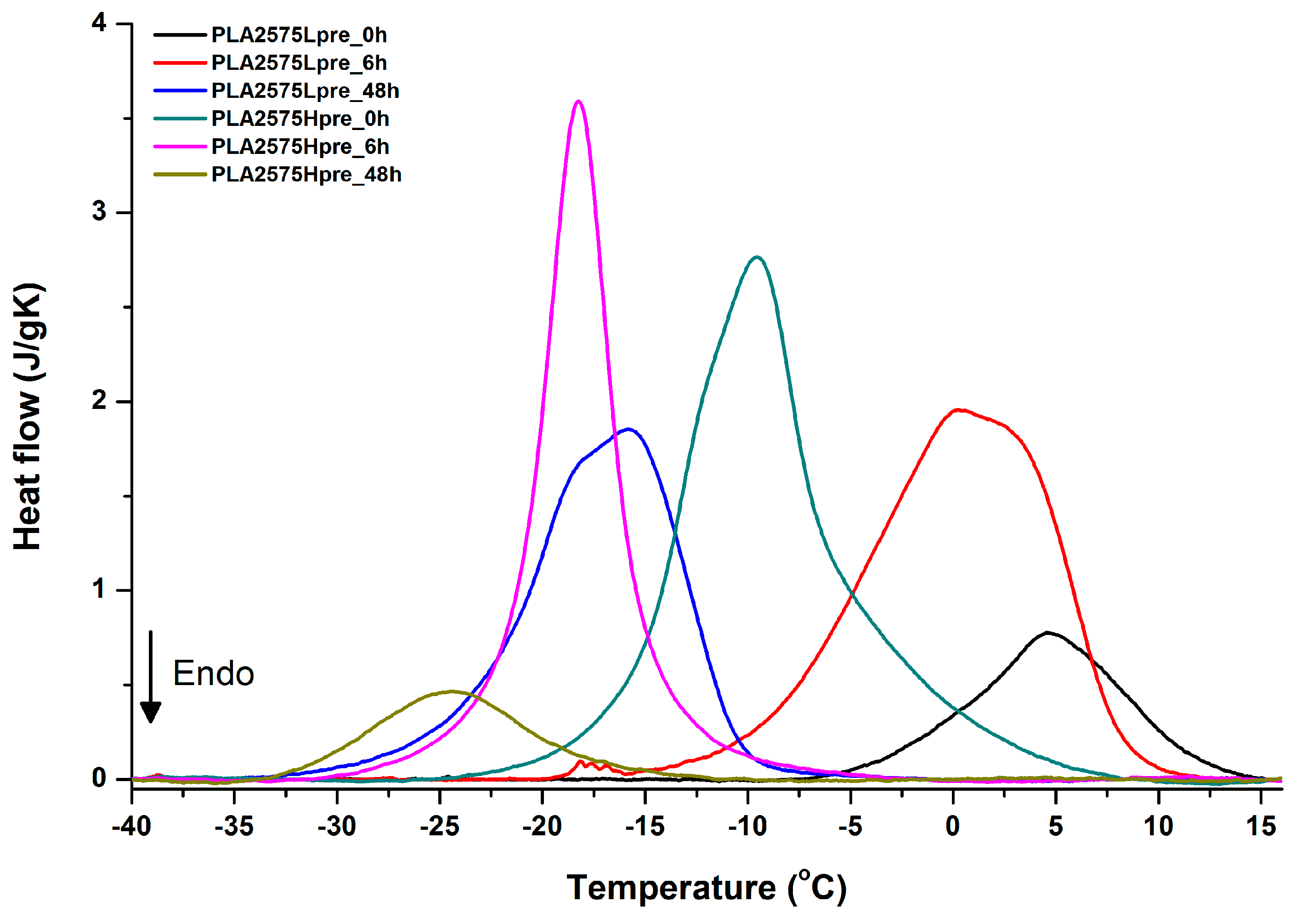
3.5. DSC Analysis
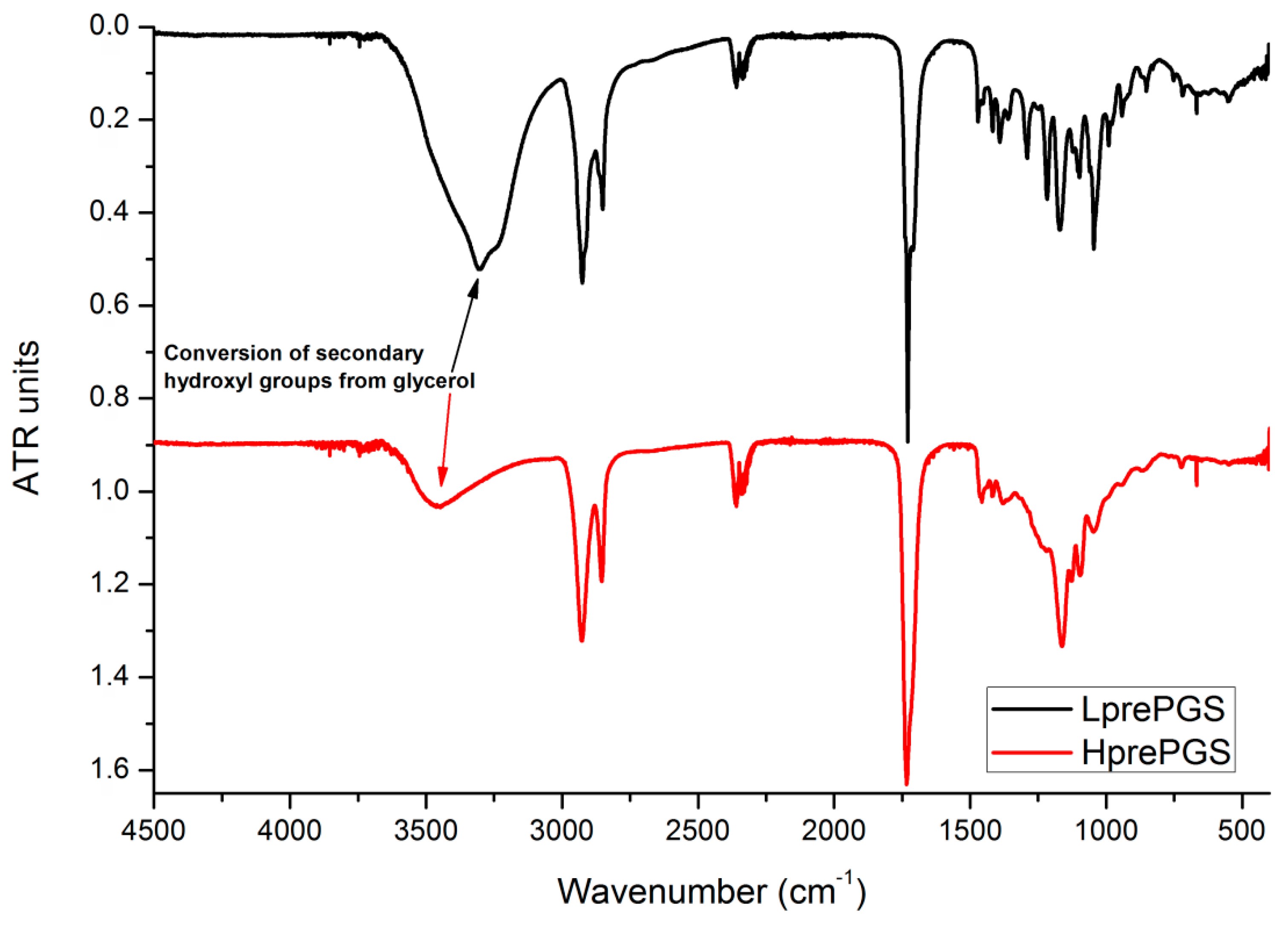
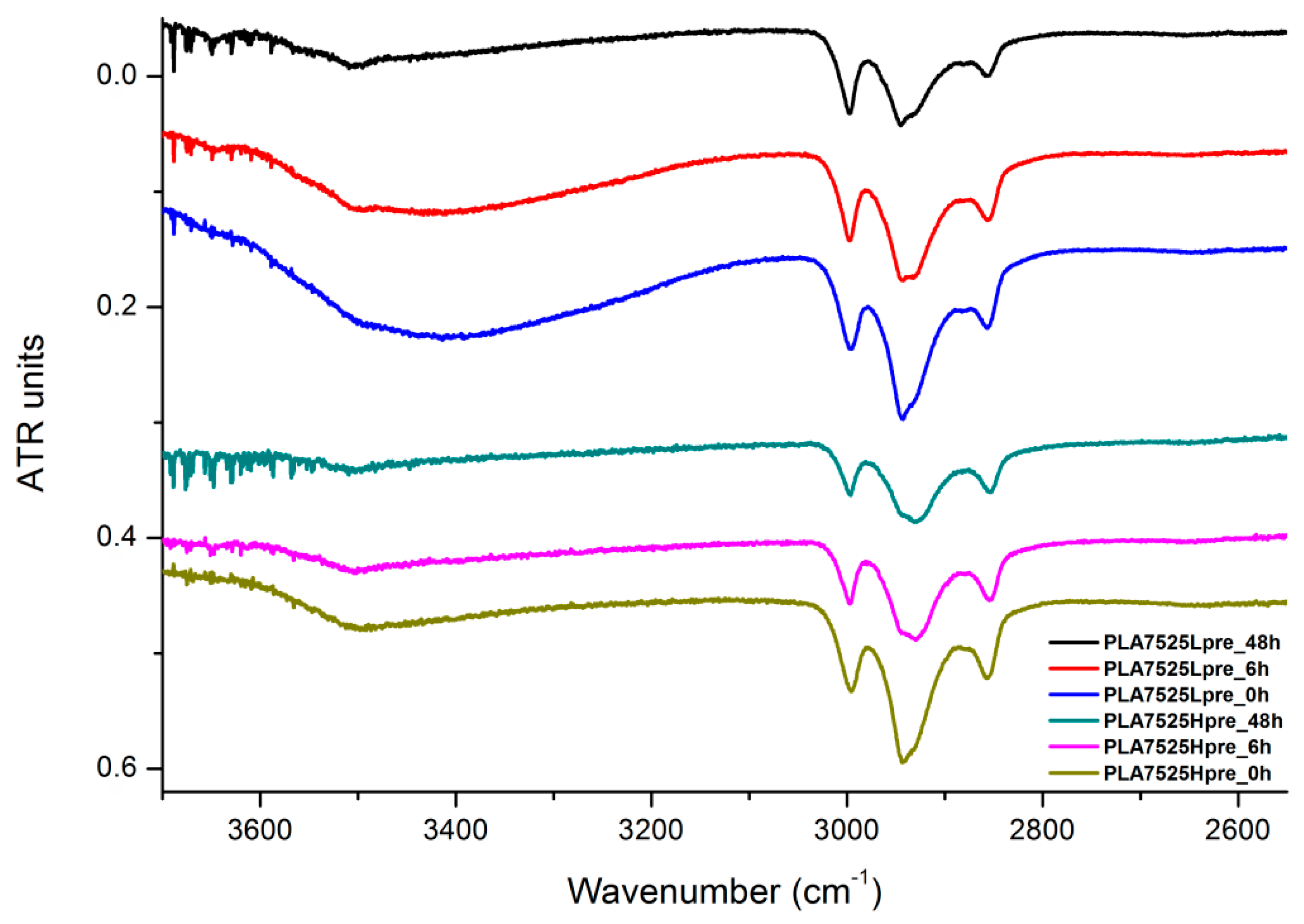
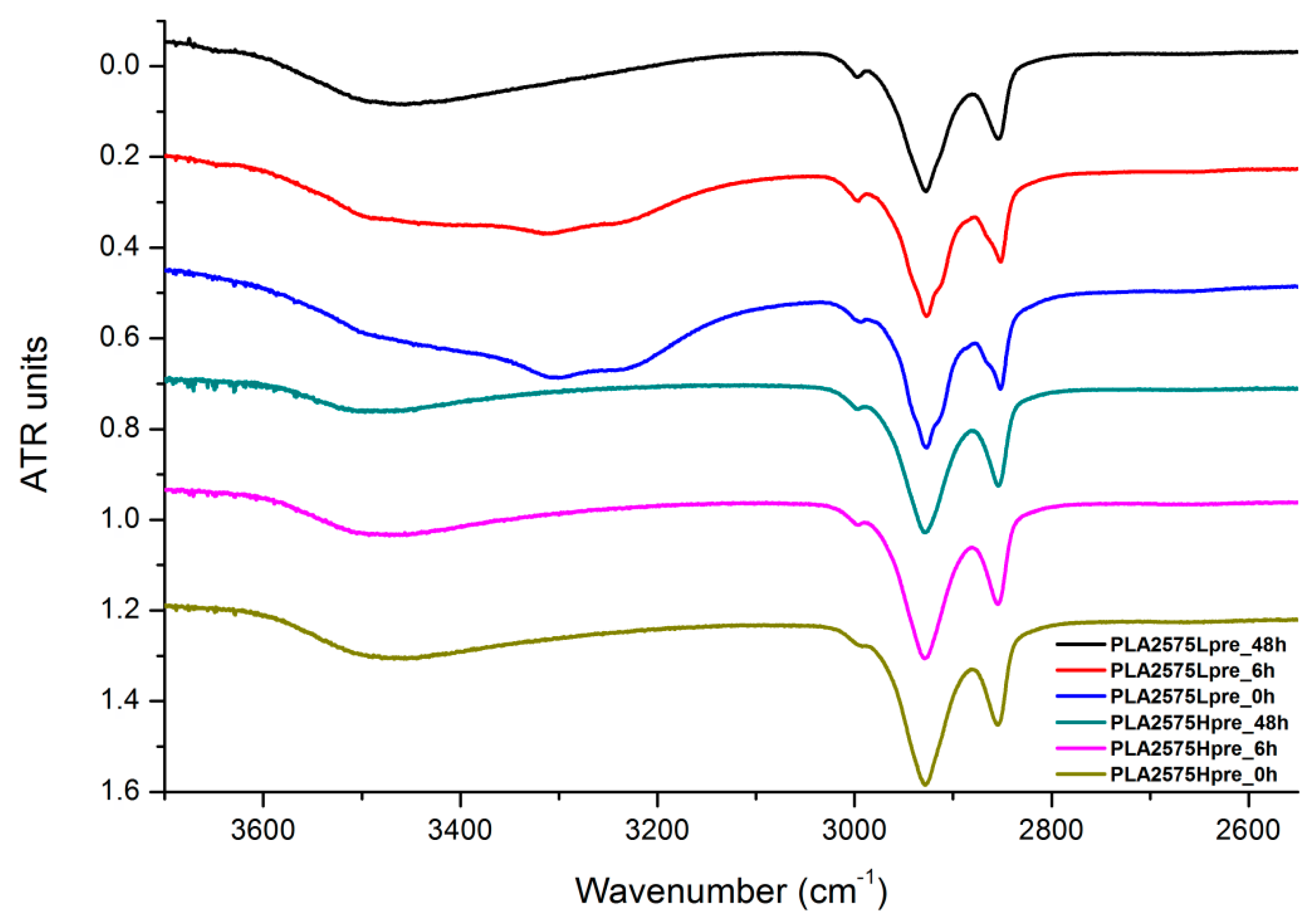
3.6. FTIR
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, Y.; Ameer, G.A.; Sheppard, B.J.; Langer, R. A tough biodegradable elastomer. Nat. Biotechnol. 2002, 20, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hong, A.T.; Naskar, N.; Chung, H.J. Criteria for Quick and Consistent Synthesis of Poly(glycerol sebacate) for Tailored Mechanical Properties. Biomacromolecules 2015, 16, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Conejero-Garcia, A.; Rivero Gimeno, H.; Moreno Saez, Y.; Vilarino-Feltrer, G.; Ortuno-Lizaran, I.; Valles-Lluch, A. Correlating synthesis parameters with physicochemical properties of poly(glycerol sebacate). Eur. Polym. J. 2017, 87, 406–419. [Google Scholar] [CrossRef]
- Wang, Y.; Kim, Y.M.; Langer, R. In vivo degradation characteristics of poly(glycerol sebacate). J. Biomed. Mater. Res. A 2003, 66, 192–197. [Google Scholar] [CrossRef]
- Sundback, C.A.; Shyu, J.Y.; Wang, Y.; Faquin, W.C.; Langer, R.S.; Vacanti, J.P.; Hadlock, T.A. Biocompatibility analysis of poly(glycerol sebacate) as a nerve guide material. Biomaterials 2005, 26, 5454–5464. [Google Scholar] [CrossRef]
- Chen, Q.Z.; Bismarck, A.; Hansen, U.; Junaid, S.; Tran, M.Q.; Harding, S.E.; Ali, N.N.; Boccaccini, A.R. Characterization of a soft elastomer poly(glycerol sebacate) designed to match the mechanical properties of myocardial tissue. Biomaterials 2008, 29, 47–57. [Google Scholar] [CrossRef]
