Crystalline Characteristics and Their Influence in the Mechanical Performance in Poly(ε-Caprolactone) / High Density Polyethylene Blends



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Blends and Films Preparation
2.3. Characterization of the Samples
2.3.1. Scanning Electron Microscopy
2.3.2. Contact Angle
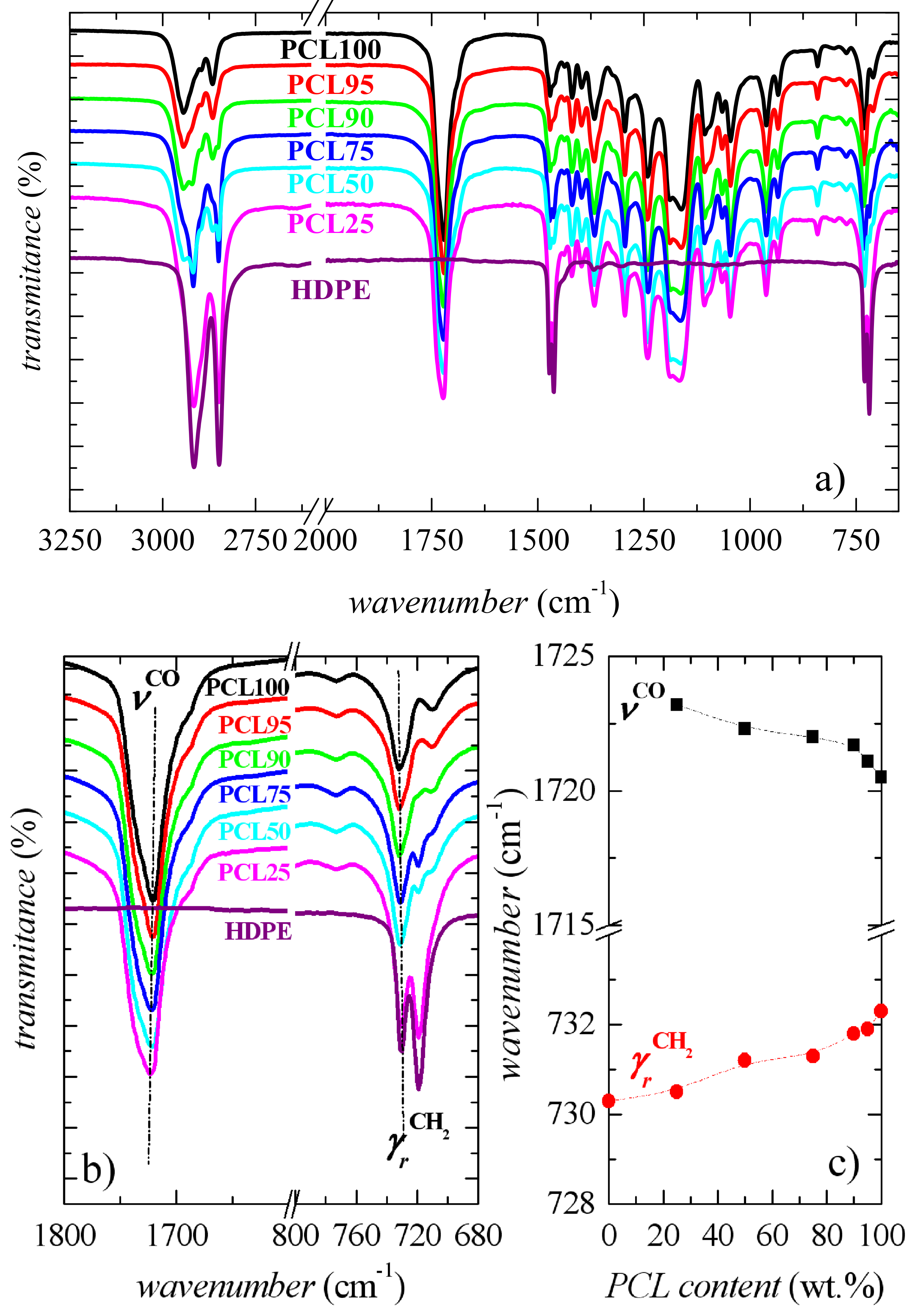
2.3.3. Fourier-Transform Infrared Spectroscopy
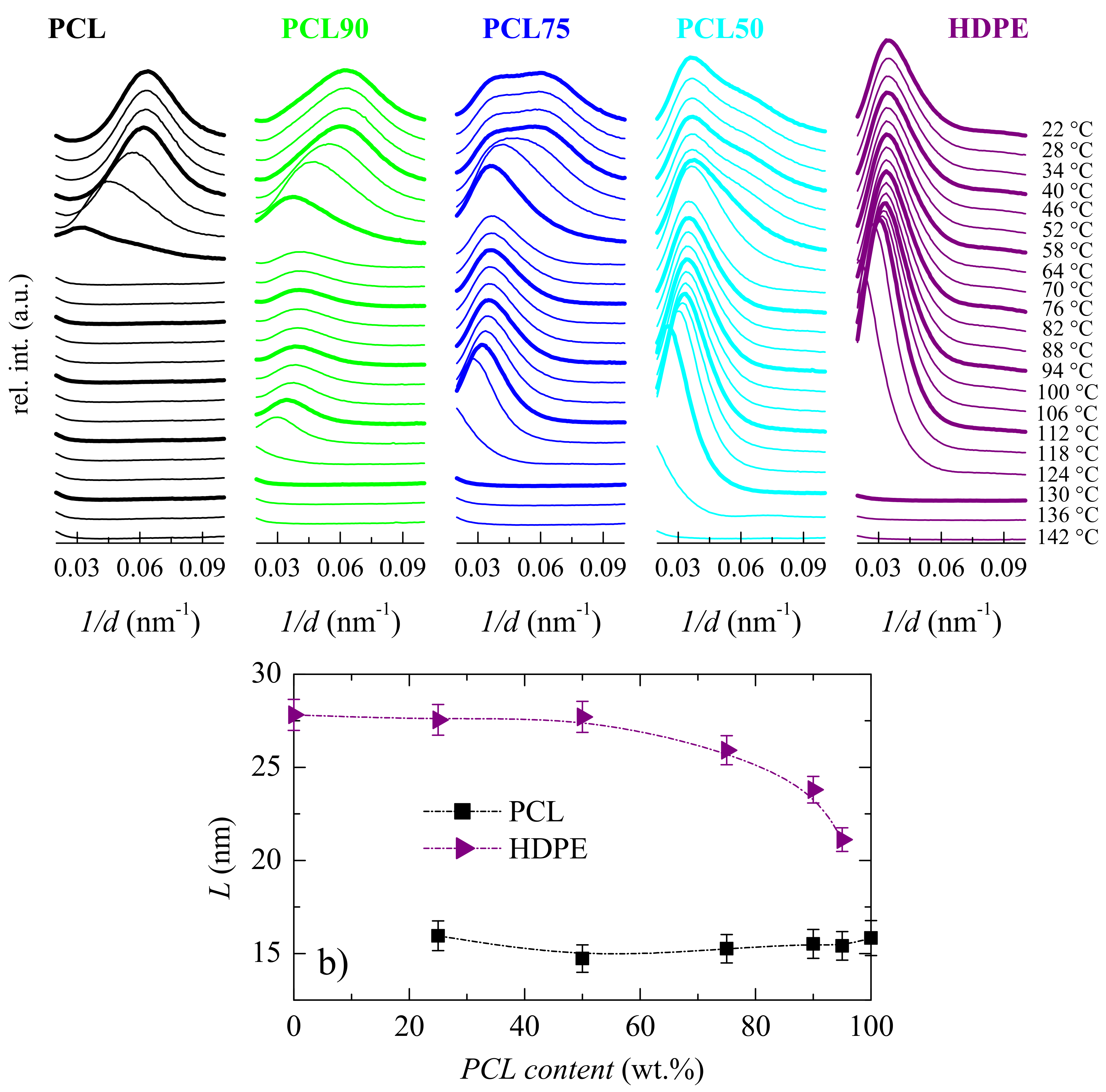
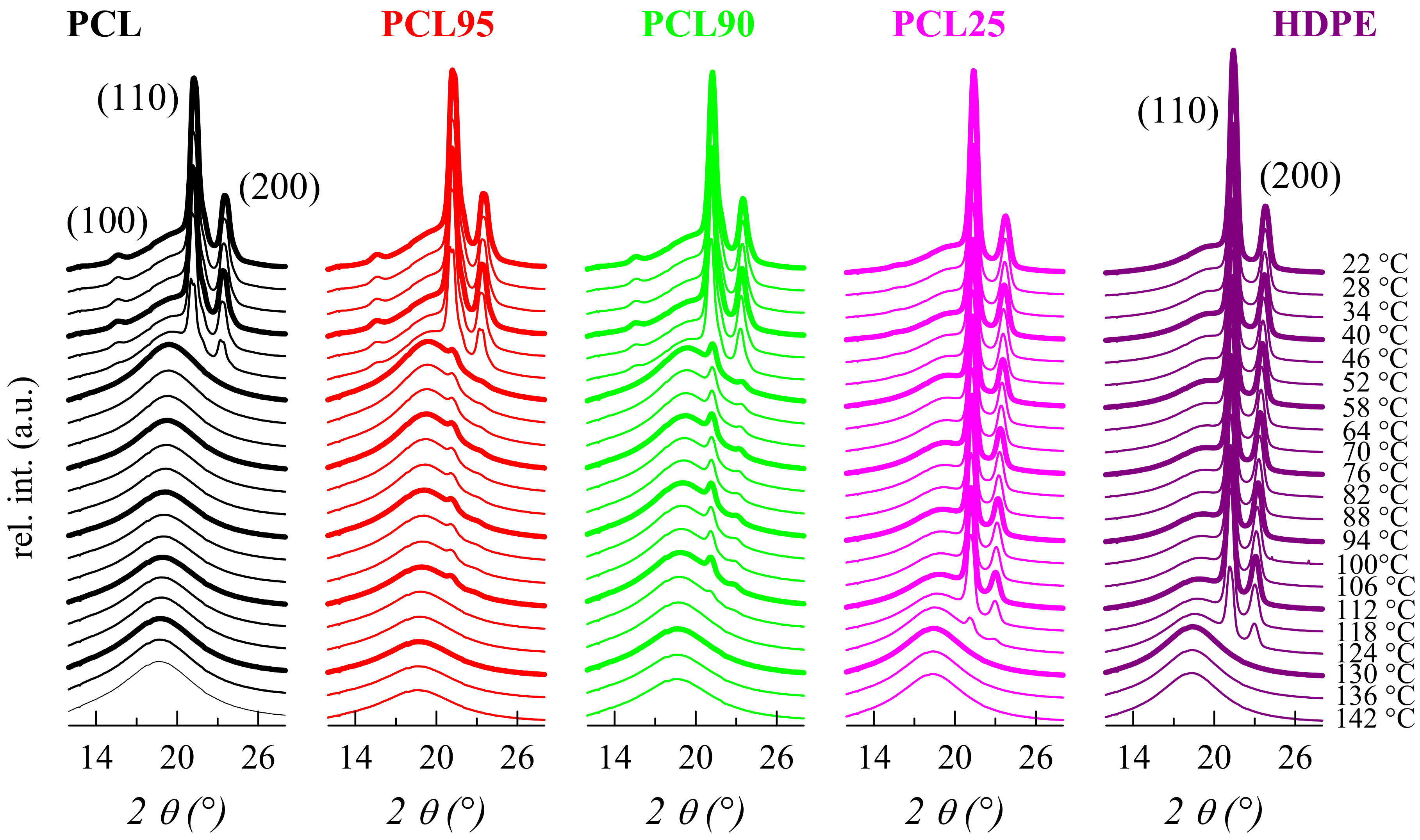
2.3.4. X-Ray Diffraction
2.3.5. Differential scanning calorimetry
2.3.6. Gas Transport Behavior
2.3.7. Stress–Strain Response
2.3.8. Dynamic Mechanical Thermal Analysis
2.4. Statistical Analysis
3. Results and Discussion
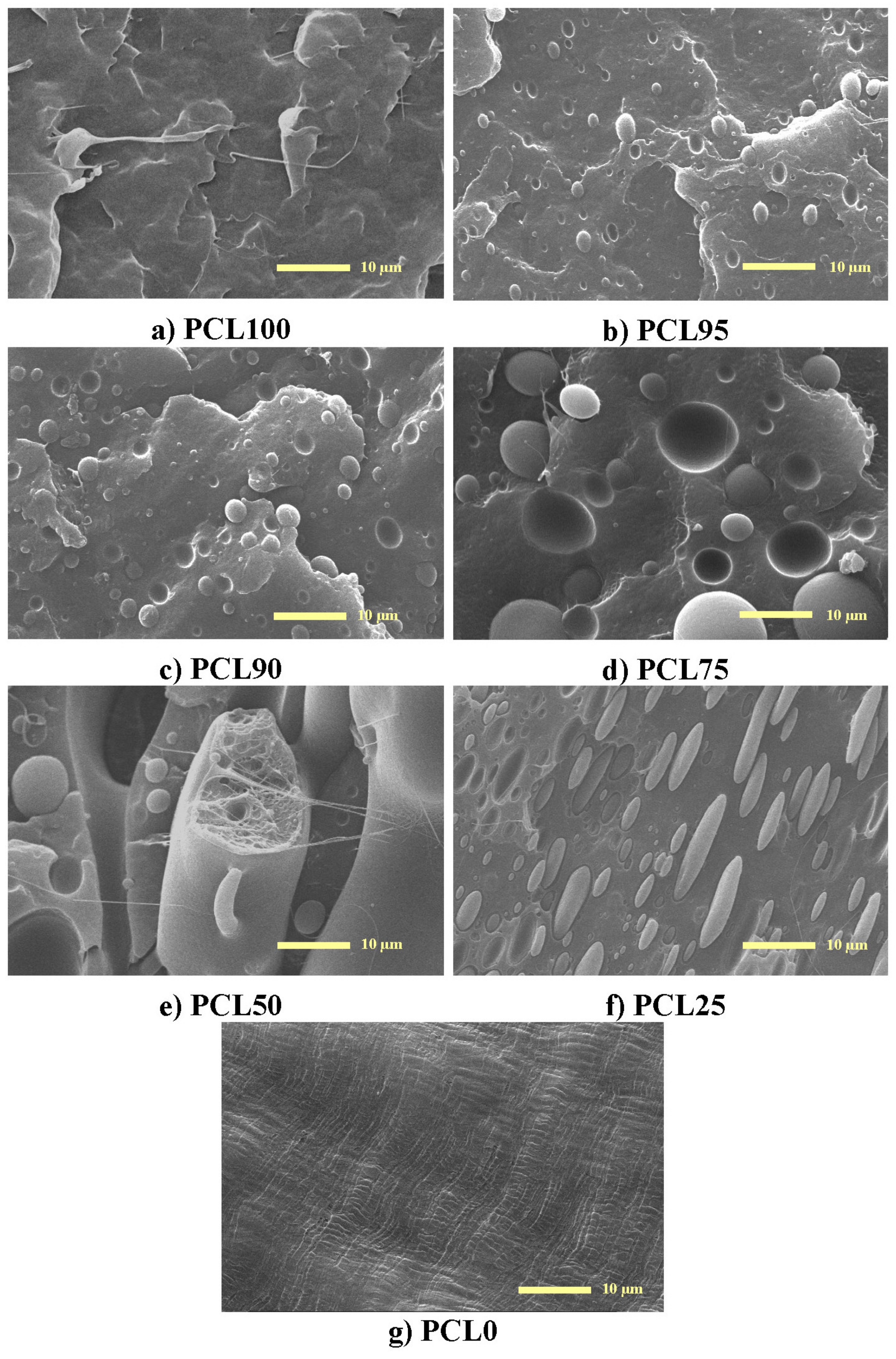
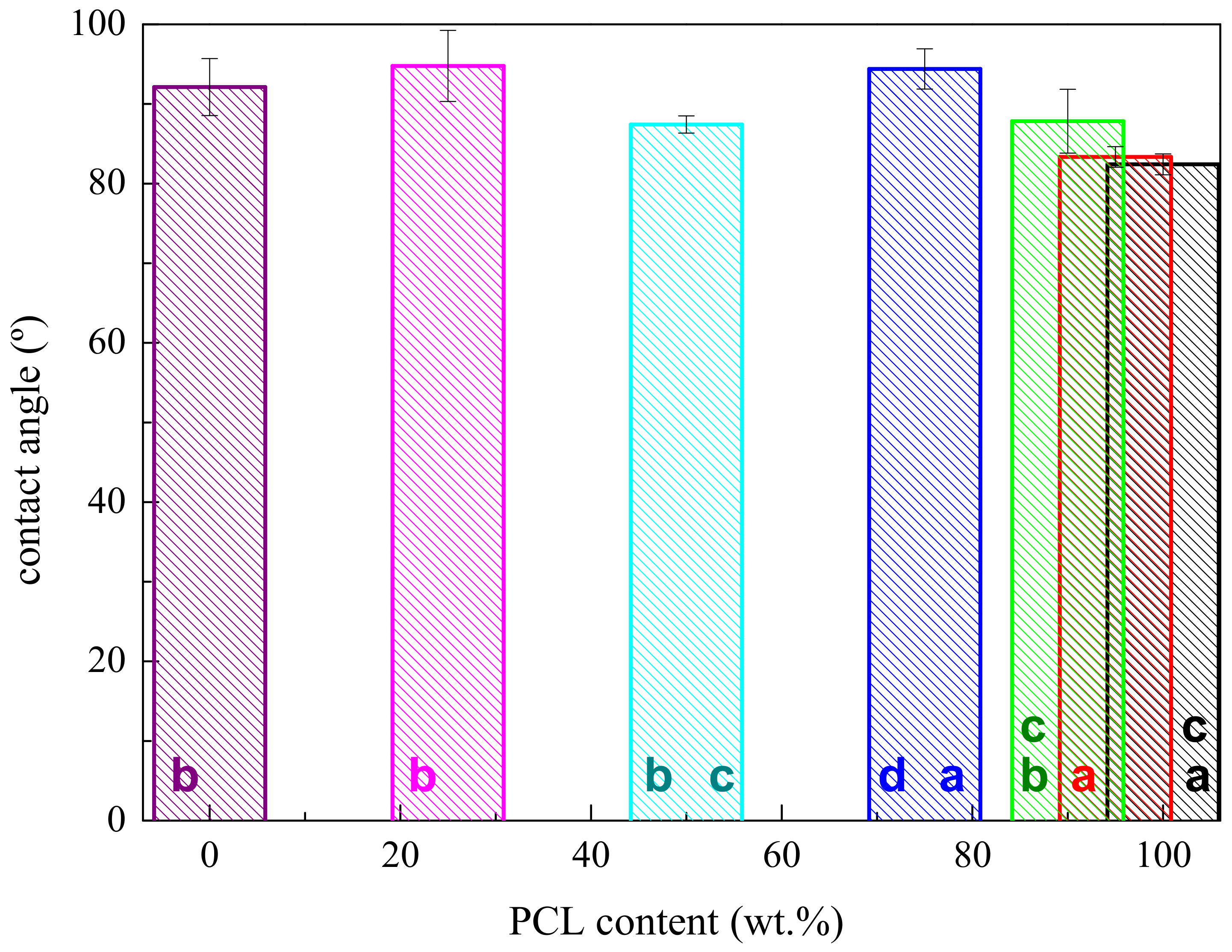
3.1. Characterization of Morphology and Interactions
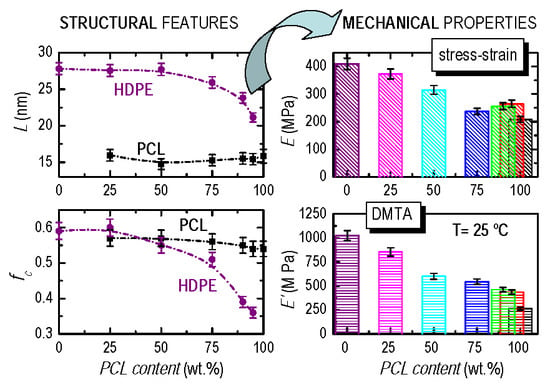
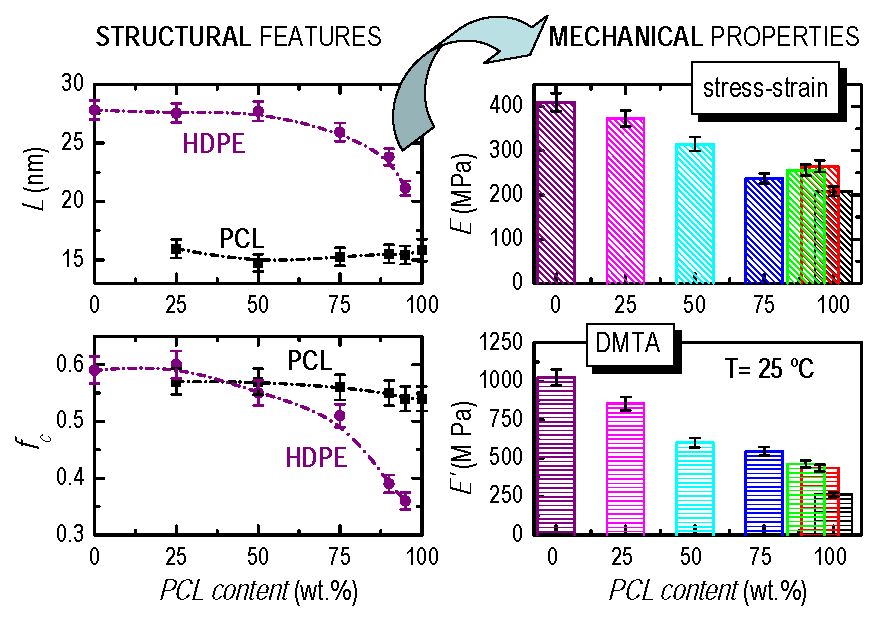
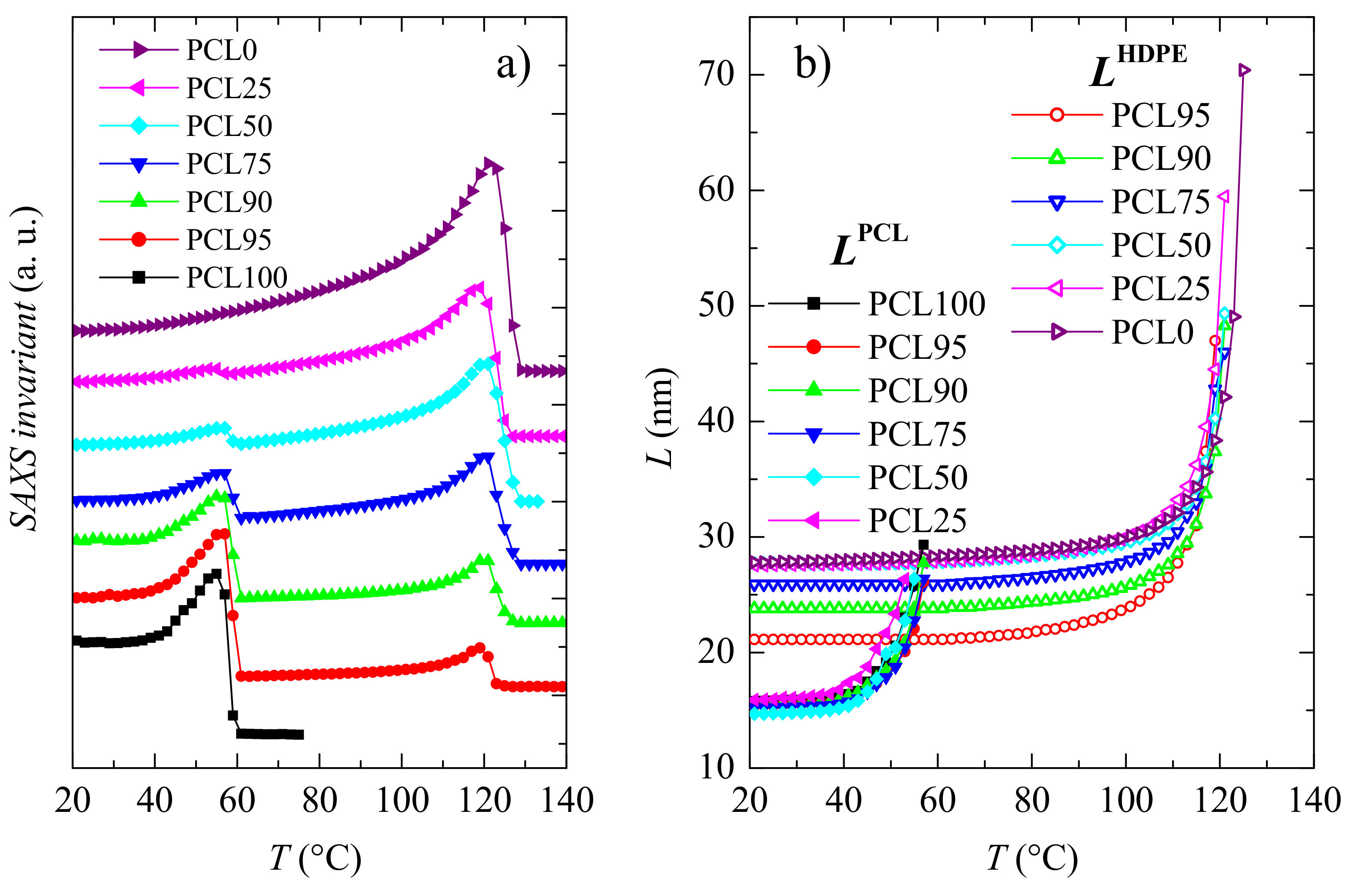
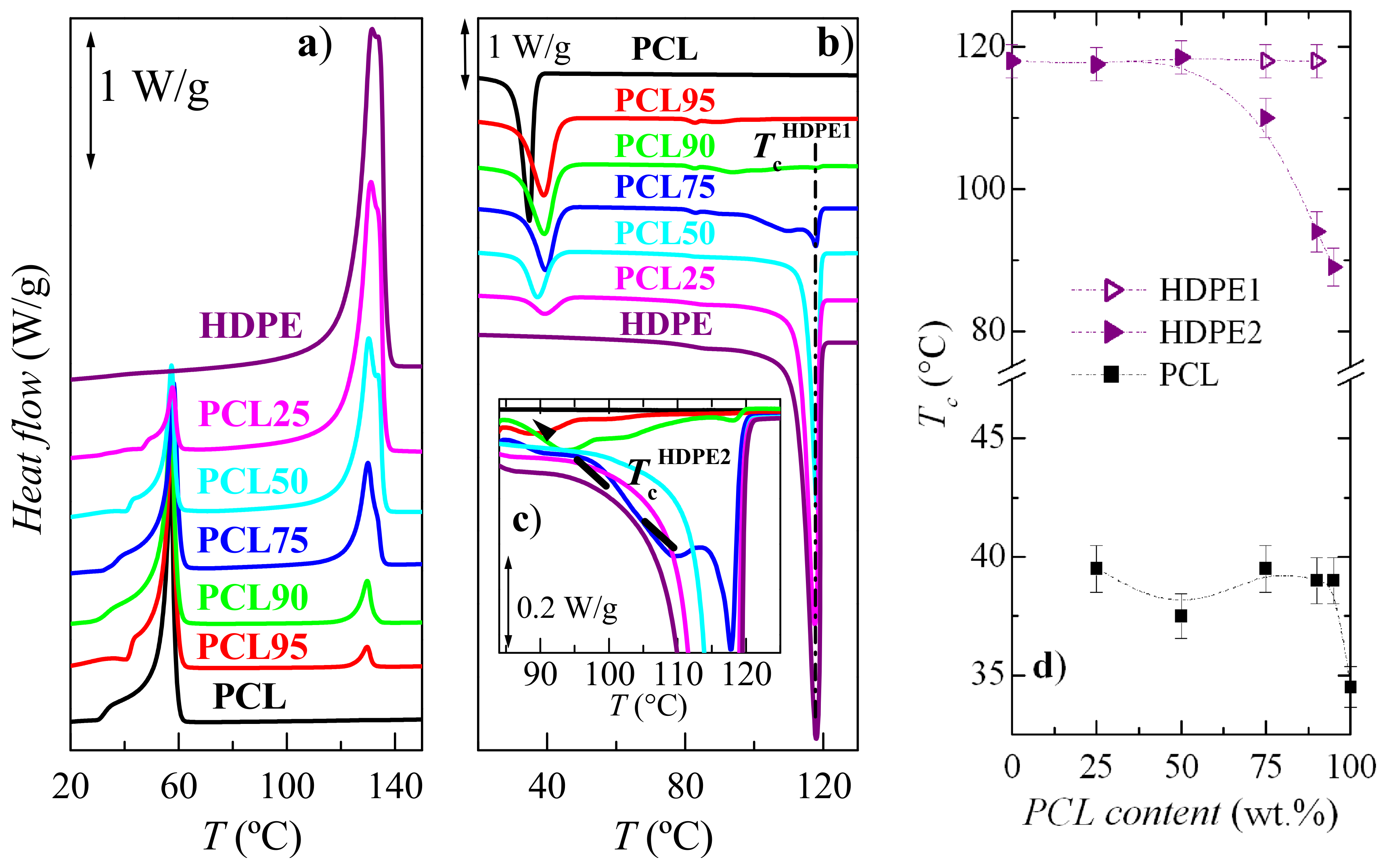
3.2. Crystalline Characteristics and Phase Transitions
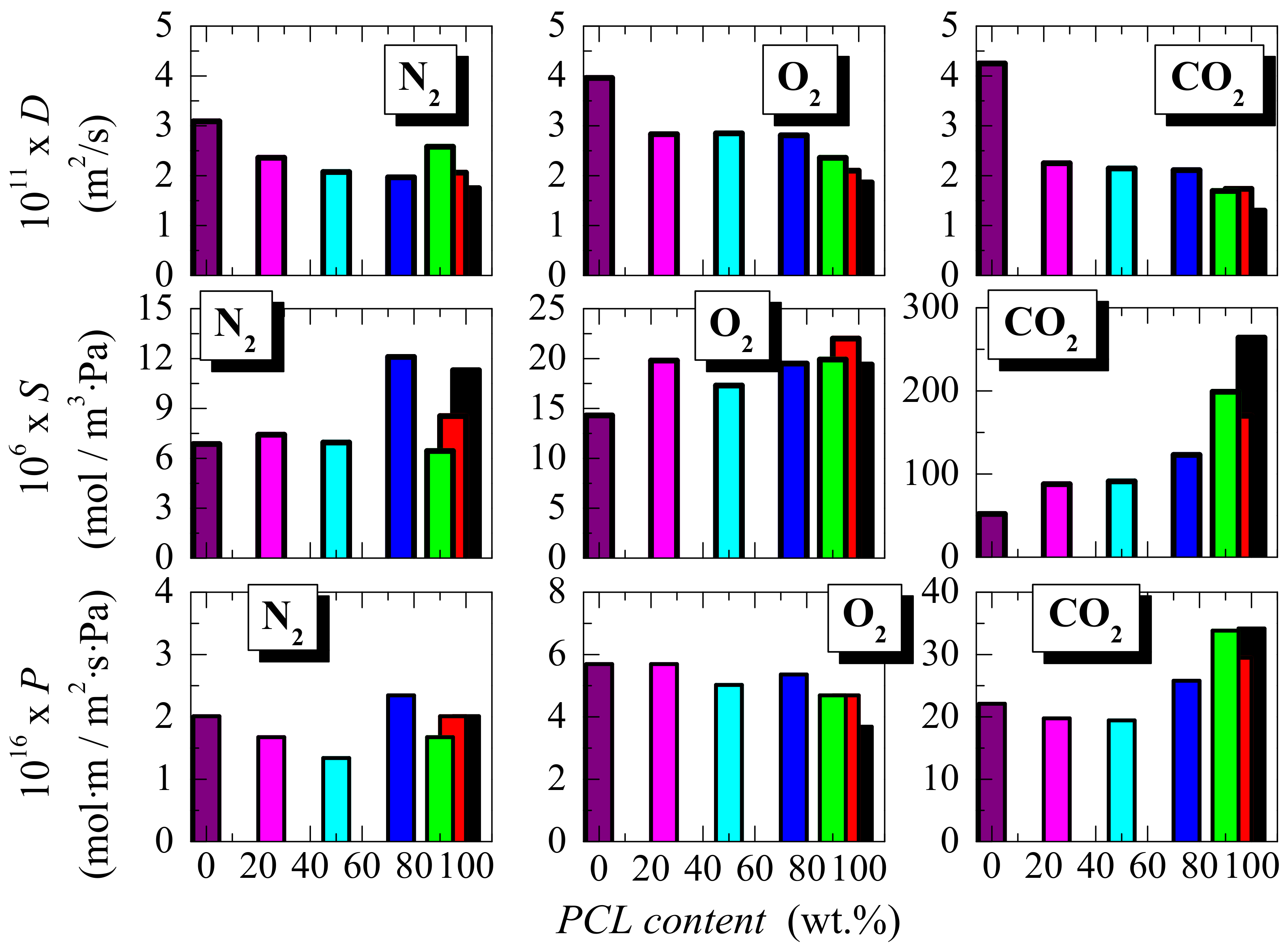
3.3. Transport Properties
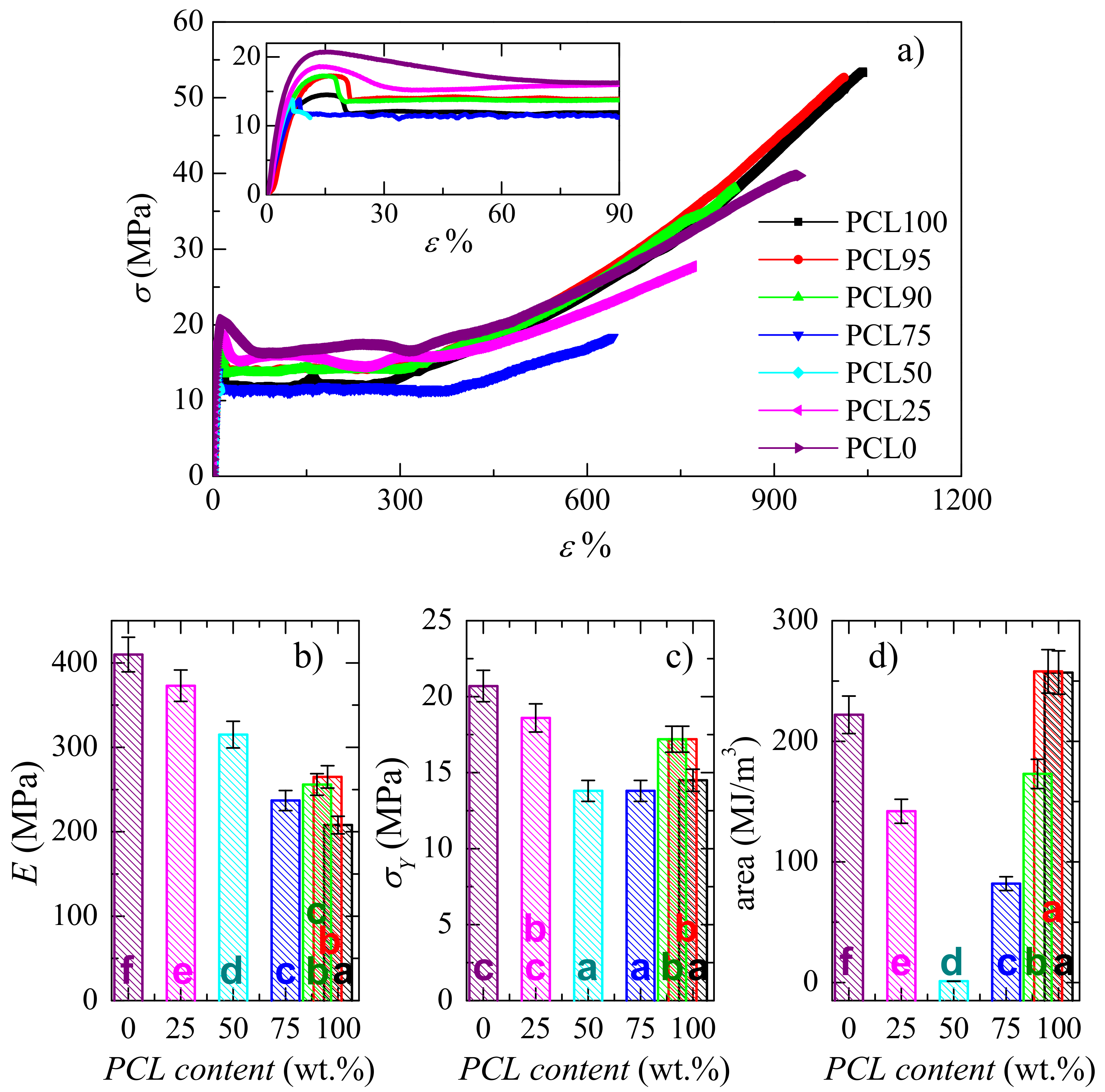
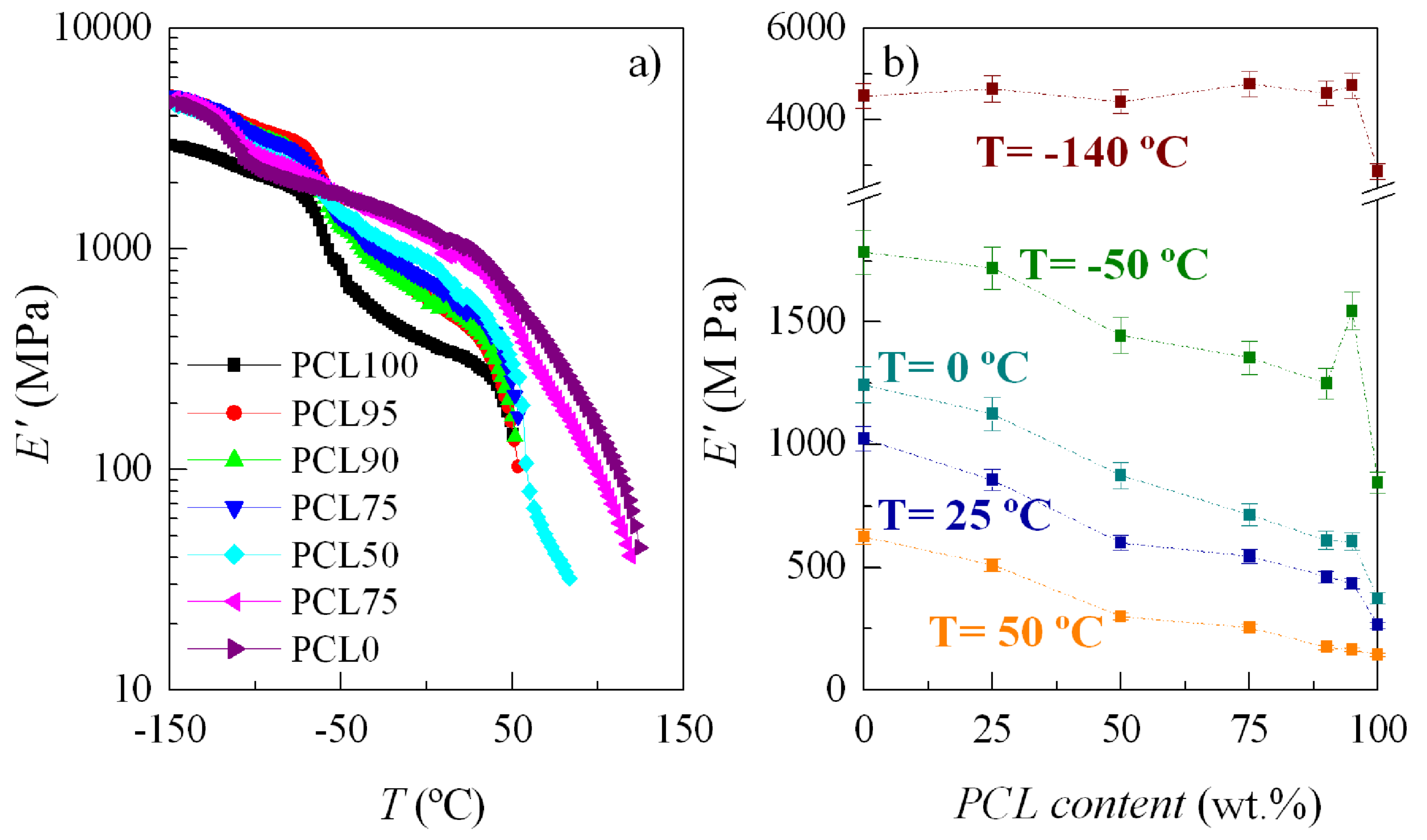
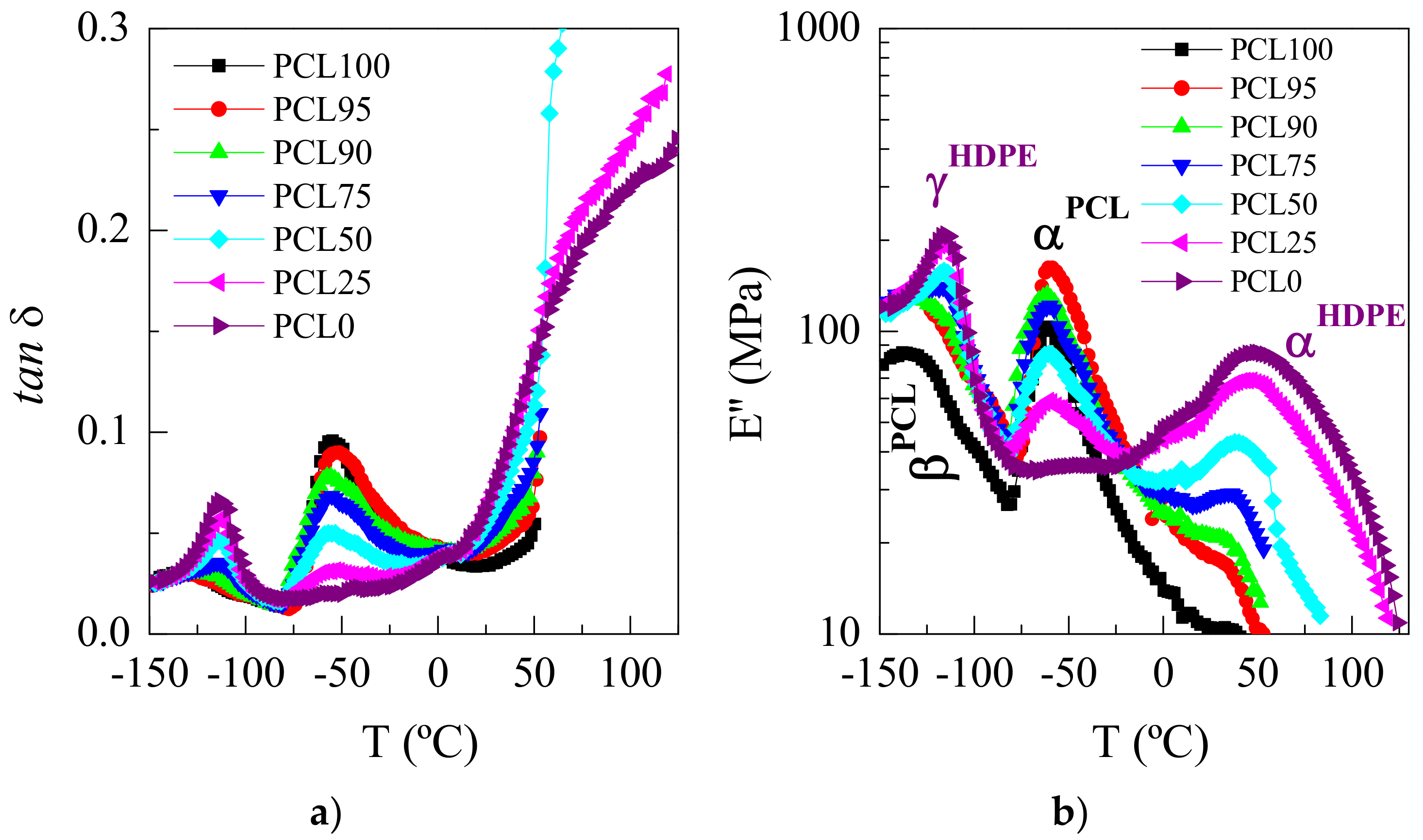
3.4. Mechanical and Viscoelastic Responses
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van Natta, F.J.; Hill, J.W.; Carothers, W.H. Polymerization and ring formation, ε-caprolactone and its polymers. J. Am. Chem. Soc. 1934, 56, 455–457. [Google Scholar] [CrossRef]
- Huang, S. Biodegradable polymers. In Encyclopedia of Polymer Science and Engineering; Mark, F., Bikales, N., Overberger, C., Menges, G., Kroshwitz, J., Eds.; John Wiley and Sons: New York, NY, USA, 1985; pp. 220–243. [Google Scholar]
- Kweona, H.Y.; Yoo, M.K.; Park, I.K.; Kim, T.H.; Lee, H.C.; Lee, H.-S.; Oh, J.-S.; Akaike, T.; Cho, C.-S. A novel degradable polycaprolactone networks for tissue engineering. Biomaterials 2003, 24, 801–808. [Google Scholar] [CrossRef]
- Zein, I.; Hutmacher, D.W.; Tan, K.C.; Teoh, S.H. Fused deposition modelling of novel scaffold architectures for tissue engineering applications. Biomaterials 2002, 23, 1169–1185. [Google Scholar] [CrossRef]
- Lee, K.H.; Kim, H.Y.; Khil, M.S.; Ra, Y.M.; Lee, D.R. Characterization of nano-structured poly(epsilon-caprolactone) nonwoven mats via electrospinning. Polymer 2003, 44, 1287–1294. [Google Scholar] [CrossRef]
