Hyperbranched Polysiloxanes Based on Polyhedral Oligomeric Silsesquioxane Cages with Ultra-High Molecular Weight and Structural Tuneability


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental
2.1. Materials and Methods
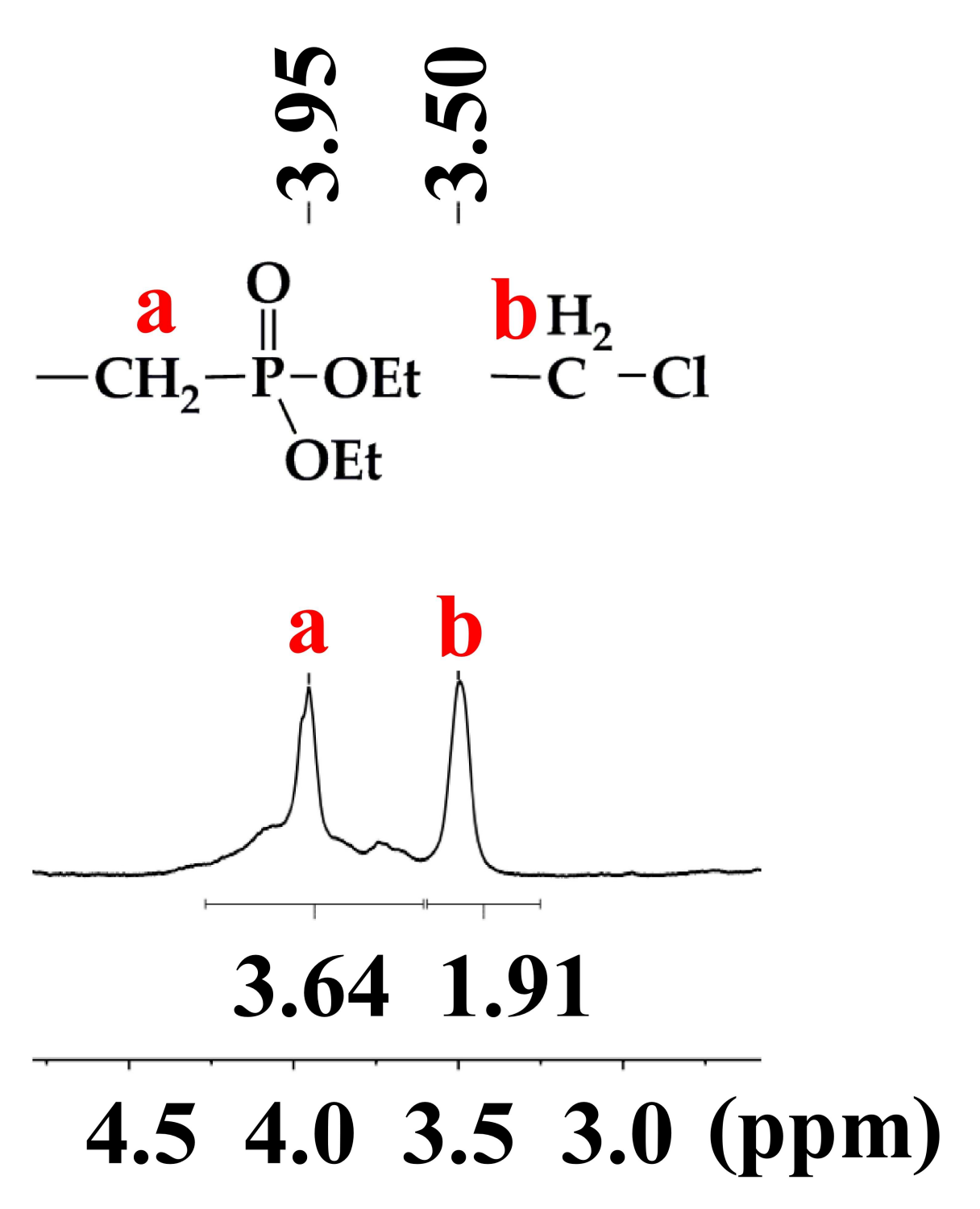
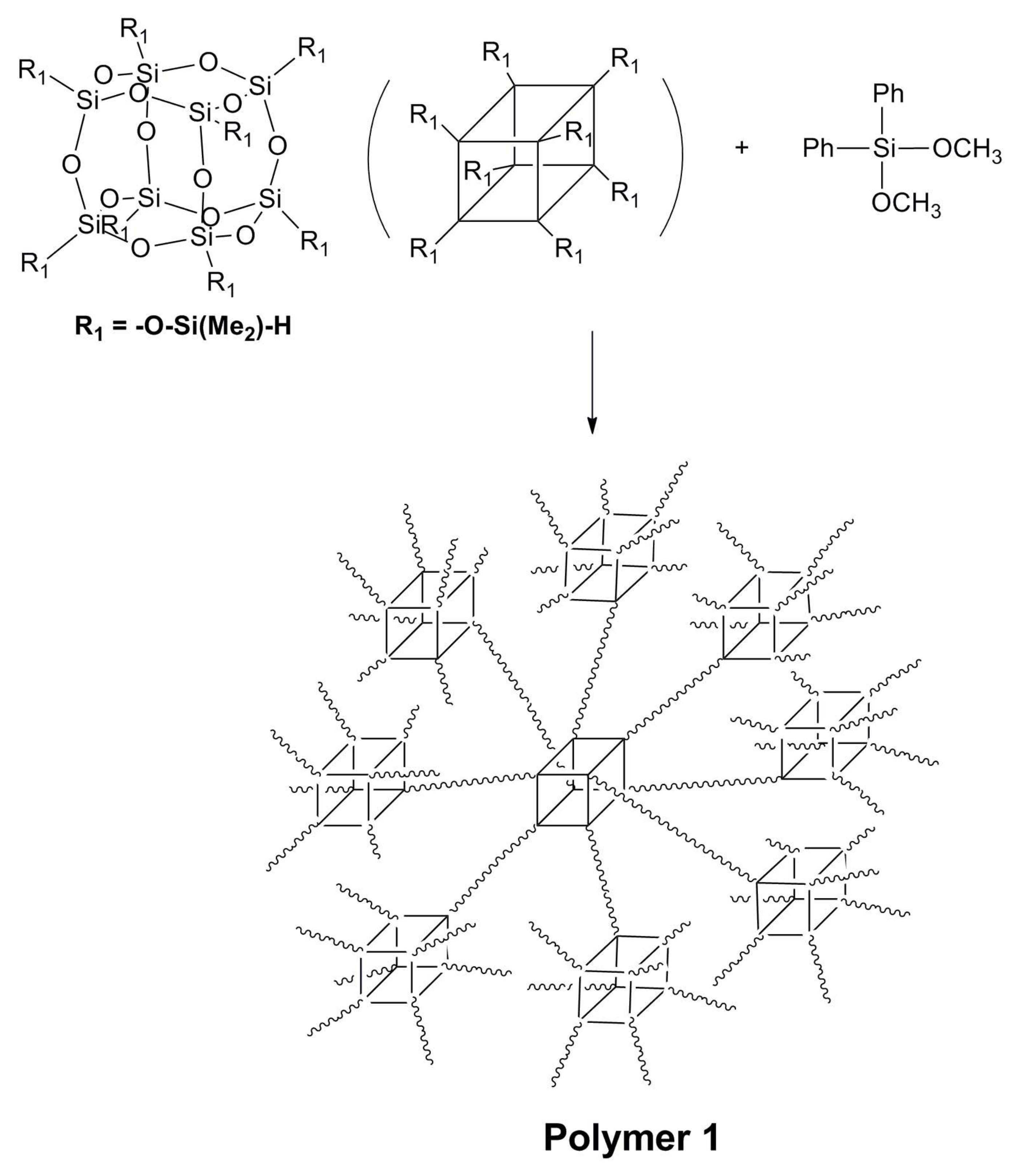