- Ravichandran, R.; Venugopal, J.R.; Sundarrajan, S.; Mukherjee, S.; Ramakrishna, S. Cardiogenic differentiation of mesenchymal stem cells on elastomeric poly (glycerol sebacate)/collagen core/shell fibers. World. J. Cardiol. 2013, 26, 28–41. [Google Scholar] [CrossRef]
- Patel, A.; Gaharwar, A.K.; Iviglia, G.; Zhang, H.; Mukundan, S.; Mihaila, S.M.; Demarchi, D.; Khademhosseini, A. Highly elastomeric poly(glycerol sebacate)-co-poly(ethylene glycol) amphiphilic block copolymers. Biomaterials 2013, 34, 3970–3983. [Google Scholar] [CrossRef]
- Rai, R.; Tallawi, M.; Grigore, A.; Boccaccini, A.R. Synthesis, properties and biomedical applications of poly (glycerol sebacate) (PGS): A review. Prog. Polym. Sci. 2012, 37, 1051–1078. [Google Scholar] [CrossRef]
- Gaharwar, A.K.; Nikkhah, M.; Sant, S.; Khademhosseini, A. Anisotropic poly (glycerol sebacate)-poly (ε-caprolactone) electrospun fibers promote endothelial cell guidance. Biofabrication 2014, 17, 7. [Google Scholar]
- Yi, F.; LaVan, D.A. Poly(glycerol sebacate) Nanofiber Scaffolds by Core/Shell Electrospinning. Macromol. Biosci. 2008, 9, 803–806. [Google Scholar] [CrossRef] [PubMed]
- Sant, S.; Hwang, C.; Lee, S.H.; Khademhosseini, A. Hybrid PGS–PCL microfibrous scaffolds with improved mechanical and biological properties. J. Tissue Eng. Regen. Med. 2011, 5, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Salehi, S.; Bahners, T.; Gutmann, J.S.; Gao, S.L.; Mader, E.; Fuchsluger, T.A. Characterization of structural, mechanical and nano-mechanical properties of electrospun PGS/ PCL fibers. RSC Adv. 2014, 4, 16951–16957. [Google Scholar] [CrossRef]