- Huang, H.; Oizumi, S.; Kojima, N.; Niino, T.; Sakai, Y. Avidin–biotin binding-based cell seeding and perfusion culture of liver-derived cells in a porous scaffold with a three-dimensional interconnected flow-channel network. Biomaterials 2007, 28, 3815–3823. [Google Scholar] [CrossRef] [PubMed]
- Marrazzo, C.; Di Maio, E.; Iannace, S. Conventional and nanometric nucleating agents in poly(epsilon-caprolactone) foaming: Crystals vs. bubbles nucleation. Polym. Eng. Sci. 2008, 48, 336–344. [Google Scholar] [CrossRef]
- Luciani, A.; Coccoli, V.; Orsi, S.; Ambrosio, L.; Netti, P.A. PCL microspheres based functional scaffolds by bottom-up approach with predefined microstructural properties and release profiles. Biomaterials 2008, 29, 4800–4807. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Mei, L.; Song, C.; Cui, X.; Wang, P. The in vivo degradation, absorption and excretion of PCL-based implant. Biomaterials 2006, 27, 1735–1740. [Google Scholar] [CrossRef] [PubMed]
- Dhanaraju, M.D.; Gopinath, D.; Ahmed, M.R.; Jayakumar, R.; Vamsadhara, C. Characterization of polymeric poly(epsilon-caprolactone) injectable implant delivery system for the controlled delivery of contraceptive steroids. J. Biomed. Mater. Res. Part A 2006, 76, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Manoukian, O.S.; Arul, M.R.; Sardashti, N.; Stedman, T.; James, R.; Rudraiah, S.; Kumbar, S.G. Biodegradable polymeric injectable implants for long-term delivery of contraceptive drugs. J. Appl. Polym. Sci. 2018, 135, 46068. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Ki, C.S.; Kim, J.W.; Lee, K.G.; Kang, S.W.; Kweon, H.Y.; Park, Y.H. Fabrication and Evaluation of Poly(epsilon-caprolactone)/Silk Fibroin Blend Nanofibrous Scaffold. Biopolymers 2012, 97, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Simoes, M.C.R.; Cragg, S.M.; Barbu, E.; De Sousa, F.B. The potential of electrospun poly(methyl methacrylate)/polycaprolactone core-sheath fibers for drug delivery applications. J. Mater. Sci. 2019, 54, 5712–5725. [Google Scholar] [CrossRef]
- Rosa, D.S.; Guedes, C.G.F.; Bardi, M.A.G. Evaluation of thermal, mechanical and morphological properties of PCL/CA and PCL/CA/PE-g-GMA blends. Polym. Test. 2007, 26, 209–215. [Google Scholar] [CrossRef]
