Abstract
Cyclic polymers exhibit unique physical and chemical properties because of the restricted chain mobility and absence of chain ends. Although many types of homopolymers and diblock copolymers possessing cyclic architectures have been synthesized to date, there are relatively few reports of cyclic triblock terpolymers because of their synthetic difficulties. In this study, a novel synthetic approach for μ-ABC tricyclic miktoarm star polymers involving t-Bu-P4-catalyzed ring-opening polymerization (ROP) of glycidyl ethers and intramolecular copper-catalyzed azido-alkyne cycloaddition (CuAAC) was developed. First, the t-Bu-P4-catalyzed ROP of decyl glycidyl ether, dec-9-enyl glycidyl ether, and 2-(2-(2-methoxyethoxy) ethoxy) ethyl glycidyl ether with the aid of functional initiators and terminators was employed for the preparation of a clickable linear triblock terpolymer precursor possessing three azido and three ethynyl groups at the selected positions. Next, the intramolecular CuAAC of the linear precursor successfully produced the well-defined tricyclic triblock terpolymer with narrow dispersity in a reasonable yield. The present strategy is useful for synthesizing model polymers for studying the topological effects on the triblock terpolymer self-assembly.
1. Introduction
Block copolymers (BCP) consisting of more than two different polymer segments (or blocks) have attracted considerable attention for their unique self-assembly properties such as microphase-separation and micellization [1,2,3]. It is well known that the molecular weight, volume fraction of each constituting block, and incompatibility between the blocks determine the dimension and morphology of the resulting self-assembled nanostructure. In addition to these classical structural parameters, macromolecular architectures such as star, comb, and cyclic polymer structures, have recently been recognized as an important factor affecting the BCP self-assembly behavior [4]. Several pioneering studies have indicated that cyclic diblock copolymers display unique self-assembly behaviors in both solution and solid states. For example, Tezuka et al. discovered that amphiphilic cyclic poly(ethylene oxide)-b-poly(butyl acrylate) formed micellar aggregates with greater thermal stability than the corresponding linear counterpart [5]. Hawker et al. reported that cyclic polystyrene-b-poly(ethylene oxide) self-assembled into a hexagonally close-packed cylindrical structure with smaller domain-spacing in the thin film state compared to its corresponding linear counterpart [6]. Thus, further studies relating to cyclic polymer synthesis and self-assembly are highly desired.
Triblock terpolymers consisting of three different polymer segments also self-assemble in both solution and solid states, producing much more complex and diversified nanostructures than those created by diblock copolymers. For example, Noda et al. investigated the morphology of polyisoprene-b-polystyrene-b-poly(2-vinylpridine) in bulk and discussed the variation in morphologies depending on the composition [7,8]. Moreover, Müller et al. investigated the solution state self-assembly of linear triblock terpolymers to produce various micellar morphologies, such as three-layer core-shell-corona spheres, footballs, and hamburgers [9,10,11] Thus, the combination of a triblock terpolymer system with branched and cyclic architectures is exciting because nanostructures with a variety of novel morphologies and functions are created. Indeed, Dotera et al. simulated the morphology of μ-ABC miktoarm star polymers and reported new self-assembled structures that could not have been attained from diblock copolymers or linear triblock terpolymer counterparts [12]. In addition, Matsushita et al. found that μ-ABC miktoarm star polymers constructed an Archimedean tiling pattern in the bulk [13]. Ree et al. also found a complex three-phased hexagonal morphology in the asymmetric nine-arm star polymer, (polystyrene)3-b-(poly(4-methoxystyrene))3-b-(polyisoprene)3 [14]. These self-assembled structures in thin films can be used for lithographic templates for fabricating complex nanopatterns [15,16]. In contrast, triblock terpolymers with cyclic architectures have received scant attention, which is mainly because of their synthetic inaccessibility. In order to study the correlation between the cyclic architecture and self-assembled nanostructures in a triblock terpolymer system, establishing a facile synthetic route toward the architecturally complex triblock terpolymers with well-defined molecular weight and composition is crucial.
Some examples of triblock terpolymers with cyclic architectures are the macrocyclic ABC triblock terpolymer and μ-ABC tricyclic miktoarm star polymer. Hadjichristidis et al. successfully synthesized a macrocyclic ABC triblock terpolymer via intramolecular Glaser coupling of a poly(isoprene-b-styrene-b-2-vinylpyridine) linear precursor [17]. The authors observed a significant influence of the cyclic architecture on the terpolymer microphase separation. Monteiro et al. reported the first synthesis of the μ-ABC tricyclic miktoarm star polymer, having macrocyclic polystyrene, poly(t-butyl acrylate), and poly(methyl acrylate) units [18]. In this synthesis, three different macrocyclic units possessing a reactive functional group were synthesized by the copper-catalyzed azido-alkyne cycloaddition (CuAAC) [19,20], and then combined via CuAAC and nitroxide radical coupling (NRC) reaction [21] to form the tricyclic structure (Scheme 1a). Although this strategy is highly sophisticated, a challenge still exists in establishing a new strategy for the μ-ABC tricyclic miktoarm star polymer without using the intermolecular coupling reaction.
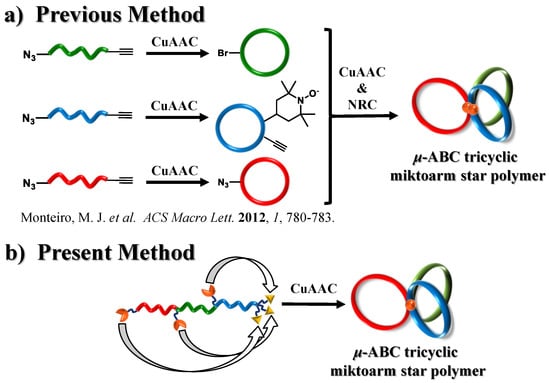
Scheme 1.
(a) Previous and (b) present synthetic approaches for μ-ABC tricyclic miktoarm star polymers.