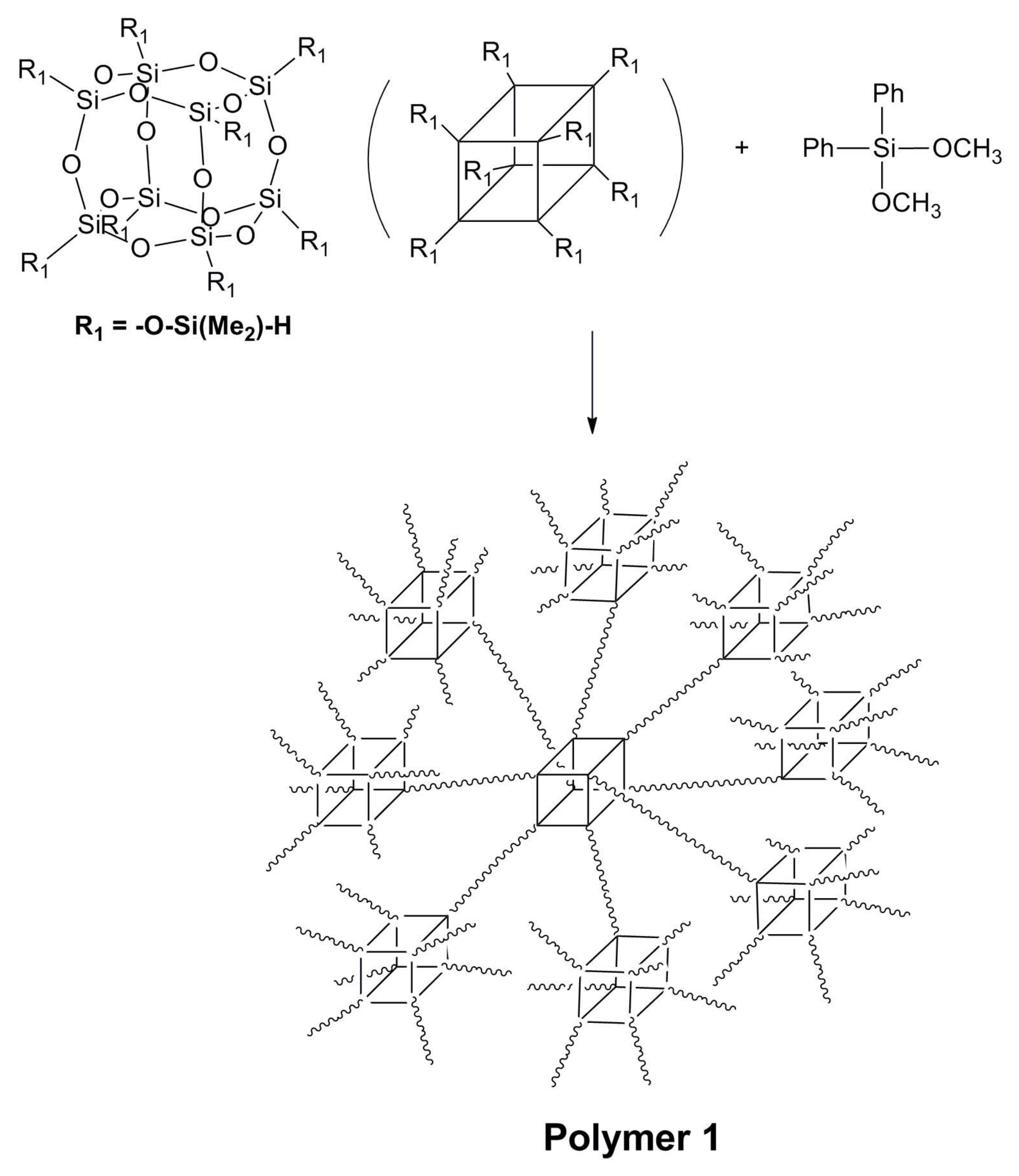
2.2. Synthesis of Polymer 1
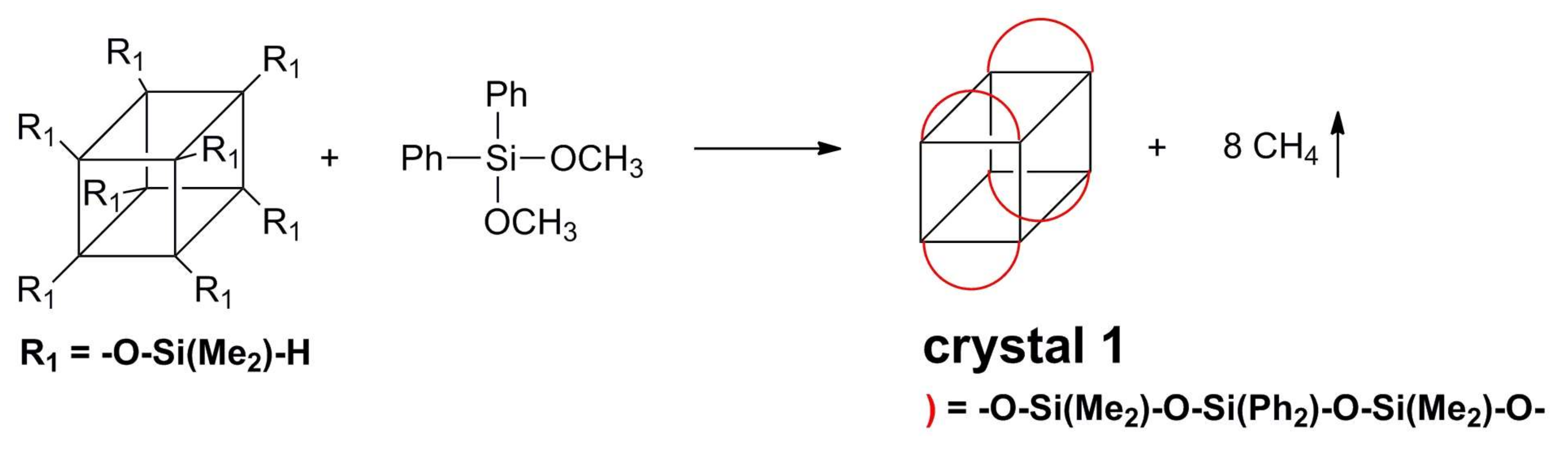
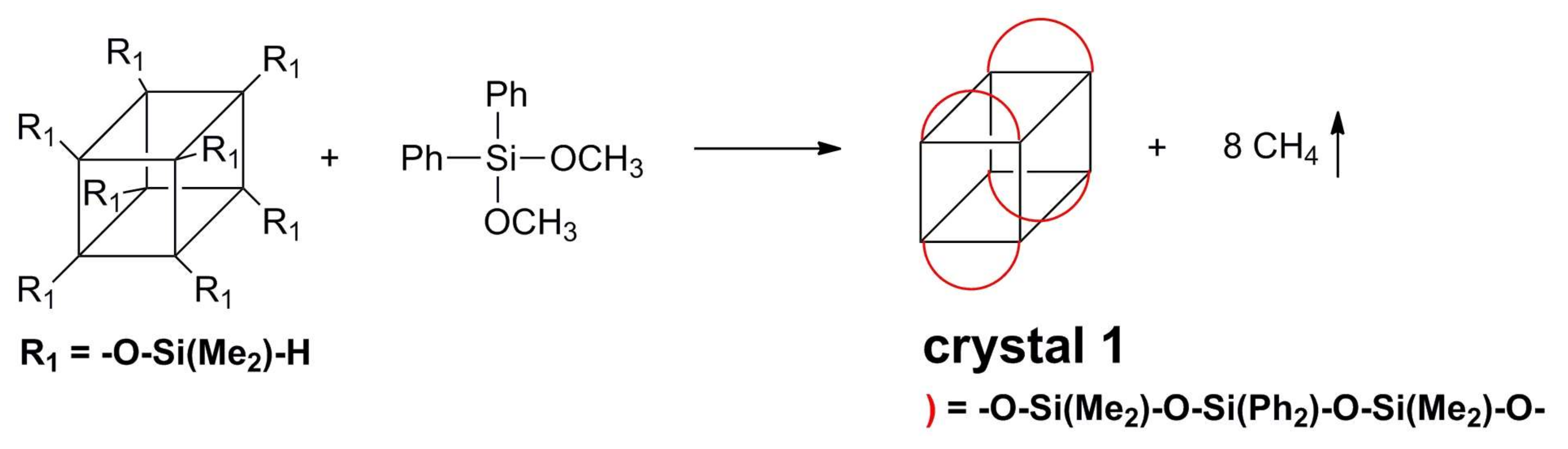