- Arcana, M.; Bundjali, B.; Yudistira, I.; Jariah, B.; Sukria, L. Study on properties of polymer blends from polypropylene with polycaprolactone and their biodegradability. Polym. J. 2007, 39, 1337–1344. [Google Scholar] [CrossRef]
- Newman, D.; Laredo, E.; Bello, A.; Grillo, A.; Feijoo, J.L.; Müler, A.J. Molecular Mobilities in Biodegradable Poly(DL-lactide)/Poly(ε-caprolactone) Blends. Macromolecules 2009, 42, 5219–5225. [Google Scholar] [CrossRef]
- Ravati, S.; Favis, B.D. Interfacial coarsening of ternary polymer blends with partial and complete wetting structures. Polymer 2013, 54, 6739–6751. [Google Scholar] [CrossRef]
- Jing, X.; Mi, H.Y.; Huang, H.X.; Turng, L.S. Shape memory thermoplastic polyurethane (TPU)/poly(epsilon-caprolactone) (PCL) blends as self-knotting sutures. J. Mech. Behav. Biomed. Mater. 2016, 64, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Seggiani, M.; Altieri, R.; Puccini, M.; Stefanelli, E.; Esposito, A.; Castellani, F.; Stanzione, V.; Vitolo, S. Polycaprolactone-collagen hydrolysate thermoplastic blends: Processability and biodegradability/compostability. Polym. Degrad. Stab. 2018, 150, 13–24. [Google Scholar] [CrossRef]
- Lee, S.-H.; Teramoto, Y.; Endo, T. Cellulose nanofiber-reinforced polycaprolactone/polypropylene hybrid nanocomposite. Compos. Part A Appl. Sci. Manuf. 2011, 42, 151–156. [Google Scholar] [CrossRef]
- Pan, L.; Pei, X.; He, R.; Wan, Q.; Wang, J. Multiwall carbon nanotubes/polycaprolactone composites for bone tissue engineering application. Colloids Surf. B Biointerfaces 2012, 93, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Boujemaoui, A.; Sanchez, C.C.; Engstrom, J.; Bruce, C.; Fogelstrom, L.; Carlmark, A.; Malmstrom, E. Polycaprolactone Nanocomposites Reinforced with Cellulose Nanocrystals Surface-Modified via Covalent Grafting or Physisorption: A Comparative Study. ACS Appl. Mater. Interfaces 2017, 9, 35305–35318. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Liu, Y.J.; Wu, H.Q.; Chen, J.H.; Yao, J. Poly(epsilon-caprolactone) filled with polydopamine-coated high aspect ratio layered double hydroxide: Simultaneous enhancement of mechanical and barrier properties. Appl. Clay Sci. 2017, 150, 202–209. [Google Scholar] [CrossRef]
- Lübbert, C.; Wendt, K.; Feisthammel, J.; Moter, A.; Lippmann, N.; Busch, T.; Mössner, J.; Hoffmeister, A.; Rodloff, A.C. Epidemiology and Resistance Patterns of Bacterial and Fungal Colonization of Biliary Plastic Stents: A Prospective Cohort Study. PLoS ONE 2016, 11, e0155479. [Google Scholar] [CrossRef] [PubMed]