- Salehi, S.; Fathi, M.; Javanmard, S.H.; Bahners, T.; Gutmann, J.S.; Ergun, S.; Steuhl, K.P.; Fuchsluger, T.A. Generation of PGS/PCL Blend Nanofibrous Scaffolds Mimicking Corneal Stroma Structure. Macromol. Mater. Eng. 2014, 299, 455–469. [Google Scholar] [CrossRef]
- Masoumi, N.; Annabi, N.; Assmann, A.; Larson, B.L.; Hjortnaes, J.; Alemdar, N.; Kharaziha, M.; Manning, K.B.; Mayer, J.E., Jr.; Khademhosseini, A. Tri-layered elastomeric scaffolds for engineering heart valve leaflets. Biomaterials 2014, 35, 7774–7785. [Google Scholar] [CrossRef]
- Rai, R.; Tallawi, M.; Frati, C.; Falco, A.; Gervasi, A.; Quaini, F.; Roether, J.A.; Hochburger, T.; Schubert, D.W.; Seik, L.; et al. Bioactive Electrospun Fibers of Poly(glycerol sebacate) and Poly(ε-caprolactone) for Cardiac Patch Application. Adv. Healthc. Mater. 2015, 4, 2012–2025. [Google Scholar] [CrossRef]
- Tallawi, M.; Dippold, D.; Rai, R.; D’Atri, D.; Roether, J.A.; Schubert, D.W.; Rosellini, E.; Engel, F.B.; Boccaccini, A.R. Novel PGS/PCL electrospun fiber mats with patterned topographical features for cardiac patch applications. Mater. Sci. Eng. C 2016, 69, 569–576. [Google Scholar] [CrossRef]
- Jeffries, E.M.; Allen, R.A.; Gao, J.; Pesce, M.; Wang, Y. Highly elastic and suturable electrospun poly(glycerol sebacate) fibrous scaffolds. Acta Biomater. 2015, 18, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, R.; Best, C.A.; Allen, R.A.; Stowell, C.E.; Onwuka, E.; Zhuang, J.J.; Lee, Y.U.; Yi, T.; Bersi, M.R.; Shinoka, T.; et al. Long-Term Functional Efficacy of a Novel Electrospun Poly(Glycerol Sebacate) Based Arterial Graft in Mice. Ann. Biomed. Eng. 2016, 44, 2402–2416. [Google Scholar] [CrossRef]