2.3. Synthesis of the Crystal 1
2.4. Synthesis of Polymer 2
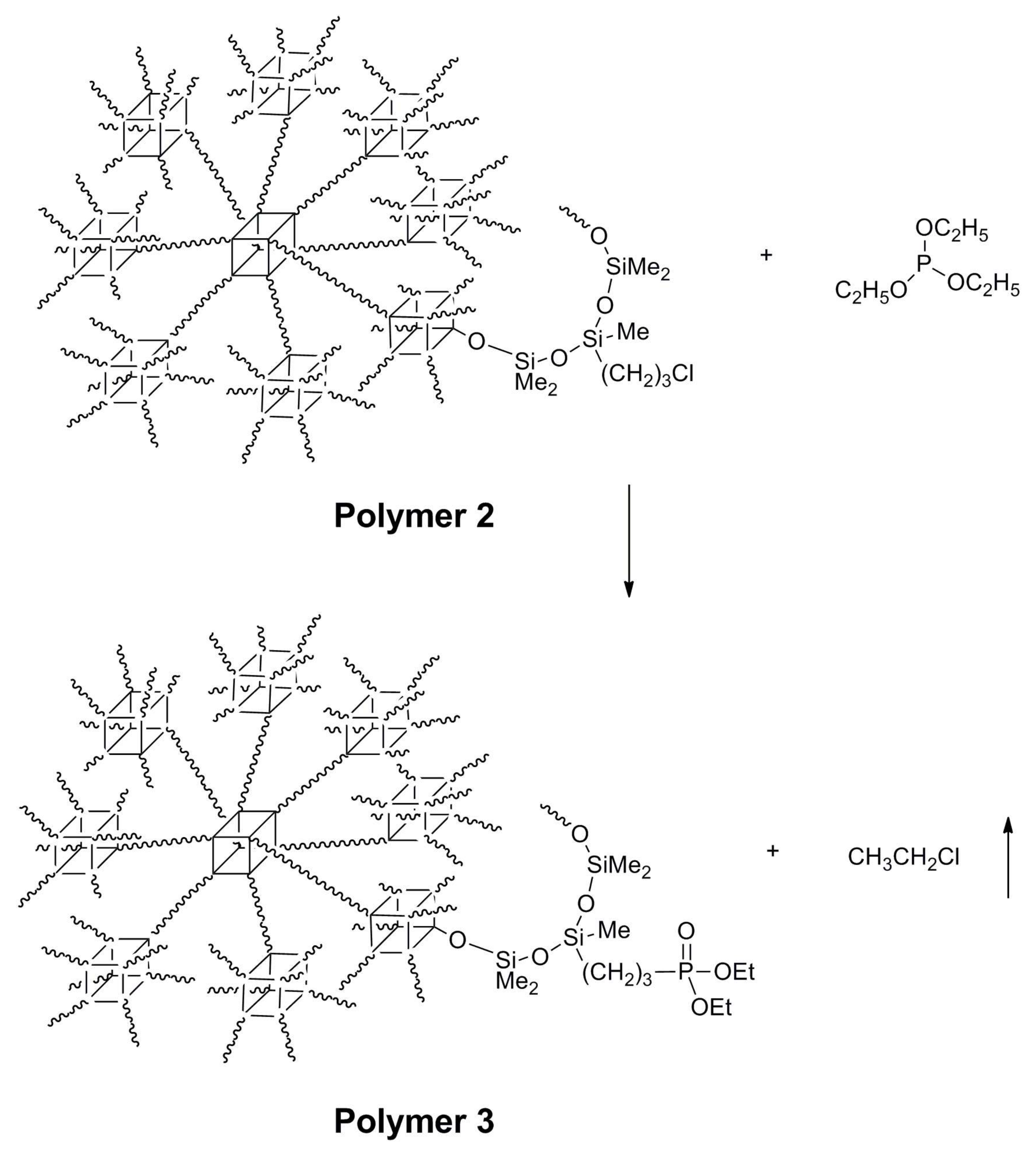
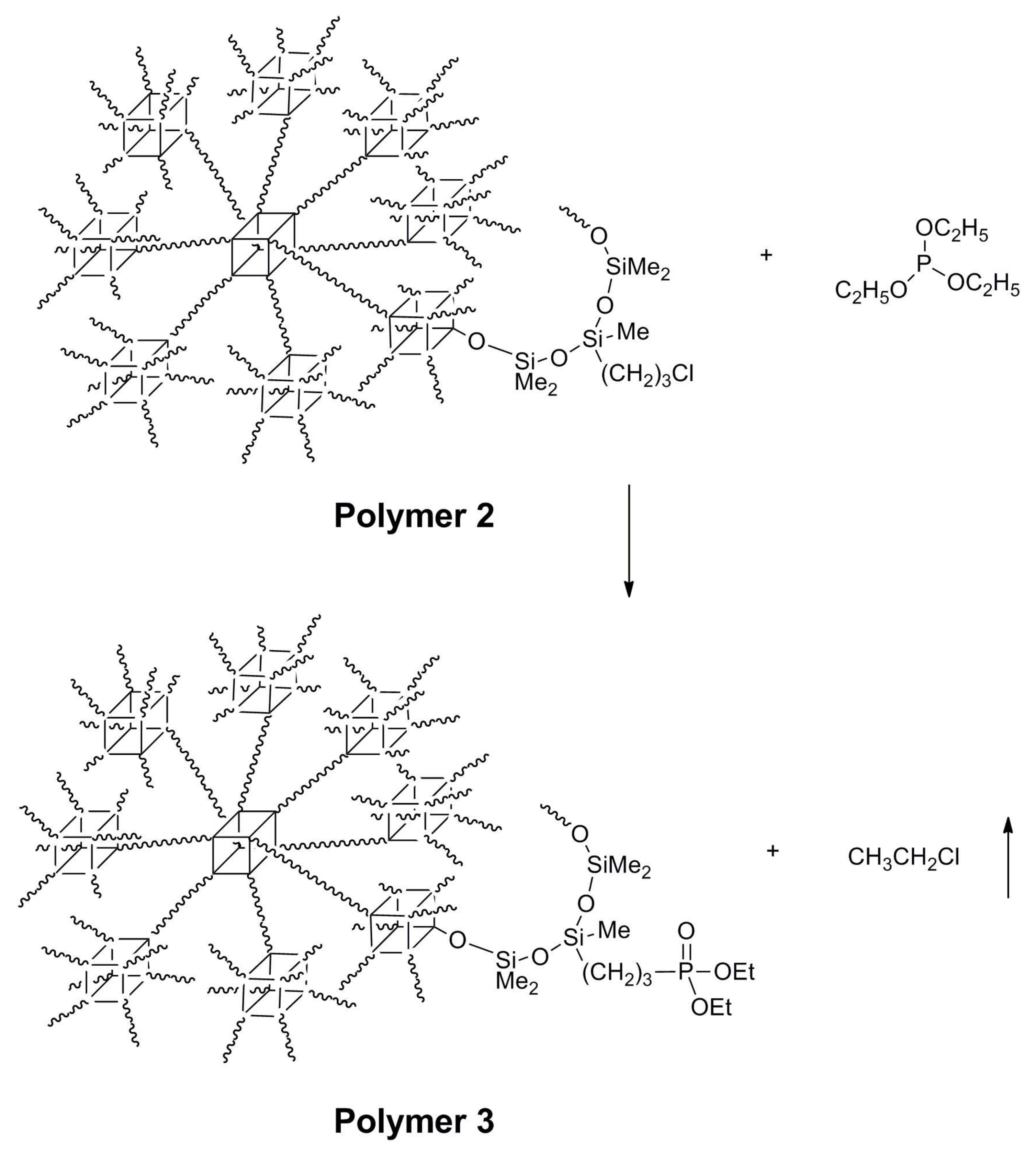