- Kwon, C.I.; Kim, G.; Jeong, S.; Lee, D.H.; Kim, K.A.; Ko, K.H.; Cho, J.Y.; Hong, S.P. The Stent Patency and Migration Rate of Different Shaped Plastic Stents in Bile Flow Phantom Model and In Vivo Animal Bile Duct Dilation Model. Dig. Dis. Sci. 2017, 62, 1246–1255. [Google Scholar] [CrossRef] [PubMed]
- Atrey, A.; Ward, S.E.; Khoshbin, A.; Hussain, N.; Bogoch, E.; Schemitsch, E.H.; Waddell, J.P. Ten-year follow-up study of three alternative bearing surfaces used in total hip arthroplasty in young patients a prospective randomised controlled trial. Bone Jt. J. 2017, 99, 1590–1595. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lew, D.H.; Roh, T.S.; Lee, W.J. Use of Acellular Allogenic Dermal Matrix (MegaDerm) in Orbital Wall Reconstruction: A Comparison with Absorbable Mesh Plate and Porous Polyethylene. J. Craniofac. Surg. 2017, 28, E644–E649. [Google Scholar] [CrossRef] [PubMed]
- Tone, S.; Hasegawa, M.; Puppulin, L.; Pezzotti, G.; Sudo, A. Surface modifications and oxidative degradation in MPC-grafted highly cross-linked polyethylene liners retrieved from short-term total hip arthroplasty. Acta Biomater. 2018, 66, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, J.A.; da Fonseca, T.S.; da Silva, G.F.; Sasso-Cerri, E.; Guerreiro-Tanomaru, J.M.; Tanomaru, M.; Cerri, P.S. Reduced interleukin-6 immunoexpression and birefringent collagen formation indicate that MTA Plus and MTA Fillapex are biocompatible. Biomed. Mater. 2018, 13, 035002. [Google Scholar] [CrossRef] [PubMed]
- Affatato, S.; Freccero, N.; Taddei, P. The biomaterials challenge: A comparison of polyethylene wear using a hip joint simulator. J. Mech. Behav. Biomed. Mater. 2016, 53, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Kalfoglou, N.K. Compatibility of Low-Density Polyethylene-Poly(ε-caprolactone) Blends. J. Appl. Polym. Sci. 1983, 28, 2541–2551. [Google Scholar] [CrossRef]
- Moura, I.; Machado, A.V.; Duarte, F.M.; Nogueira, R. Biodegradability Assessment of Aliphatic Polyesters-Based Blends Using Standard Methods. J. Appl. Polym. Sci. 2011, 119, 3338–3346. [Google Scholar] [CrossRef]