In this study, a novel synthetic approach toward μ-ABC tricyclic miktoarm star polymers consisting of a polyether backbone based on intramolecular click cyclization has been proposed (Scheme 1b). The most important feature of the proposed approach is that the three multicyclic units can be constructed in a single click reaction step. In previous studies, figure-eight-, trefoil-, and quatrefoil-shaped block copolymers were synthesized via the intramolecular click reaction [22,23]. Thus, the present strategy should be highly feasible as long as a well-defined clickable precursor can be synthesized. For the synthesis of the clickable precursor, t-Bu-P4-catalyzed ring-opening polymerization (ROP) of glycidyl ethers was employed as it enabled a precise control over the end group structure and molecular weight. Scheme 2 describes a detailed synthetic pathway for constructing a μ-ABC tricyclic miktoarm star polymer (P9) consisting of cyclic units of poly(decyl glycidyl ether) (M1), poly(dec-9-enyl glycidyl ether) (M2), and poly[2-(2-(2-methoxyethoxy) ethoxy) ethyl glycidyl ether] (M3). The linear triblock terpolymer possessing three azido groups and three ethynyl groups (P8) was synthesized by combining t-Bu-P4-catalyzed ROP and ω-end functionalization. It was then subjected to intramolecular click cyclization to produce P9. To the best of our knowledge, this is the first example of the construction of a μ-ABC tricyclic miktoarm star polymer via intramolecular coupling.
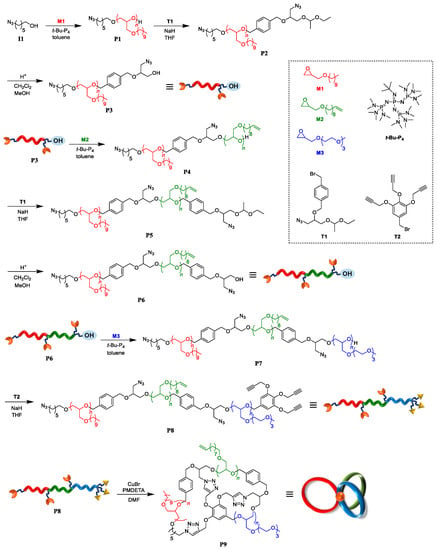
Scheme 2.
Synthetic pathway for μ-ABC tricyclic miktoarm star polymer (P9) based on intramolecular click cyclization.
2. Materials and Methods
Materials. Decyl glycidyl ether (M1) [24], dec-9-enyl glycidyl ether (M2) [24], and 2-(2-(2-methoxyethoxy) ethoxy) ethyl glycidyl ether (M3) [25] were synthesized according to reported methods, purified by distillation under vacuum over calcium hydride, and stored under an argon atmosphere. 1-(((1-Azido-3-(1-ethoxyethoxy)propan-2-yl)oxy)-methyl)-4-(bromomethyl)benzene (T1) [22], 5-(bromomethyl)-1,2,3-tris(prop-2-yn-1-yloxy)benzene (T2) [26], 6-azide-1-hexanol (I1) [27], and propargyl-functionalized Wang resin (PSt-C≡CH) [28] were synthesized according to reported methods. All other chemicals were used as received. Calcium hydride (CaH2) and benzoic acid were purchased from Kanto Chemical Co., Ltd. (Tokyo, Japan), and sodium hydride (NaH, 60%) and N,N,N’,N’’,N’’-pentamethyldiethylenetriamine (PMDETA) were purchased from Tokyo Chemical Industry Co., Ltd. (TCI, Tokyo, Japan). t-Bu-P4 (1.0 mol·L−1 in n-hexane), copper (I) bromide (CuBr), aluminum oxide (alumina), and DOWEX® 50WX2 were procured from the Sigma-Aldrich Chemicals Co. (St. Louis, MI, USA). Dry dimethylformamide (DMF, >99.5%; water content, <0.005%), dry toluene (>99.5%; water content, <0.001%), and dry tetrahydrofuran (THF, >99.5%; water content, <0.001%) were purchased from Kanto Chemical Co., Inc.
Instruments. The instruments used in this study, such as the glovebox (an MBRAUN stainless steel glovebox, Garching, Germany), size exclusion chromatography (SEC) (a Shodex GPC-101 gel permeation chromatography system, Tokyo, Japan), NMR (a JEOL ECS400 instrument, Tokyo, Japan, 400 MHz), and FT-IR (a PerkinElmer Frontier MIR spectrometer, Waltham, MA, USA) are the same as reported in our previous paper [29].
Synthesis of azido poly(M1) (P1). A typical polymerization procedure is as follows (method A): M1 (1.11 mL, 4.67 mmol) was added to a stirred solution of I1 (141 µL, 141 µmol) and t-Bu-P4 (141 µL of 1.0 mol·L−1 solution in n-hexane, 141 µmol) in toluene (478 µL), and then the entire mixture was stirred at r.t. (room temperature) for 20 h. Subsequently, an excess of benzoic acid was added to the mixture to terminate the polymerization, and the product was purified by passing it through a pad of alumina using THF to obtain P1 as a colorless viscous liquid (558 mg). Yield: 56.5%. 1H NMR (400 MHz, CDCl3): δ (ppm) 3.29–3.75 (m, N3(CH2)5CH2OCH2–, N3(CH2)6OCH2–, –OCH2CH(CH2OCH2(CH2)8CH3)O–), 3.85–3.95 (s, –OCH2CH(CH2OCH2(CH2)8CH3)OH), 3.26 (t, N3CH2–), 1.10–1.69 (m, N3CH2(CH2)4CH2O–, –OCH2CH(CH2OCH2CH2(CH2)7CH3)O–), and 0.78–0.95 (t, –OCH2CH(CH2OCH2(CH2)8CH3)O–). Mn,NMR = 7000 g·mol−1 (CDCl3); Mn,SEC = 7400 g·mol−1 (THF); Đ = 1.03.