- Hu, J.; Kai, D.; Ye, H.; Tian, L.; Ding, X.; Ramakrishna, S.; Loh, X.J. Electrospinning of poly(glycerol sebacate)-based nanofibers for nerve tissue engineering. Mater. Sci. Eng. C 2017, 70, 1089–1094. [Google Scholar] [CrossRef]
- Liverani, L.; Piegat, A.; Niemczyk, A.; El Fray, M.; Boccaccini, A.R. Electrospun fibers of poly(butylene succinate–co–dilinoleic succinate) and its blend with poly(glycerol sebacate) for soft tissue engineering applications. Eur. Polym. J. 2016, 81, 295–306. [Google Scholar] [CrossRef]
- Nadim, A.; Khorasani, S.N.; Kharaziha, M.; Davoodi, S.M. Design and characterization of dexamethasone-loaded poly (glycerol sebacate)-poly caprolactone/gelatin scaffold by coaxial electro spinning for soft tissue engineering. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 1, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Salehi, S.; Czugala, M.; Stafiej, P.; Fathi, M.; Bahners, T.; Gutmann, J.S.; Singer, B.B.; Fuchsluger, T.A. Poly (glycerol sebacate)-poly (ε-caprolactone) blend nanofibrous scaffold as intrinsic bio- and immunocompatible system for corneal repair. Acta Biomater. 2017, 50, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Vogt, L.; Liverani, L.; Roether, J.A.; Boccaccini, A.R. Electrospun Zein Fibers Incorporating Poly(glycerol sebacate) for Soft Tissue Engineering. Nanomaterials 2018, 8, 150. [Google Scholar] [CrossRef]