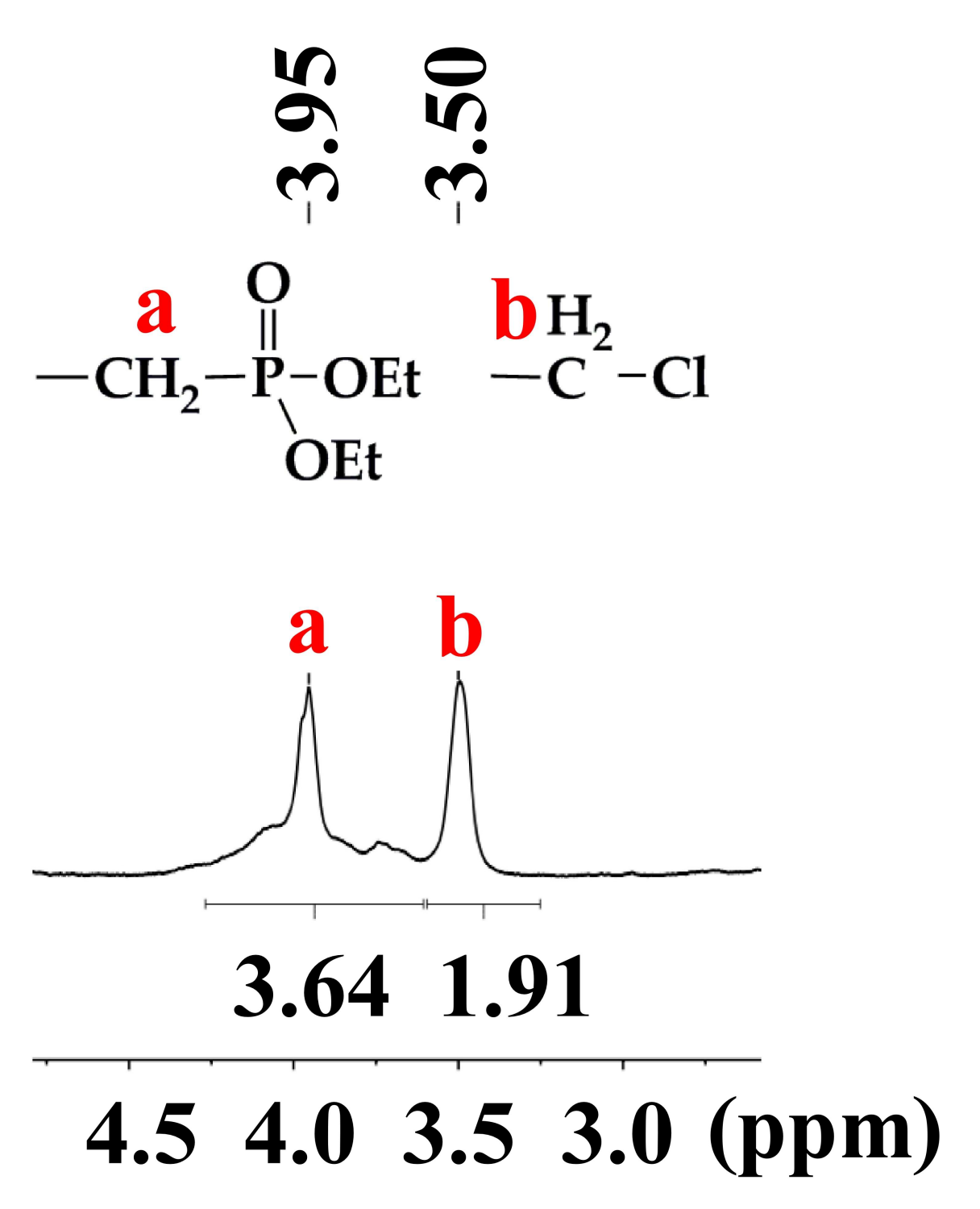
2.5. Synthesis of Polymer 3
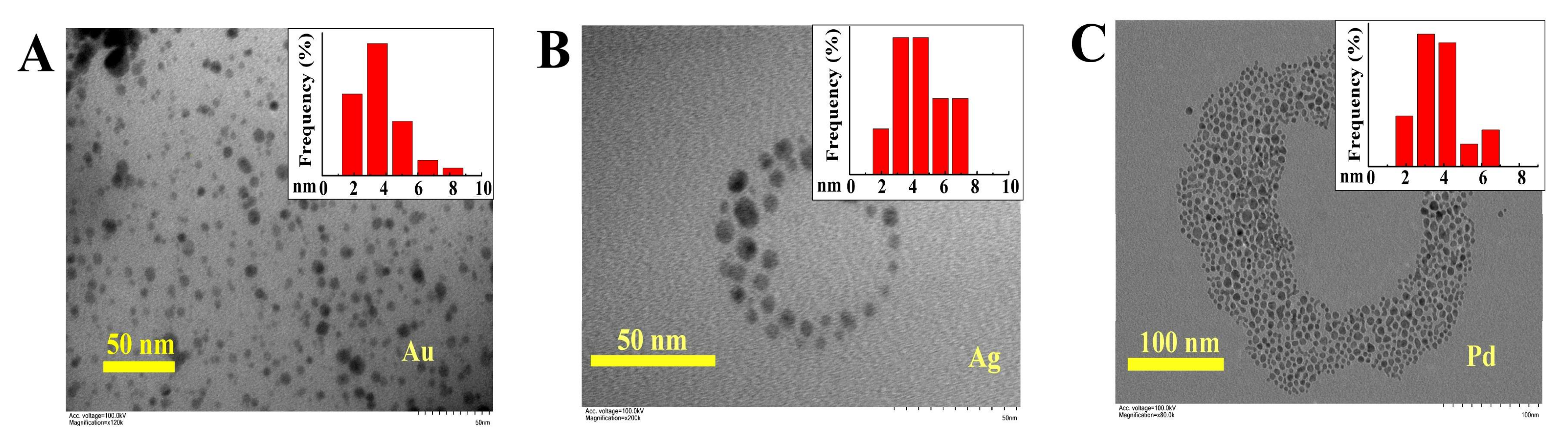
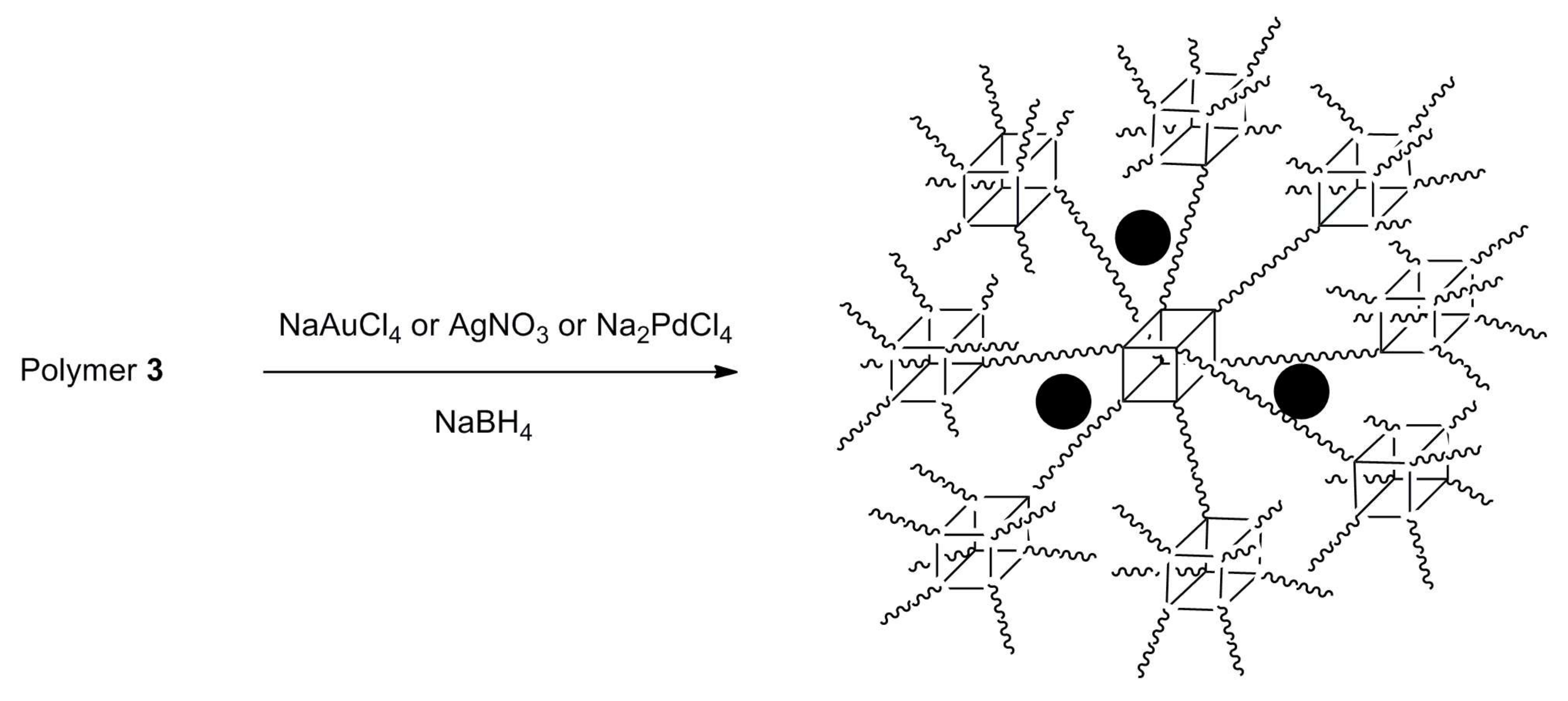