Synthesis of diazido poly(M1) (P2). A typical procedure for the ω-chain end functionalization is as follows (method B): Under nitrogen atmosphere, NaH (47.8 mg, 1.19 mmol) was added to a stirred solution of P1 (Mn,NMR = 7000 g·mol−1, 558 mg) in THF (5.0 mL) and the entire mixture was stirred at 40 °C for 30 min. T1 (296 mg, 796 µmol) was then added to the reaction mixture, which was stirred at r.t. for 48 h under nitrogen. The product was purified by passing it through a pad of alumina to obtain P2 as a pale yellow viscous liquid (489 mg). Yield: 75.4%. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.36 (aromatic), 4.70 (m, –CH2PhCH2O–), 4.64 (s, –OCH(CH3)O–), 3.29–3.75 (m, N3(CH2)5CH2OCH2−, –OCH2CH(CH2OCH2(CH2)8CH3)O–, N3CH2CH(O–)CH2–, –CH2OCH(CH3)OCH2CH3), 3.26 (t, N3CH2CH2–), 1.10–1.69 (m, N3CH2(CH2)4CH2O–, –OCH2CH(CH2OCH2CH2(CH2)7CH3)O–), and 0.78–0.95 (t, –OCH2CH(CH2OCH2(CH2)8CH3)O–). Mn,NMR = 7950 g mol−1 (CDCl3); Mn,SEC = 8000 g mol−1 (THF); Đ = 1.03.
Synthesis of diazido-hydroxyl poly(M1) (P3). A typical procedure for the deprotection of ethoxyethyl group is as follows (method C): DOWEX® (100 mg) and distilled water (100 µL) were added to the stirred solution of P2 (Mn,NMR = 7950 g·mol−1, 489 mg) in CH2Cl2/MeOH (10 mL, v/v = 4/1), and the entire mixture was stirred at r.t. for 48 h. The product was purified by passing it through a pad of alumina to obtain P3 as a pale yellow viscous liquid (331 mg). Yield: 70.6%. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.36 (aromatic), 4.70 (m, –CH2PhCH2O–), 3.29–3.75 (m, N3(CH2)5CH2OCH2–, –OCH2CH(CH2OCH2(CH2)8CH3)O–, N3CH2CH(O–)CH2–, –CH2OH), 3.26 (t, N3CH2–), 1.10–1.69 (m, N3CH2(CH2)4CH2O–, –OCH2CH(CH2OCH2CH2(CH2)7CH3)O–), and 0.78–0.95 (t, –OCH2CH(CH2OCH2(CH2)8CH3)O–). Mn,NMR = 7810 g·mol−1 (CDCl3); Mn,SEC = 7500 g·mol−1 (THF); Đ = 1.03.
Synthesis of poly(M1)-b-poly(M2) (P4). Method A was used for the polymerization of M2 (273 µL, 35.6 µmol) with P3 (Mn,NMR = 7810 g·mol−1, 331 mg) and t-Bu-P4 (35.6 µL of 1.0 mol·L−1 solution in n-hexane, 35.6 µmol) in toluene (478 µL) to obtain P4 as a pale yellow viscous liquid (533 mg). Yield: 84.7%. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.36 (aromatic), 5.71–5.90 (q, –OCH2CH(CH2OCH2(CH2)6CH=CH2)O–), 4.86–5.07 (d, –OCH2CH(CH2OCH2(CH2)6CH=CH2)O–), 4.56–4.77 (m, –CH2PhCH2O–), 3.29–3.85 (m, N3(CH2)5CH2OCH2–, –OCH2CH(CH2OCH2(CH2)8CH3)O–, –OCH2CH(CH2OCH2(CH2)6CH=CH2)O–, N3CH2CH(O–)CH2–), 3.21–3.28 (t, N3CH2–), 1.14–1.80 (m, N3CH2(CH2)4CH2O–, –OCH2CH(CH2OCH2CH2(CH2)7CH3)O–, –OCH2CH(CH2OCH2(CH2)6CH=CH2)O–), and 0.78–0.98 (t, –OCH2CH(CH2OCH2(CH2)8CH3)O–). Mn,NMR = 14,600 g·mol−1 (CDCl3); Mn,SEC = 12,500 g·mol−1 (THF); Đ = 1.10.
Synthesis of triazido poly(M1)-b-poly(M2) (P5). Method B was used for the ω-chain end functionalization of P4 (Mn,NMR = 14,600 g·mol−1, 533 mg) with NaH (21.2 mg, 530 µmol) and T1 (131 mg, 353 µmol) in THF (5.0 mL) to obtain P5 as a pale yellow viscous liquid (485 mg). Yield: 90.8%. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.36 (aromatic), 5.72–5.85 (q, –OCH2CH(CH2OCH2(CH2)6CH=CH2)O–), 4.87–5.02 (d, –OCH2CH(CH2OCH2(CH2)6CH=CH2)O–), 4.55–4.77 (m, –CH2PhCH2O–), 3.29–3.74 (m, N3(CH2)5CH2OCH2–, –OCH2CH(CH2OCH2(CH2)8CH3)O–, –OCH2CH(CH2OCH2(CH2)6CH=CH2)O–, N3CH2CH(O–)CH2–), 3.21–3.28 (t, N3CH2–), 1.14–1.75 (m, N3CH2(CH2)4CH2O–, –OCH2CH(CH2OCH2CH2(CH2)7CH3)O–, –OCH2CH(CH2OCH2(CH2)6CH=CH2)O–), and 0.78–0.96 (t, –OCH2CH(CH2OCH2(CH2)8CH3)O–). Mn,NMR = 14,800 g·mol−1 (CDCl3); Mn,SEC = 12,700 g·mol−1 (THF); Đ = 1.08.
Synthesis of triazido-hydroxyl poly(M1)-b-poly(M2) (P6). Method C was used for the deprotection of P5 (Mn,NMR = 14,800 g·mol−1, 485 mg) with DOWEX® (100 mg) and distilled water (100 µL) in CH2Cl2/MeOH (10 mL, v/v = 4/1). The residue was purified by preparative SEC to obtain P6 as a pale yellow viscous liquid (168 mg). Yield: 34.5%. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.36 (aromatic), 5.72–5.85 (q, –OCH2CH(CH2OCH2(CH2)6CH=CH2)O–), 4.87–5.02 (d, –OCH2CH(CH2OCH2(CH2)6CH=CH2)O–), 4.55–4.77 (m, –CH2PhCH2O–), 3.29–3.74 (m, N3(CH2)5CH2OCH2–, –OCH2CH(CH2OCH2(CH2)8CH3)O–, –OCH2CH(CH2OCH2(CH2)6CH=CH2)O–, N3CH2CH(O–)CH2–, –CH2OH), 3.21–3.28 (t, N3CH2–), 1.14–1.75 (m, N3CH2(CH2)4CH2O–, –OCH2CH(CH2OCH2CH2(CH2)7CH3)O–, –OCH2CH(CH2OCH2(CH2)6CH=CH2)O–), and 0.78–0.96 (t, –OCH2CH(CH2OCH2(CH2)8CH3)O–). Mn,NMR = 14,400 g·mol−1 (CDCl3); Mn,SEC = 12,600 g·mol−1 (THF); Đ = 1.04.