- Saudi, A.; Rafienia, M.; Kharazi, A.Z.; Salehi, H.; Zarrabi, A.; Karevan, M. Design and fabrication of poly (glycerol sebacate)-based fibers for neural tissue engineering: Synthesis, electrospinning, and characterization. Polym. Adv. Technol. 2019, 30, 1427–1440. [Google Scholar] [CrossRef]
- Yan, Y.; Sencadas, V.; Zhang, J.; Wei, D.; Jiang, Z. Superomniphilic Poly(glycerol sebacate)–Poly(l-lactic acid) Electrospun Membranes for Oil Spill Remediation. Adv. Mater. Interfaces 2017, 4, 1700484. [Google Scholar] [CrossRef]
- You, Z.R.; Hu, M.H.; Tuan-Mu, H.Y.; Hu, J.J. Fabrication of poly(glycerol sebacate) fibrous membranes by coaxial electrospinning: Influence of shell and core solutions. J. Mech. Behav. Biomed. Mater. 2016, 63, 220–231. [Google Scholar] [CrossRef]
- Yan, Y.; Sencadas, V.; Jin, T.; Huang, X.-F.; Chen, J.; Wei, D.; Jiang, Z. Tailoring the wettability and mechanical properties of electrospun poly(l-lactic acid)-poly(glycerol sebacate) core-shell membranes for biomedical applications. J. Colloid Interface Sci. 2017, 508, 87–94. [Google Scholar] [CrossRef]
- Jiang, L.; Jiang, Y.; Stiadle, J.; Wang, X.; Wang, L.; Li, Q.; Shen, C.; Thibeault, S.L.; Turng, L.-S. Electrospun nanofibrous thermoplastic polyurethane/poly(glycerol sebacate) hybrid scaffolds for vocal fold tissue engineering applications. Mater. Sci. Eng. C 2019, 94, 740–749. [Google Scholar] [CrossRef]
- Wu, H.-J.; Hu, M.-H.; Tuan-Mu, H.-Y.; Hu, J.-J. Preparation of aligned poly(glycerol sebacate) fibrous membranes for anisotropic tissue engineering. Mater. Sci. Eng. C 2019, 100, 30–37. [Google Scholar] [CrossRef]