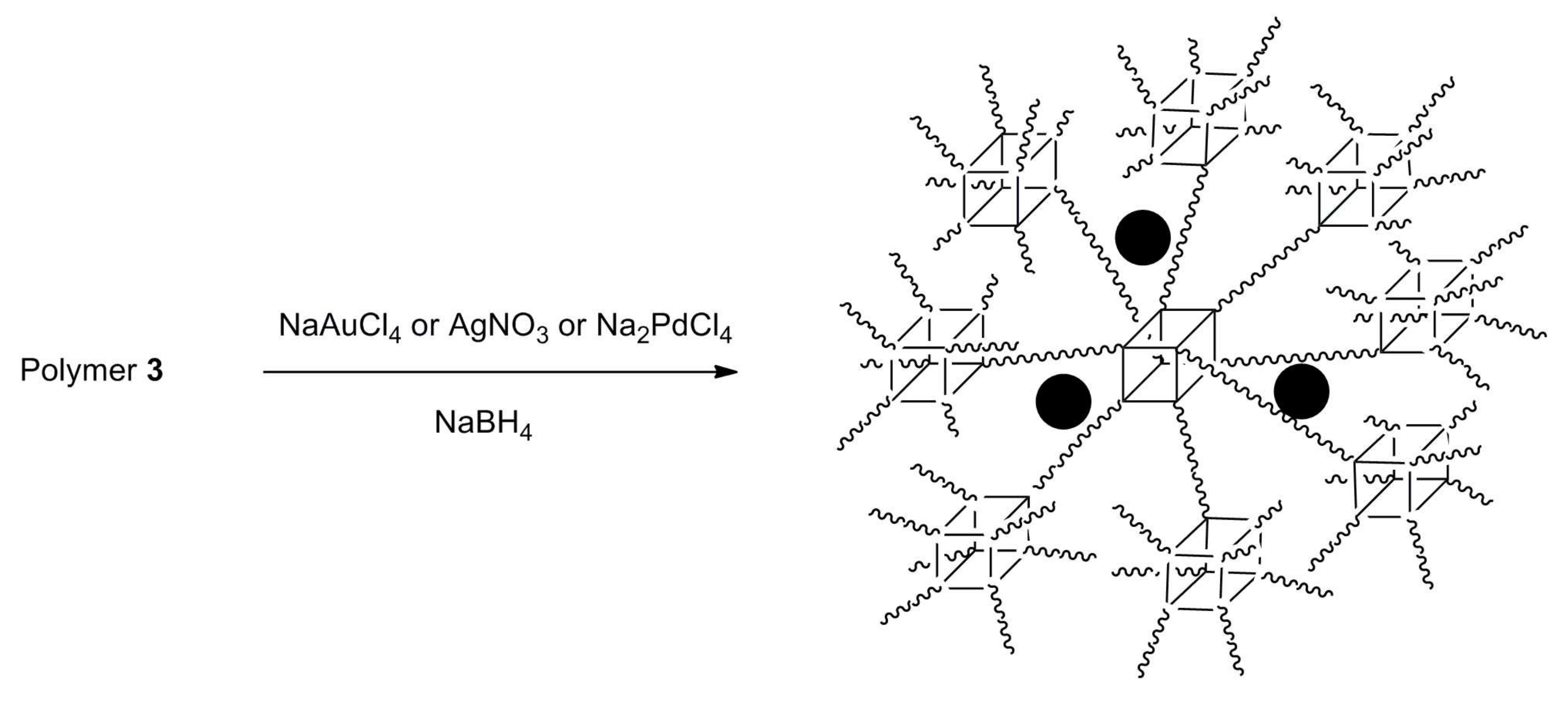
2.6. Transition Metal Nanoparticle Encapsulation by Polymer 3
3. Results and Discussion
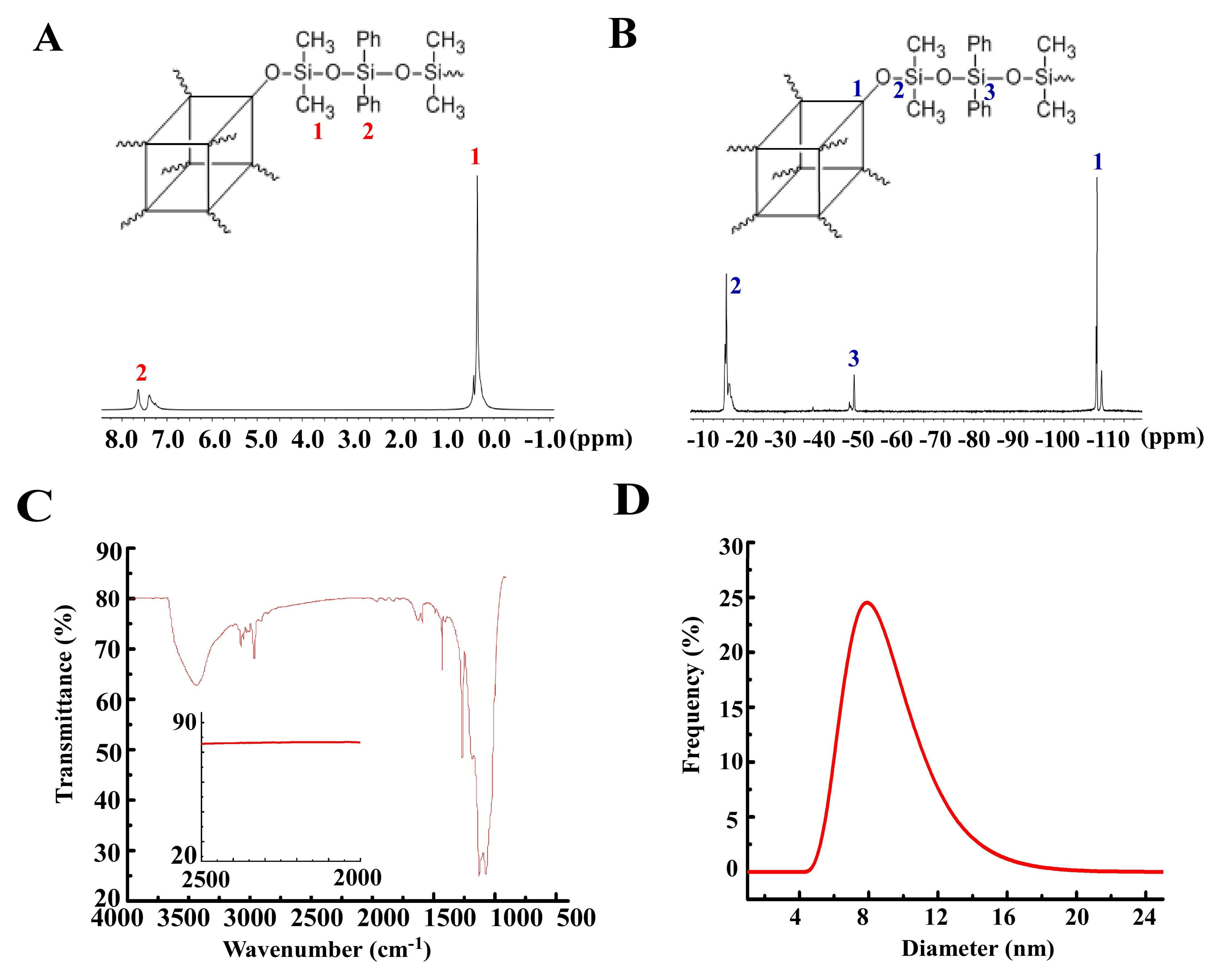