- Chang, H.; Zhang, J.; Li, L.; Wang, Z.; Yang, C.; Takahashi, I.; Ozaki, Y.; Yan, S. A Study on the Epitaxial Ordering Process of the Polycaprolactone on the Highly Oriented Polyethylene Substrate. Macromolecules 2010, 43, 362–366. [Google Scholar] [CrossRef]
- Cerrada, M.L.; Benavente, R.; Peña, B.; Pérez, E. The effect of thermal treatment on the structure and relaxation processes of olefinic polymers synthesized with metallocene catalysts. Polymer 2000, 41, 5957–5965. [Google Scholar] [CrossRef]
- Nakagawa, S.; Kadena, K.; Ishizone, T.; Nojima, S.; Shimizu, T.; Yamaguchi, K.; Nakahama, S. Crystallization Behavior and Crystal Orientation of Poly(ε-caprolactone) Homopolymers Confined in Nanocylinders: Effects of Nanocylinder Dimension. Macromolecules 2012, 45, 1892–1900. [Google Scholar] [CrossRef]
- Muñoz-Bonilla, A.; Cerrada, M.L.; Fernández-García, M.; Kubacka, A.; Ferrer, M.; Fernández-García, M. Biodegradable Polycaprolactone-Titania Nanocomposites: Preparation, Characterization And Antimicrobial Properties. Int. J. Mol. Sci. 2013, 14, 9249–9266. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, B. Macromolecular Physics; Academic Press: New York, NY, USA, 1980; Volume 3, p. 42. [Google Scholar]
- Ward, I.M. Mechanical Properties of Solid Polymers; Wiley: Chichester, UK, 1990. [Google Scholar]
- Barton, A.F.M. Handbook of Polymer-Liquid Interaction Parameters and Solubility Parameters; CRC Press: Boca Ratón, FL, USA, 1990. [Google Scholar]
- Zhang, Y.; Guo, W.; Zhang, H.; Wu, C. Influence of chain extension on the compatibilization and properties of recycled poly(ethylene terephthalate)/linear low density polyethylene blends. Polym. Degrad. Stab. 2009, 94, 1135–1141. [Google Scholar] [CrossRef]
- Chen, Z.; Hay, J.N.; Jenkins, M.J. FTIR spectroscopic analysis of poly(ethylene terephthalate) on crystallization. Eur. Polym. J. 2012, 48, 1586–1611. [Google Scholar] [CrossRef]
- Gulmine, J.V.; Janissek, P.R.; Heise, H.M.; Akcelrud, L. Polyethylene characterization by FTIR. Polym. Test. 2002, 21, 557–563. [Google Scholar] [CrossRef]
- Phillipson, K.; Hay, J.; Jenkins, M. Thermal analysis FTIR spectroscopy of poly(ε-caprolactone). Thermochim. Acta 2014, 595, 74–82. [Google Scholar] [CrossRef]
- Ryan, A.J.; Stanford, J.L.; Bras, W.; Nye, T.M.W. Biodegradable Polycaprolactone-Titania Nanocomposites: Preparation, Characterization and Antimicrobial Properties. Polymer 1997, 38, 759–768. [Google Scholar] [CrossRef]