Synthesis of poly(M1)-b-poly(M2)-b-poly(M3) (P7). Method A was used for polymerization of M3 (62 µL, 299 µmol) with P6 (Mn,NMR = 14,400 g·mol−1, 131 mg) and t-Bu-P4 (9.1 µL of 1.0 mol·L−1 solution in n-hexane, 9.1 µmol) in toluene (98 µL). The residue was purified by preparative SEC to obtain P7 as a pale yellow viscous liquid (127 mg). Yield: 80.4%. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.36 (aromatic), 5.72–5.85 (q, –OCH2CH(CH2OCH2(CH2)6CH=CH2)O–), 4.87–5.02 (d, –OCH2CH(CH2OCH2(CH2)6CH=CH2)O–), 4.55–4.72 (m, –CH2PhCH2O–), 3.29–3.74 (m, N3(CH2)5CH2OCH2–, –OCH2CH(CH2OCH2(CH2)8CH3)O–, –OCH2CH(CH2OCH2(CH2)6CH=CH2)O–, –OCH2CH(CH2O(CH2CH2O)3CH3)O–, N3CH2CH(O–)CH2–), 3.21–3.28 (t, N3CH2–), 1.15–1.75 (m, N3CH2(CH2)4CH2O–, –OCH2CH(CH2OCH2CH2(CH2)7CH3)O–, –OCH2CH(CH2OCH2(CH2)6CH=CH2)O–), and 0.75–0.93 (t, –OCH2CH(CH2OCH2(CH2)8CH3)O–). Mn,NMR = 19,600 g·mol−1 (CDCl3); Mn,SEC = 14,200 g·mol−1 (THF); Đ = 1.03.
Synthesis of triazido-triethynylpoly(M1)-b-poly(M2)-b-poly(M3) (P8). Method B was used for the ω-chain end functionalization of P7 (Mn, NMR = 19,600 g·mol−1, 50 mg) with NaH (1.51 mg, 37.8 µmol) and T2 (8.39 mg, 25.2 µmol) in THF (5.0 mL). The residue was purified by preparative SEC to obtain P8 as a pale yellow viscous liquid (43 mg). Yield: 90.8%. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.36 (aromatic), 5.72–5.85 (q, –OCH2CH(CH2OCH2(CH2)6CH=CH2)O–), 4.87–5.02 (d, –OCH2CH(CH2OCH2(CH2)6CH=CH2)O–), 4.55–4.77 (m, –CH2PhCH2O–, –CH2OPhOCH2C≡CH), 3.28–3.75 (m, N3(CH2)5CH2OCH2–, –OCH2CH(CH2OCH2(CH2)8CH3)O–,–OCH2CH(CH2OCH2(CH2)6CH=CH2)O–, –OCH2CH(CH2O(CH2CH2O)3CH3)O–, N3CH2CH(O–)CH2–), 3.21–3.28 (t, N3CH2–), 2.57–2.64 (s, m–OCH2C≡CH), 2.44–2.49 (s, p–OCH2C≡CH), 1.15–1.75 (m, N3CH2(CH2)4CH2O–, –OCH2CH(CH2OCH2CH2(CH2)7CH3)O–, –OCH2CH(CH2OCH2(CH2)6CH=CH2)O–), and 0.75–0.93 (t, –OCH2CH(CH2OCH2(CH2)8CH3)O–). Mn,NMR = 19,900 g·mol−1 (CDCl3); Mn,SEC = 14,700 g·mol−1 (THF); Đ = 1.04.
Synthesis ofμ-ABC tricyclic miktoarm star polymer (P9). A solution of P8 (Mn,NMR = 19,900 g·mol−1, 43 mg) in degassed DMF (2.25 mL) was added to a stirred solution of CuBr (30.8 mg, 215 µmol) and PMDETA (89.8 µL, 430 µmol) in degassed DMF (100 mL) using a syringe pump at the rate of 0.3 mL·h−1 at 100 °C under a nitrogen atmosphere. Next, PSt-C≡CH (10 mg), CuBr (6.2 mg, 43 µmol), and PMDETA (18.0 µL, 86 µmol) were added to the reaction mixture. After stirring at 100 °C for 24 h, the solvent was removed by evaporation, and the residue was purified by passing it through a pad of alumina. This was followed by preparative SEC to obtain P9 as a pale yellow viscous liquid (22 mg). Yield: 50.7%. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.64–7.94 (triazole methine), 7.19–7.32, 6.62–6.86 (aromatic), 5.72–5.85 (q, –OCH2CH(CH2OCH2(CH2)6CH=CH2)O–), 5.05–5.26 (m, –triazole ring–CH2OPh–), 4.87–5.02 (d, –OCH2CH(CH2OCH2(CH2)6CH=CH2)O–), 4.37–4.73 (m, –CH2PhCH2O–, –CH2OPhOCH2–triazole ring–, –OCH(CH2–triazole ring–)CH2O–), 4.22–4.34 (m, –triazole ring–CH2(CH2)4CH2O–), 3.88–4.03 (m, –OCH(CH2–triazole ring–)CH2O–), 3.28–3.75 (m, –triazole ring–CH2(CH2)4CH2O–, –OCH2CH(CH2OCH2(CH2)8CH3)O–, –OCH2CH(CH2OCH2(CH2)6CH=CH2)O–, –OCH2CH(CH2O(CH2CH2O)3CH3)O–, –OCH(CH2–triazole ring–)CH2O–), 1.15–1.75 (m, –triazole ring–CH2(CH2)4CH2O–, –OCH2CH(CH2OCH2CH2(CH2)7CH3)O–, –OCH2CH(CH2OCH2(CH2)6CH=CH2)O–), and 0.75–0.93 (t, –OCH2CH(CH2OCH2(CH2)8CH3)O–). Mn,NMR = 20,100 g·mol−1 (CDCl3); Mn,SEC = 11,500 g·mol−1 (THF); Đ = 1.02.