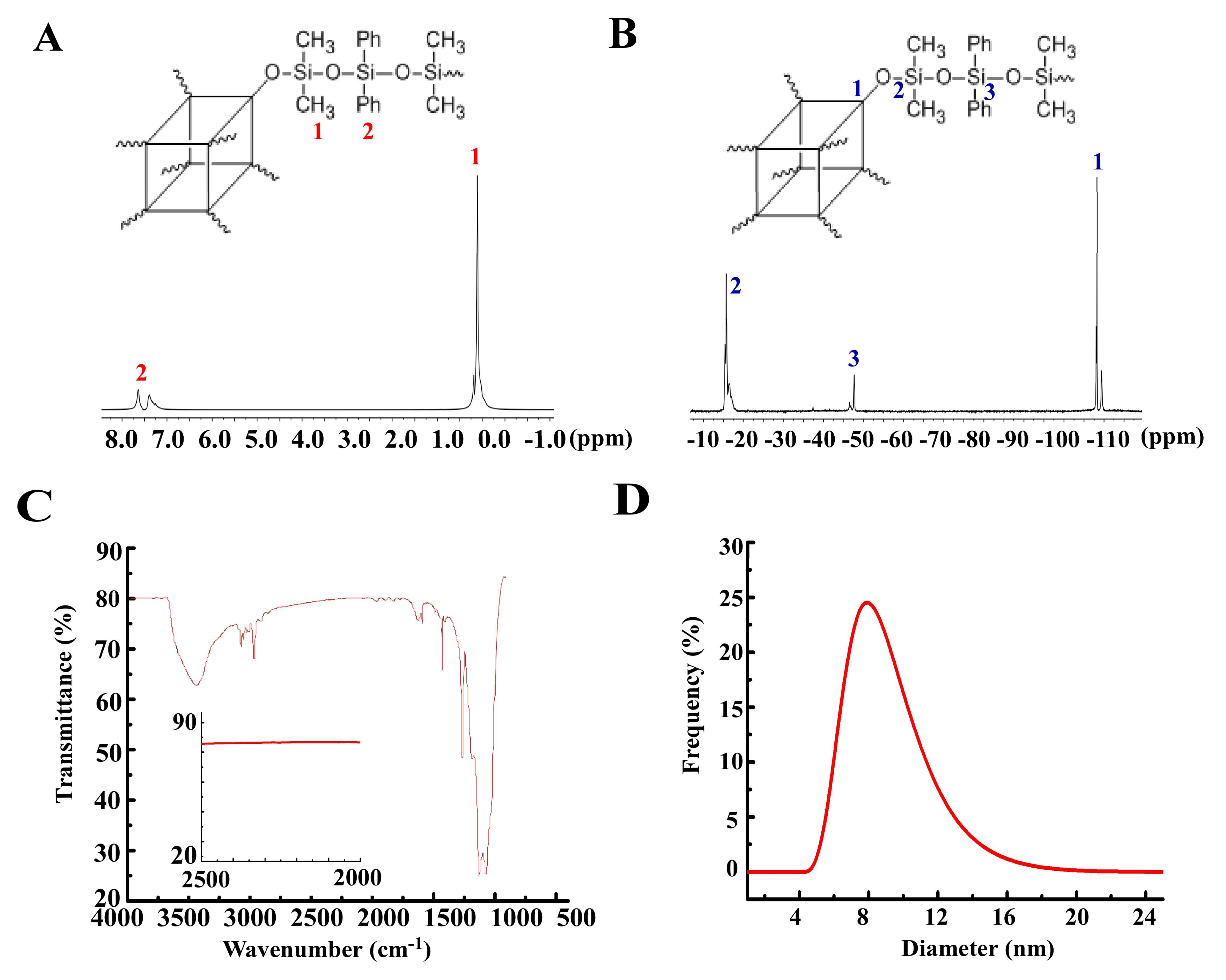
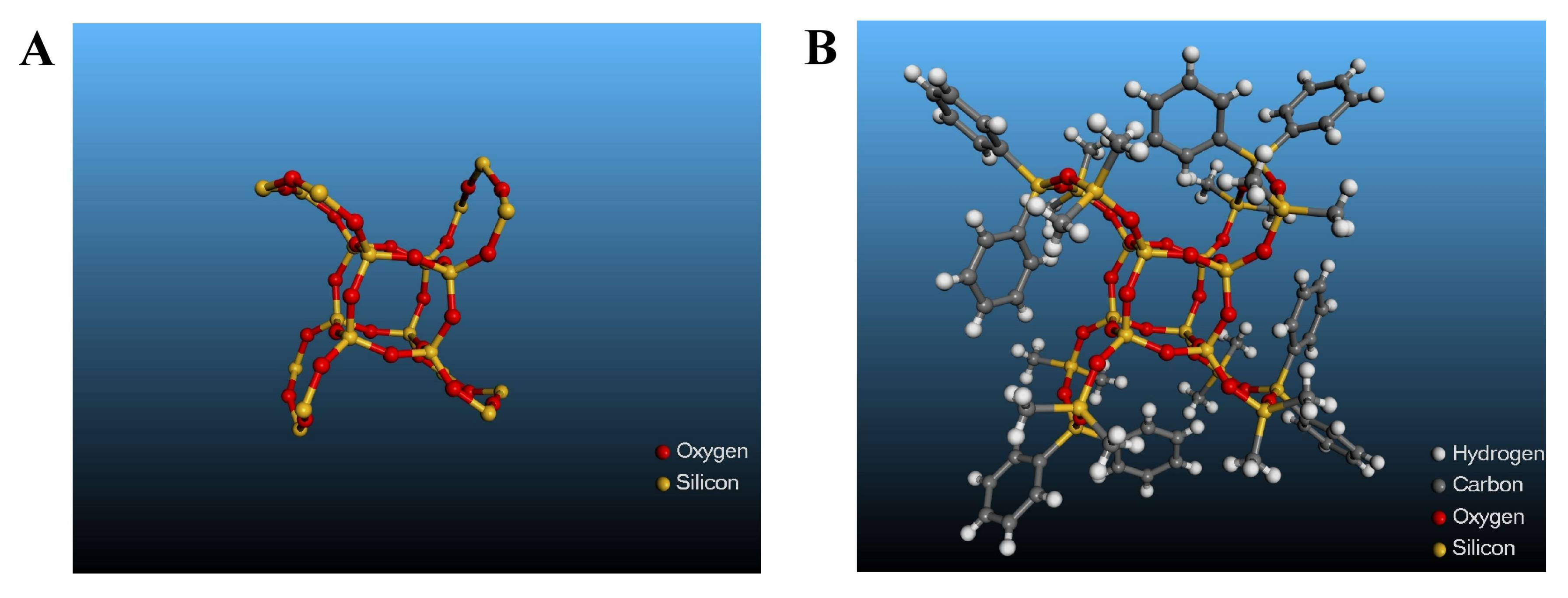
3.1. Synthesis of Polymer 1
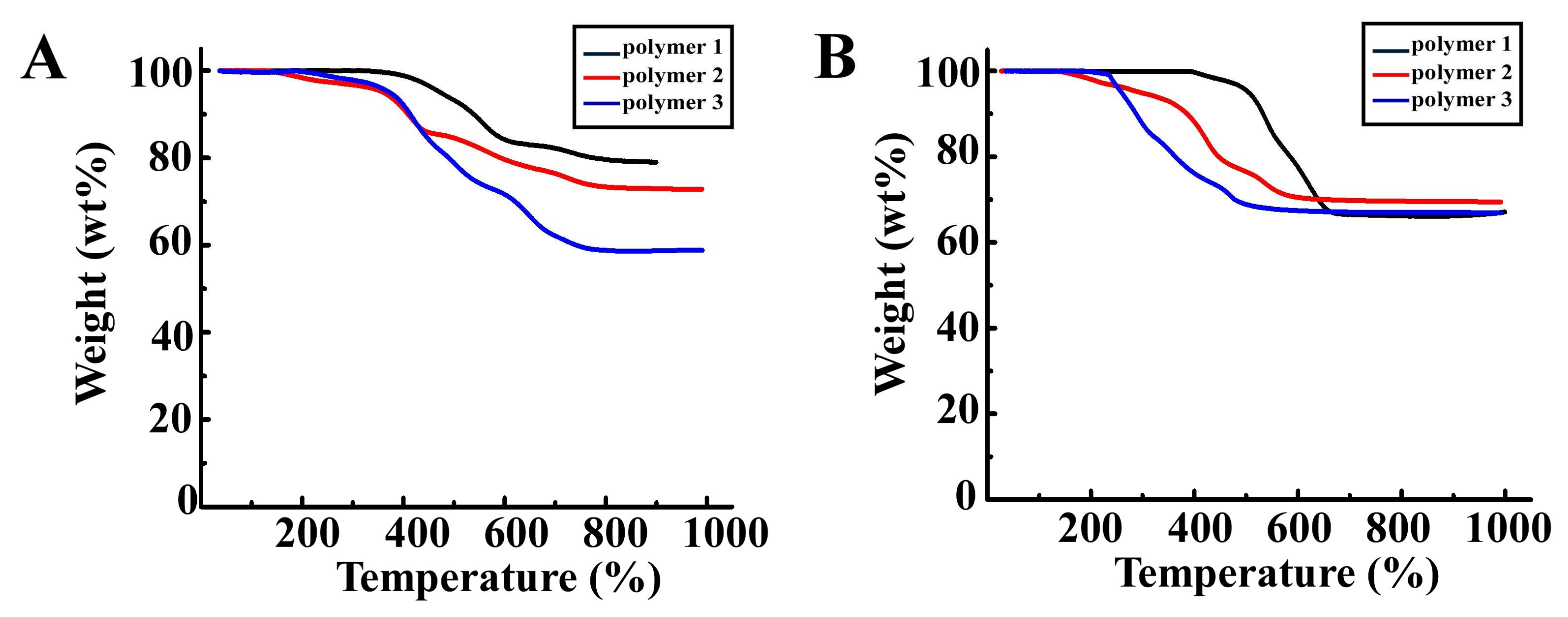