- Baltá-Calleja, F.J.; Vonk, C.G. X-ray Scattering of Synthetic Polymers; Elsevier: Amsterdam, The Netherlands, 1989. [Google Scholar]
- Cerrada, M.L.; Benavente, R.; Pérez, E. Influence of thermal history on morphology and viscoelastic behavior of ethylene-1-octene copolymers synthesized with metallocene catalysts. J. Mater. Res. 2001, 16, 1103–1111. [Google Scholar] [CrossRef]
- Cerrada, M.L.; Benavente, R.; Pérez, E. Crystalline structure and viscoelastic behavior in composites of a metallocenic ethylene-1-octene copolymer and glass fiber. Macromol. Chem. Phys. 2002, 203, 718–726. [Google Scholar] [CrossRef]
- Cerrada, M.L.; Pérez, E.; Lourenço, J.P.; Campos, J.M.; Ribeiro, M.R. Hybrid HDPE/MCM-41 Nanocomposites: Crystalline Structure and Viscoelastic Behaviour. Microporous Microporous Mater. 2010, 130, 215–223. [Google Scholar] [CrossRef]
- Cerrada, M.L.; Bento, A.; Pérez, E.; Lorenzo, V.; Lourenço, J.P.; Ribeiro, M.R. Hybrid Materials Based on Polyethylene and MCM-41 Particles Functionalized with Silanes: Catalytic Aspects of In Situ Polymerization, Crystalline Features and Mechanical Properties. Microporous Microporous Mater. 2016, 232, 86–96. [Google Scholar] [CrossRef]
- Pérez, E.; Cerrada, M.L.; Benavente, R.; Gómez-Elvira, J.M. Enhancing the formation of the new trigonal polymorph in isotactic propene-1-pentene copolymers: Determination of the X-ray crystallinity. Macromol. Res. 2011, 19, 1179–1185. [Google Scholar] [CrossRef]
- Pérez, E.; Gómez-Elvira, J.M.; Benavente, R.; Cerrada, M.L. Tailoring the Formation Rate of the Mesophase in Random Propylene-co-1-Pentene Copolymers. Macromolecules 2012, 45, 6481–6490. [Google Scholar] [CrossRef]
- García-Peñas, A.; Gómez-Elvira, J.M.; Barranco-García, R.; Pérez, E.; Cerrada, M.L. Trigonal form as a tool for tuning mechanical behavior in poly(propylene-co-1-pentene-co-1-heptene) terpolymers. Polymer 2016, 99, 112–121. [Google Scholar] [CrossRef]
- Wijmans, J.G.; Baker, R.W. The solution-diffusion model: A review. J. Membr. Sci. 1995, 107, 1–21. [Google Scholar] [CrossRef]
- Rutherford, S.W.; Do, D.D. Review of time lag permeation technique as a method for characterisation of porous media and membranes. Adsorption 1997, 3, 283–312. [Google Scholar] [CrossRef]
- Michaels, A.S.; Bixler, H.J. Flow of gases through polyethylene. J. Polym. Sci. 1961, 50, 413–439. [Google Scholar] [CrossRef]