3. Results and Discussion
Synthesis of diazido-hydroxyl poly(M1) (P3). As the first step of the synthetic route, diazido-hydroxyl poly(M1) (P3) was synthesized in three steps, namely, the polymerization of decyl glycidyl ether (M1) with 6-azido-1-hexanol (I1), end group modification with 1-(((1-azido-3-(1-ethoxyethoxy)propan-2-yl)oxy)-methyl)-4-(bromomethyl)benzene (T1), and deprotection of the ethoxyethyl group (Scheme 2). Following a previous report [22], the t-Bu-P4-catalyzed ROP of M1 using I1 as an initiator was carried out at the [M1]0/[I1]0/[t-Bu-P4] ratio of 33/1/1 to produce azido poly(M1) (P1; Mn,NMR = 7000 g·mol−1, degree of polymerization for block: DP1 = 33, molecular weight dispersion: Đ = 1.03) in 56.5% isolated yield. The 1H NMR spectrum of P1 showed the characteristic signals corresponding to the poly(M1) backbone along with minor signals of the initiator residue, such as the methylene groups adjacent to the azido groups (A: 3.26 ppm in Figure 1d), verifying that the ROP of M1 was initiated from I1. The number-average molecular weight determined from NMR analysis (Mn,NMR) of P1 was in good agreement with the Mn value (Mn,theo) calculated by the monomer conversion and the initial monomer-to-initiator ratio (Mn,theo = 7220) (Table 1). Next, P1 was treated with an excess amount of T1 in the presence of sodium hydride to obtain diazido poly(M1) (P2; Mn,NMR = 7,950 g·mol−1, DP1 = 33, Đ = 1.03). After ω-end functionalization, the ethoxyethyl group of P2 was deprotected under acidic conditions to give diazido-hydroxyl poly(M1) (P3; Mn,NMR = 7810 g·mol−1, DP = 33, Đ = 1.03). After thorough screening of the deprotection conditions, it was found that the cation exchange resin (DOWEX® hydrogen form) was the best suited for a clean reaction without undesired side reactions. There was no significant difference between the SEC traces of the P1, P2, and P3, suggesting the absence of any side reactions (Figure 2a–c). The 1H NMR spectrum of P3 showed the signals of the benzyl protons (protons f in Figure 2e,f) at 4.70 ppm as well as methylene protons adjacent to the azido group (protons a) at 3.26 ppm, based on which the introduction of an azido and a hydroxyl group at the ω-chain end was verified.
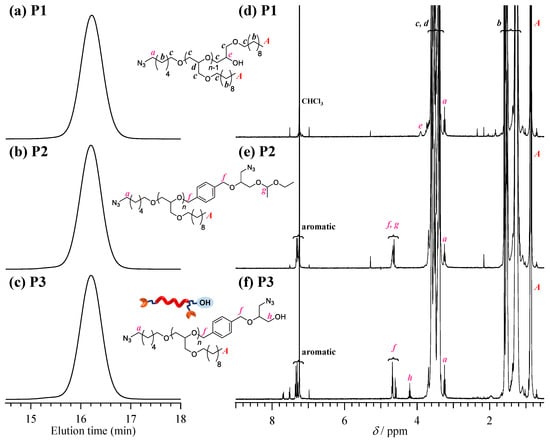
Figure 1.
(a–c) Size exclusion chromatography (SEC) traces of P1, P2, and P3. (d–f) 1H NMR spectra of P1, P2, and P3 in CDCl3 (400 MHz).

Table 1.
Characteristics of P1, P2, and P3.
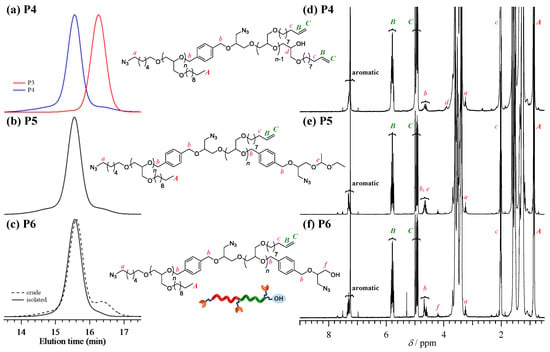
Figure 2.
(a–c) SEC traces of P4 (red line, first polymerization of M1; blue line, after the block copolymerization of M2), P5, and P6 (dotted line, before purification; solid line, after purification using preparative SEC). (d–f) 1H NMR spectra of P1, P2, and P3 in CDCl3 (400 MHz).
Synthesis of triazido-hydroxyl poly(M1)-b-poly(M2) (P6). After rigorous dehydration, P3 was utilized as a macroinitiator for the synthesis of poly(M1)-b-poly(M2) (P4; Mn,NMR = 14,600 g·mol−1, DP1/DP2 = 33/33, Đ = 1.10). The t-Bu-P4-catalyzed ROP of dec-9-enyl glycidyl ether (M2) with P3 macroinitiator was carried out at the [M2]0/[P3]0/[t-Bu-P4] ratio of 33/1/1 to obtain P4 in 84.7% yield. The SEC trace of P3 shifted to the higher molecular weight region after polymerization (Figure 2a), which confirmed that the polymerization reaction was initiated from the hydroxyl group of P3. The 1H NMR spectrum of P3 showed characteristic signals of both the poly(M2) and poly(M1) backbones, verifying successful post polymerization (Figure 2d). In a similar fashion to the synthesis of P3, P4 was treated with T1 in the presence of sodium hydride, and the ethoxyethyl group was deprotected under acidic conditions to give triazido-hydroxyl poly(M1)-b-poly(M2) (P6). It should be noted that a non-negligible amount of a lower molecular weight byproduct was observed in the SEC trace of crude P6 (Figure 2c). It was expected that the residue of macroinitiator P3, which was not completely deprotected, would correspond to this shoulder peak. According to the SEC profile, 5.0% of the macroinitiator did not participate in the polymerization reaction (Figure 2a). To remove the unreacted macroinitiator, the crude product was subjected to preparative SEC and pure P6 was isolated in 34.5% yield (Mn,NMR = 14,400 g·mol−1, DP1/DP2 = 33/33, Đ = 1.04) (Table 2).