- O’Brien, D.; Hankins, A.; Golestaneh, N.; Paranjape, M. Highly aligned and geometrically structured poly(glycerol sebacate)-polyethylene oxide composite fiber matrices towards bioscaffolding applications. Biomed. Microdevices 2019, 21, 53. [Google Scholar] [CrossRef]
- Turner, J.F.; Riga, A.; O’Connor, A.; Zhang, J.; Collins, J. Characterization of drawn and undrawn poly-L-lactide films by differential scanning calorimetry. J. Therm. Anal. Calorim. 2004, 75, 257–268. [Google Scholar] [CrossRef]
- Stoclet, G.; Seguela, R.; Lefebvre, J.-M.; Rochas, C. New Insights on the Strain-Induced Mesophase of Poly(D,L-lactide): In Situ WAXS and DSC Study of the Thermo-Mechanical Stability. Macromolecules 2010, 43, 7228–7237. [Google Scholar] [CrossRef]
- Misztal-Faraj, B.; Pęcherski, R.B.; Denis, P.; Jarecki, L. Modeling of oriented crystallization kinetics of polymers in the entire range of uniaxial molecular orientation. Polymer 2019, 173, 141–157. [Google Scholar] [CrossRef]
- Russo, V.; Tammaro, L.; Di Marcantonio, L.; Sorrentino, A.; Ancora, M.; Valbonetti, L.; Turriani, M.; Martelli, A.; Cammà, C.; Barboni, B. Amniotic epithelial stem cell biocompatibility for electrospun poly(lactide-co-glycolide), poly(ε-caprolactone), poly(lactic acid) scaffolds. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 69, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Celli, A.; Scandola, M. Thermal properties and physical ageing of poly (l-lactic acid). Polymer 1992, 33, 2699–2703. [Google Scholar] [CrossRef]