- Michaels, A.S.; Parker, R.B., Jr. Sorption and flow of gases in polyethylene. J. Polym. Sci. 1959, 41, 53–71. [Google Scholar] [CrossRef]
- Laguna, M.F.; Cerrada, M.L.; Benavente, R.; Pérez, E. Effect of the comonomer content on the permeation behavior in polyolefin films synthesized with metallocene catalysts. J. Membr. Sci. 2003, 212, 167–176. [Google Scholar] [CrossRef]
- Seguela, R.; Rietsch, F. Double yield point in polyethylene under tensile loading. J. Mater. Sci. Lett. 1990, 9, 46–47. [Google Scholar] [CrossRef]
- Brooks, N.W.; Duckett, R.A.; Ward, I.M. Investigation into double yield points in polyethylene. Polymer 1992, 33, 1872–1880. [Google Scholar] [CrossRef]
- Fonseca, C.; Pereña, J.M.; Benavente, R.; Cerrada, M.L.; Bello, A.; Pérez, E. Microhardness and thermal study of the annealing effects in vinyl alcohol-ethylene copolymers. Polymer 1995, 36, 1887–1892. [Google Scholar] [CrossRef]
- Cerrada, M.L.; de la Fuente, J.L.; Fernández-García, M.; Madruga, E.L. Viscoelastic and mechanical properties of poly(butyl acrylate-g-styrene) copolymers. Polymer 2001, 42, 4647–4655. [Google Scholar] [CrossRef]
- Cerrada, M.L.; Pereña, J.M.; Benavente, R.; Pérez, E. Viscoelastic processes in vinyl alcohol-ethylene copolymers. Influence of composition and thermal treatment. Polymer 2000, 41, 6655–6661. [Google Scholar] [CrossRef]
- de la Fuente, J.L.; Fernández-García, M.; Cerrada, M.L. Viscoelastic behavior of a HTPB gum and its highly filled composites. Effect of the type of filler on the relaxation processes. J. Appl. Polym. Sci. 2003, 88, 1705–1712. [Google Scholar] [CrossRef]
- Prieto, O.; Pereña, J.M.; Benavente, R.; Pérez, E.; Cerrada, M.L. Viscoelastic relaxation mechanisms of conventional polypropylene toughened by a plastomer. J. Polym. Sci. Part B Polym. Phys. 2003, 41, 1878–1888. [Google Scholar] [CrossRef]
- De la Fuente, J.L.; Wilhelm, M.; Spiess, H.W.; Madruga, E.L.; Fernández Garcia, M.; Cerrada, M.L. Thermal, Morphological and Rheological Characterization of Poly(Acrylic Acid–g–Styrene) Amphiphilic Graft Copolymers. Polymer 2005, 46, 4544–4553. [Google Scholar] [CrossRef]
- Schatzki, T.F. Statistical computation of distribution functions of dimensions of macromolecules. J. Polym. Sci. 1962, 57, 337–356. [Google Scholar] [CrossRef]