Table 2.
Characteristics of P4, P5, and P6.
Synthesis of triazido-triethynylpoly(M1)-b-poly(M2)-b-poly(M3) (P8). In a similar fashion to the synthesis of P4, the t-Bu-P4-catalyzed ROP of 2-(2-(2-methoxyethoxy) ethoxy) ethyl glycidyl ether (M3) was carried out at the [M3]0/[P6]0/[t-Bu-P4] ratio of 33/1/1 using P6 as a macroinitiator for the synthesis of poly(M1)-b-poly(M2)-b-poly(M3) (P7). The success of the extension of a poly(M3) block from the macroinitiator P6 was confirmed by the fact that the elution peak maximum of P6 shifted to the higher molecular weight region (Figure 3a). However, the SEC trace of the obtained crude product P7 showed a non-negligible amount of higher and lower molecular weight byproducts, along with the main product. The population of the higher and lower molecular weight byproducts was calculated to be 4.4% and 15.2%, respectively, based on the SEC elution peak area. The low molecular weight byproduct corresponded to the poly(M3) homopolymer that was produced by the ROP of M3 initiated from water contaminant in the monomer or macroinitiator. On the other hand, the high molecular weight byproduct was possibly one of the intermolecularly cross-linked products formed through the reaction between the growing oxyanion and side chain olefin [30]. Thus, the crude product was purified by preparative SEC and pure P7 was isolated in 80.4% yield (Mn,NMR = 19,600 g·mol−1, DP1/DP2/DP3 = 33/33/25, Đ = 1.03) (Table 3). After purification, P7 was treated with an excess amount of 5-(bromomethyl)-1,2,3-tris(prop-2-yn-1-yloxy)benzene (T2) in the presence of sodium hydride to obtain triazido-triethynyl poly(M1)-b-poly(M2)-b-poly(M3) (P8; Mn,NMR = 19,900 g·mol−1, DP1/DP2/DP3 = 33/33/25, Đ = 1.04). The proton signal corresponding to the terminal alkynes (protons d and e) was observed at 2.44–2.64 ppm in the 1H NMR spectrum (Figure 3d), and the quantitative introduction of the propargyl groups was verified by comparing the peak areas of the ethynyl protons d and e (2.44–2.64 ppm) with the benzyl proton b (4.45 ppm).
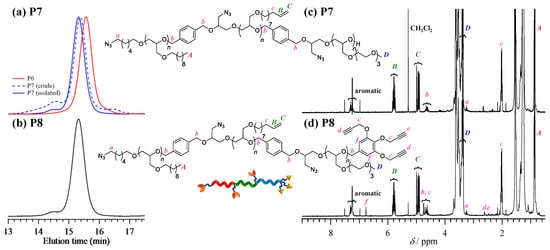
Figure 3.
(a,b) SEC traces of P7 (dotted line, before purification; solid line, after purification using preparative SEC) and P8. (c,d) 1H NMR spectra of P7 and P8 in CDCl3 (400 MHz).
Table 3.
Characteristics of P7 and P8.
Synthesis ofμ-ABC tricyclic miktoarm star polymer (P9). Finally, the intramolecular multiple click cyclization of P8 was performed to obtain the target μ-ABC tricyclic miktoarm star polymer (P9) using the CuBr/N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDETA) catalyst system in DMF at 100 °C. To avoid the intermolecular click reaction, the slow addition technique was employed. Thus, the P8 solution in DMF (19.1 mg·mL−1) was added slowly to the catalyst solution using the syringe pump at the rate of 0.3 mL·h−1. After complete addition, the reaction was continued for another 24 h at 100 °C. Finally, an alkyne-functionalized Wang resin was added to the reaction mixture, by which the unreacted P8 and any other possible byproducts possessing azido groups were removed by the click reaction. FT-IR analysis of the crude product obtained after the alumina column revealed the complete disappearance of the azido groups (Figure 4b). Notably, the absorption band corresponding to the side chain vinyl groups remained after the click reaction, which indicated that there was no significant side reaction. The crude product was then subjected to SEC analysis to confirm the progress of the cyclization reaction (Figure 4a). The elution peak maximum of the product was observed in the lower molecular weight region as compared to the linear precursor P8, which strongly supported the expected decrease in the hydrodynamic volume by the intramolecular cyclization reaction. On the other hand, small broad peaks were visible in the higher molecular weight region, which could be attributed to the oligomeric byproducts formed by the intermolecular click reaction. The population of the intramolecularly cyclized product was calculated to be 85.7% based on the SEC elution peak area. Further purification was then performed by the preparative SEC to remove the high molecular weight byproducts, giving pure product in 53.3% yield. The isolated product displayed a unimodal SEC trace with the Đ value of 1.02 (Figure 4a). The ratio between the Mn,SECs at the SEC peak top of P9 and P8, that is, Mn,p(P9)/Mn,p(P8) = <G>, was calculated to be 0.79 (Table 4). In the 1H NMR spectrum, new signals (a′, 4.22–4.34 ppm; c′, 5.05−5.26 ppm; d′, 7.64–7.94 ppm in Figure 5a,b) assignable to the triazole rings formed by the click reaction appeared, while the signals corresponding to the ethynyl and methylene groups adjacent to the azido groups completely disappeared. As results of SEC, FT-IR, and 1H NMR analyses all indicated a successful intramolecular click reaction, the product was identified as P9. On the basis of end group analysis by 1H NMR, the Mn,NMR and DP1/DP2/DP3 were calculated to be 20,100 g·mol−1 and 33/33/25, respectively.