- Kairong, L.; Daping, Q.; Zejian, L. Effects of physical aging on glass transition behavior of poly(dl-lactide). Eur. Polym. J. 2002, 38, 157–162. [Google Scholar]
- Mano, J.F.; Gomez Ribelles, J.L.; Alves, N.M.; Salmeron Sanchez, M. Glass transition dynamics and structural relaxation of PLLA studied by DSC: Influence of crystallinity. Polymer 2005, 46, 8258–8265. [Google Scholar] [CrossRef]
- Pan, P.; Zhu, B.; Inoue, Y. Enthalpy Relaxation and Embrittlement of Poly(l-lactide) during Physical Aging. Macromolecules 2007, 40, 9664–9671. [Google Scholar] [CrossRef]
- Kwon, M.; Lee, S.C.; Jeong, Y.G. Influences of physical aging on enthalpy relaxation behavior, gas permeability, and dynamic mechanical property of polylactide films with various D-isomer contents. Macromol. Res. 2010, 18, 346–351. [Google Scholar] [CrossRef]
- Rai, R.; Tallawi, M.; Barbani, N.; Frati, C.; Madeddu, D.; Cavalli, S.; Graiani, G.; Quaini, F.; Roether, J.A.; Schubert, D.W.; et al. Biomimetic poly(glycerol sebacate) (PGS) membranes for cardiac patch application. Mater. Sci. Eng. C Mater. Biol. Appl. 2013, 33, 3677–3687. [Google Scholar] [CrossRef] [PubMed]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Elastomer (E) or Thermoplastic (T) | Young Modulus (Or Stiffness) | Tensile Strength | Degradation (Month) |
| PGA | T | 7–10 GPa | 70 MPa | 2–12 |
| PLLA or PDLLA | T | 1–4 GPa | 30–80 MPa | 2–12 |
| PHB | E | 2–3 GPa | 36 MPa | Degradable |
| PPD (also called PDS) | E | 0.6 GPa | 12 MPa | 6 |