- Sabater i Serra, R.; Escobar Ivirico, J.L.; Meseguer Dueñas, J.M.; Andrio Balado, A.; Gómez Ribelles, J.L.; Salmerón Sánchez, M. Dielectric relaxation spectrum of poly (ε-caprolactone) networks hydrophilized by copolymerization with 2-hydroxyethyl acrylate. Eur. Phys. J. E 2007, 22, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Harrison, K.L.; Jenkins, M.J. The effect of crystallinity and water absorption on the dynamic mechanical relaxation behaviour of polycaprolactone. Polym. Int. 2004, 53, 1298–1304. [Google Scholar] [CrossRef]
- Nikolic, M.S.; Mitric, M.; Dapcevic, A.; Djonlagic, J. Viscoelastic properties of poly(ε-caprolactone)/clay nanocomposites in solid and in melt state. J. Appl. Polym. Sci. 2016, 133, 42896. [Google Scholar] [CrossRef]
- McCrum, B.; Read, B.; Williams, G. Anelastic and Dielectric Effects in Polymeric Solids; Dover Publications: New York, NY, USA, 1991. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Specimen | fcPCL | TmPCL (°C) | fcHDPE | TmHDPE (°C) |
|---|---|---|---|---|
| PCL100 | 0.54 | 57.0 | − | − |
| PCL95 | 0.54 | 57.5 | 0.36 | 129.5 |
| PCL90 | 0.55 | 57.5 | 0.39 | 129.5 |
| PCL75 | 0.56 | 58.0 | 0.51 | 130.0 |
| PCL50 | 0.57 | 57.5 | 0.55 | 130.5 |
| PCL25 | 0.57 | 57.5 | 0.60 | 131.5 |
| PCL0 | − | − | 0.59 | 131.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blázquez-Blázquez, E.; Pérez, E.; Lorenzo, V.; Cerrada, M.L. Crystalline Characteristics and Their Influence in the Mechanical Performance in Poly(ε-Caprolactone) / High Density Polyethylene Blends. Polymers 2019, 11, 1874. https://doi.org/10.3390/polym11111874
Blázquez-Blázquez E, Pérez E, Lorenzo V, Cerrada ML. Crystalline Characteristics and Their Influence in the Mechanical Performance in Poly(ε-Caprolactone) / High Density Polyethylene Blends. Polymers. 2019; 11(11):1874. https://doi.org/10.3390/polym11111874
Chicago/Turabian StyleBlázquez-Blázquez, Enrique, Ernesto Pérez, Vicente Lorenzo, and María L. Cerrada. 2019. "Crystalline Characteristics and Their Influence in the Mechanical Performance in Poly(ε-Caprolactone) / High Density Polyethylene Blends" Polymers 11, no. 11: 1874. https://doi.org/10.3390/polym11111874
APA StyleBlázquez-Blázquez, E., Pérez, E., Lorenzo, V., & Cerrada, M. L. (2019). Crystalline Characteristics and Their Influence in the Mechanical Performance in Poly(ε-Caprolactone) / High Density Polyethylene Blends. Polymers, 11(11), 1874. https://doi.org/10.3390/polym11111874