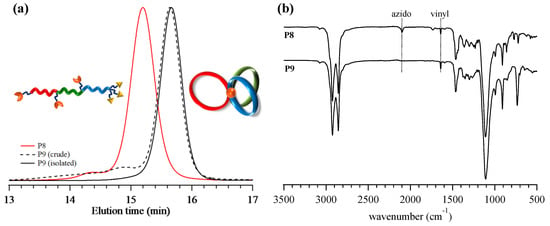
Figure 4.
(a) SEC traces of P8 (red line) and P9 (dotted line, before purification; solid line, after purification using preparative SEC). (b) FT-IR spectra of P8 (upper) and P9 (lower).
Table 4.
Characteristics of P8 and P9.
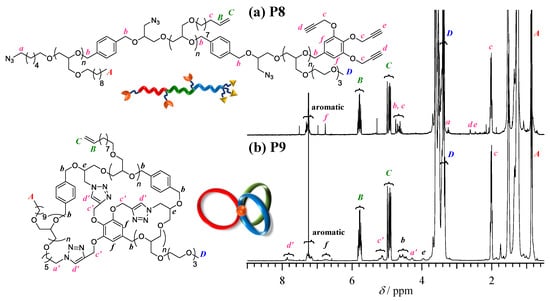
Figure 5.
(a,b) 1H NMR spectra of P8 and P9 in CDCl3 (400 MHz).
4. Conclusions
A new synthetic strategy for the μ-ABC tricyclic miktoarm star polymer comprising three different cyclic units of polyethers, namely, poly(decyl glycidyl ether), poly(dec-9-enyl glycidyl ether), and poly[2-(2-(2-methoxyethoxy) ethoxy) ethyl glycidyl ether], has been developed. The t-Bu-P4-catalyzed ROP of glycidyl ethers was employed for the preparation of a clickable linear triblock terpolymer precursor possessing three azido and three ethynyl groups at the selected positions. The intramolecular multiple click cyclization of the linear precursors successfully produced the well-defined tricyclic triblock terpolymer with narrow dispersity in a reasonable yield. Given the functional group loading capacity of the poly(glycidyl ether), the present strategy can provide model polymers suitable for studying the topological effects on the triblock terpolymer self-assembly. Indeed, the poly(dec-9-enyl glycidyl ether) segment has a reactive olefinic side chain [31] that can be transformed into a variety of functionalities via thiol-ene reaction, epoxidation, hydroboration, and hydrosilylation. Such side chain modification would permit the present terpolymer to self-assemble into three-phase microphase-separated structures, which can be used as templates for constructing complex nanopatterns. Efforts toward the synthesis of a series of triblock terpolymers with various architectures, including linear, star, and cyclic structures, are currently underway in order to comprehensively understand the correlation between the macromolecular architecture and microphase separation in triblock terpolymer systems.
Author Contributions
T.S. conducted the polymer syntheses and authored the manuscript. T.I. and T.S. designed the experiments and assisted in writing the article. T.Y. and K.T. also contributed by co-authoring the manuscript. All authors contributed to the scientific discussions.
Funding
This work was financially supported by a MEXT Grant-in-Aid for Challenging Exploratory Research (16K140000) and a MEXT Grant-in-Aid for Scientific Research (B) (16H04152).
Acknowledgments
We thank Yusuke Satoh for technical assistance with the synthesis experiments.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kim, J.K.; Yang, S.Y.; Lee, Y.; Kim, Y. Functional nanomaterials based on block copolymer self-assembly. Prog. Polym. Sci. 2010, 35, 1325–1349. [Google Scholar] [CrossRef]
- Lodge, T.P. Block copolymers: Past successes and future challenges. Macromol. Chem. Phys. 2003, 204, 265–273. [Google Scholar] [CrossRef]
- Luo, M.; Epps, T.H., III. Directed block copolymer thin film self-assembly: Emerging trends in nanopattern fabrication. Macromolecules 2013, 46, 7567–7579. [Google Scholar] [CrossRef]
- Sun, H.; Kabb, C.P.; Dai, Y.; Hill, M.R.; Ghiviriga, I.; Bapat, A.P.; Sumerlin, B.S. Macromolecular metamorphosis via stimulus-induced transformations of polymer architecture. Nat. Chem. 2017, 9, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Honda, S.; Yamamoto, T.; Tezuka, Y. Topology-directed control on thermal stability: Micelles formed from linear and cyclized amphiphilic block copolymers. J. Am. Chem. Soc. 2010, 132, 10251–10253. [Google Scholar] [CrossRef] [PubMed]
- Poelma, J.E.; Ono, K.; Miyama, D.; Aida, T.; Satoh, K.; Hawker, C.J. Cyclic block copolymers for controlling feature sizes in block copolymer lithography. ACS Nano 2012, 6, 10845–10854. [Google Scholar] [CrossRef] [PubMed]
- Mogi, Y.; Kotsuji, H.; Kaneko, Y.; Mori, K.; Matsushita, Y.; Noda, I. Preparation and morphology of triblock copolymers of the ABC type. Macromolecules 1992, 25, 5408–5411. [Google Scholar] [CrossRef]
- Mogi, Y.; Nomura, M.; Kotsuji, H.; Ohnishi, K.; Matsushita, Y.; Noda, I. Superlattice structures in morphologies of the ABC triblock copolymers. Macromolecules 1994, 27, 6755–6760. [Google Scholar] [CrossRef]
- Schacher, F.; Walther, A.; Ruppel, M.; Drechsler, M.; Müller, A.H.E. Multicompartment core micelles of triblock terpolymers in organic media. Macromolecules 2009, 42, 3540–3548. [Google Scholar] [CrossRef]
- Gröschel, A.H.; Schacher, F.H.; Schmalz, H.; Borisov, O.V.; Zhulina, E.B.; Walther, A.; Müller, A.H.E. Precise hierarchical self-assembly of multicompartment micelles. Nat. Commun. 2012, 3, 710. [Google Scholar] [CrossRef] [PubMed]
- Löbling, T.I.; Borisov, O.; Haataja, J.S.; Ikkala, O.; Gröschel, A.H.; Müller, A.H.E. Rational design of ABC triblock terpolymer solution nanostructures with controlled patch morphology. Nat. Commun. 2016, 7, 12097. [Google Scholar] [CrossRef] [PubMed]