| TMC | E | 6 MPa | 12 MPa | Degradable |
| TMC-PDLLA (50:50) | E | 16 MPa | 10 MPa | Degradable |
| POC | T | 1-16 MPa | 6.7 MPa | Degradable |
| PGS | E | 0.04–1.2 MPa | 0.2–0.5 MPa | Degradable |
| Collagen fibre (Tendon/cartilage/ligament/bone) | E | 2–46 MPa | 1–7 MPa | Degradable |
| Collagen gel (calf skin) | E | 0.002–0.022 MPa | 1–9 kPa | Degradable |
| Myocardium of rat | E | 0.001–0.14 MPa | 30–70 kPa | NA |
| Myocardium of human | E | 0.02–0.5 MPa | 3–15 kPa | NA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Denis, P.; Wrzecionek, M.; Gadomska-Gajadhur, A.; Sajkiewicz, P. Poly(Glycerol Sebacate)–Poly(l-Lactide) Nonwovens. Towards Attractive Electrospun Material for Tissue Engineering. Polymers 2019, 11, 2113. https://doi.org/10.3390/polym11122113
Denis P, Wrzecionek M, Gadomska-Gajadhur A, Sajkiewicz P. Poly(Glycerol Sebacate)–Poly(l-Lactide) Nonwovens. Towards Attractive Electrospun Material for Tissue Engineering. Polymers. 2019; 11(12):2113. https://doi.org/10.3390/polym11122113
Chicago/Turabian StyleDenis, Piotr, Michał Wrzecionek, Agnieszka Gadomska-Gajadhur, and Paweł Sajkiewicz. 2019. "Poly(Glycerol Sebacate)–Poly(l-Lactide) Nonwovens. Towards Attractive Electrospun Material for Tissue Engineering" Polymers 11, no. 12: 2113. https://doi.org/10.3390/polym11122113
APA StyleDenis, P., Wrzecionek, M., Gadomska-Gajadhur, A., & Sajkiewicz, P. (2019). Poly(Glycerol Sebacate)–Poly(l-Lactide) Nonwovens. Towards Attractive Electrospun Material for Tissue Engineering. Polymers, 11(12), 2113. https://doi.org/10.3390/polym11122113