- Gemma, T.; Hatano, A.; Dotera, T. Monte Carlo simulations of the morphology of ABC star polymers using the diagonal bond method. Macromolecules 2002, 35, 3225–3237. [Google Scholar] [CrossRef]
- Takano, A.; Kawashima, W.; Noro, A.; Isono, Y.; Tanaka, N.; Dotera, T.; Matsushita, Y. A mesoscopic archimedean tiling having a new complexity in an ABC star polymer. Polymer Sci. 2005, 43, 2427–2432. [Google Scholar] [CrossRef]
- Rho, Y.; Kim, C.; Higashihara, T.; Jin, S.; Jung, J.; Shin, T.J.; Hirao, A.; Ree, M. Complex self-assembled morphologies of thin films of an asymmetric A3B3C3 star polymer. ACS Macro Lett. 2013, 2, 849–855. [Google Scholar] [CrossRef]
- Ruiz, R.; Dobisz, E.; Albrecht, T.R. Rectangular patterns using block copolymer directed assembly for high bit aspect ratio patterned media. ACS Nano 2011, 5, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.-B.; Choi, H.K.; Hannon, A.F.; Alexander-Katz, A.; Ross, C.A.; Berggren, K.K. Design rules for self-assembled block copolymer patterns using tiled templates. Nat. Commun. 2014, 5, 3305. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, G.; Bilalis, P.; Hadjichristidis, N. Well-defined cyclic triblock terpolymers: A missing piece of the morphology puzzle. ACS Macro Lett. 2016, 5, 1242–1246. [Google Scholar] [CrossRef]
- Jia, Z.; Lonsdale, D.E.; Kulis, J.; Monteiro, M.J. Construction of a 3-miktoarm star from cyclic polymers. ACS Macro Lett. 2012, 1, 780–783. [Google Scholar] [CrossRef]
- Huisgen, R.; Szemies, G.; Möbius, L. 1.3-dipolare cycloadditionen, XXXII kinetik der additionen organischer azide an CC-mehrfachbindungen. Chem. Ber. 1967, 100, 2494–2507. [Google Scholar] [CrossRef]
- Binder, W.H.; Sachsenhofer, R. ’Click’ chemistry in polymer and materials science. Macromol. Rapid Commun. 2007, 28, 15–54. [Google Scholar] [CrossRef]
- Kulis, J.; Bell, C.A.; Micallef, A.S.; Jia, Z.; Monteiro, M.J. Rapid, selective, and reversible nitroxide radical coupling (NRC) reactions at ambient temperature. Macromolecules 2009, 42, 8218–8227. [Google Scholar] [CrossRef]
- Isono, T.; Satoh, Y.; Miyachi, K.; Chen, Y.; Sato, S.-I.; Tajima, K.; Satoh, T.; Kakuchi, T. Synthesis of linear, cyclic, figure-eight-shaped, and tadpole-shaped amphiphilic block copolyethers via t-Bu-P4-catalyzed ring-opening polymerization of hydrophilic and hydrophobic glycidyl ethers. Macromolecules 2014, 47, 2853–2863. [Google Scholar] [CrossRef]
- Satoh, Y.; Matsuno, H.; Yamamoto, T.; Tajima, K.; Isono, T.; Satoh, T. Synthesis of well-defined three- and four-armed cage-shaped polymers via “topological conversion” from trefoil- and quatrefoil-shaped polymers. Macromolecules 2017, 50, 97–106. [Google Scholar] [CrossRef]
- Karuna, M.S.L.; Devi, B.L.A.P.; Prasad, P.S.S.; Prasad, R.B.N. Synthesis of sulfated sodium salts of 1-alkylamino-3-alkyloxy-2-propanols and N,N-Di-(2-hydroxy-3-alkyloxy propyl) alkylamines as potential surfactants. J. Surfact. Deterg. 2009, 12, 117–123. [Google Scholar] [CrossRef]
- Diaz-Quijada, G.A.; Farrahi, S.; Clarke, J.; Tonary, A.M.; Pezacki, J.P. Picoliter wells from selective growth of HEK293 cells on chemically modified PDMS surfaces. J. Biomater. Appl. 2007, 21, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Camponovo, J.; Ruiz, J.; Cloutet, E.; Astruc, D. New polyalkynyl dendrons and dendrimers: “Click” chemistry with azidomethylferrocene and specific anion and cation electrochemical sensing properties of the 1,2,3-Triazole-containing dendrimers. Chem. Eur. J. 2009, 15, 2990–3002. [Google Scholar] [CrossRef] [PubMed]
- Malkoch, M.; Schleicher, K.; Drockenmuller, E.; Hawker, C.J.; Russell, T.P.; Fokin, V.V. Structurally diverse dendritic libraries: A highly efficient functionalization approach using click chemistry. Macromolecules 2005, 38, 3663–3678. [Google Scholar] [CrossRef]
- Rao, J.; Zhang, Y.; Zhang, J.; Liu, S.F. Facile preparation of well-defined AB2 Y-shaped miktoarm star polypeptide copolymer via the combination of ring-opening polymerization and click chemistry. BioMacromolecules 2008, 9, 2586–2593. [Google Scholar] [CrossRef] [PubMed]
- Satoh, Y.; Miyachi, K.; Matsuno, H.; Isono, T.; Tajima, K.; Kakuchi, T.; Satoh, T. Synthesis of well-defined amphiphilic star-block and miktoarm star copolyethers via t-Bu-P4-catalyzed ring-opening polymerization of glycidyl ethers. Macromolecules 2016, 49, 499–509. [Google Scholar] [CrossRef]
- Luo, C.; Bandar, J.S. Superbase-catalyzed anti-markovnikov alcohol addition reactions to aryl alkenes. J. Am. Chem. Soc. 2018, 140, 3547–3550. [Google Scholar] [CrossRef] [PubMed]
- Isono, T.; Lee, H.; Miyachi, K.; Satoh, Y.; Kakuchi, T.; Ree, M.; Satoh, T. Synthesis, thermal properties, and morphologies of amphiphilic brush block copolymers with tacticity-controlled polyether main chain. Macromolecules 2018, 51, 2939–2950. [Google Scholar] [CrossRef]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).