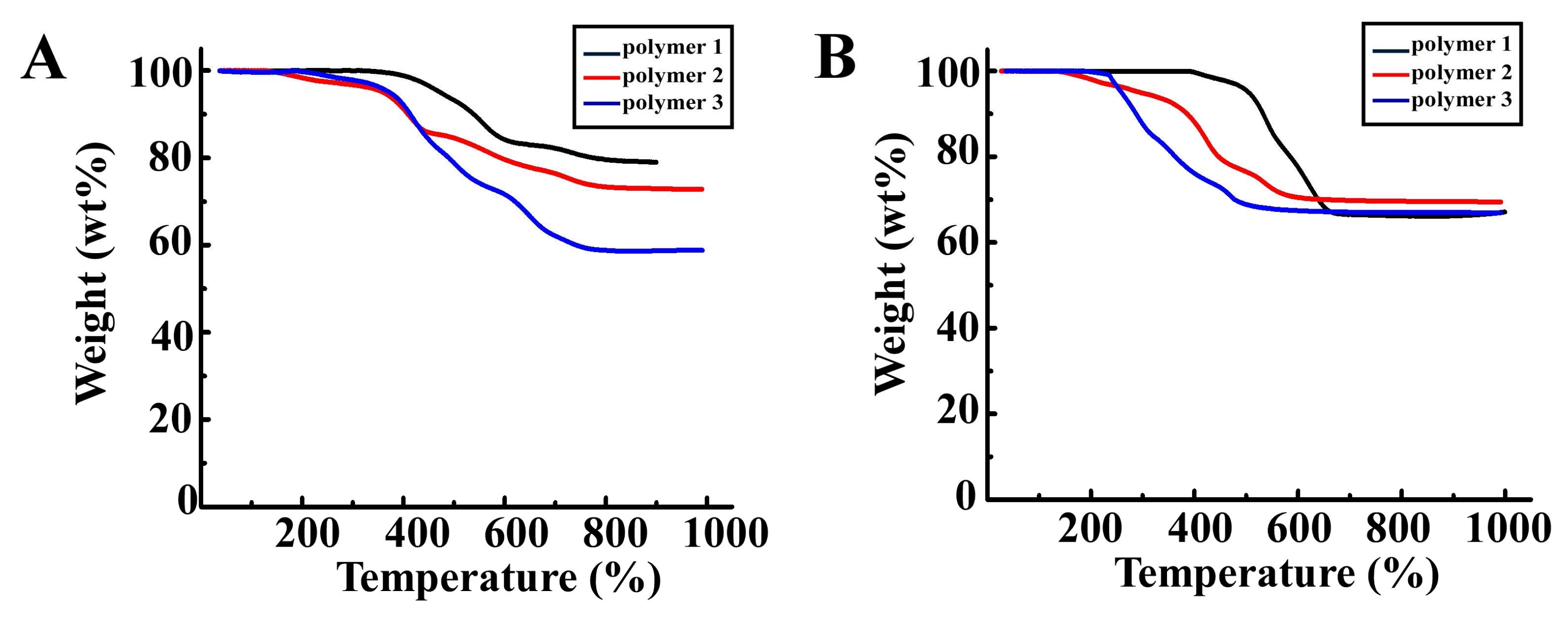
3.2. TGA of Polymer 1
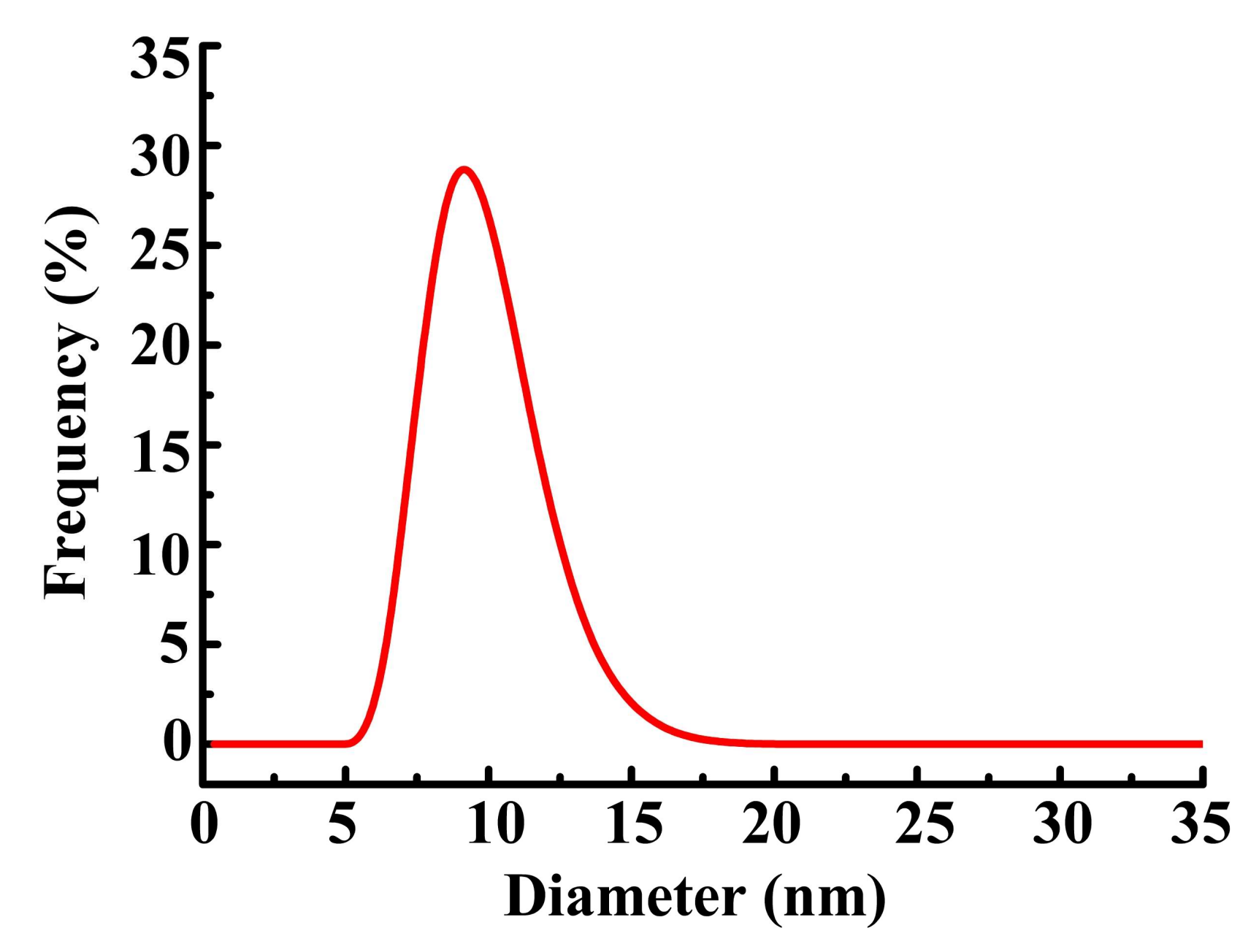
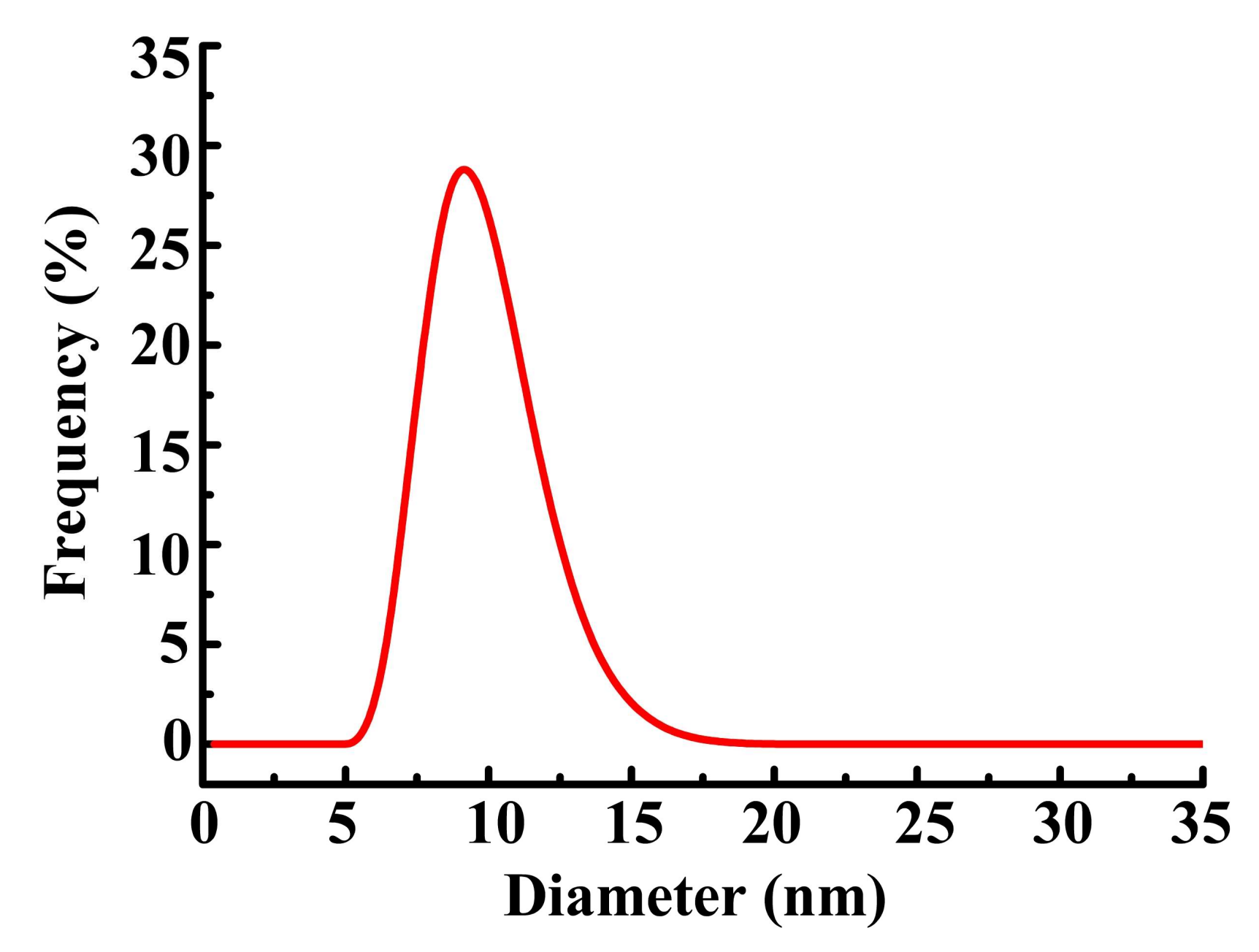
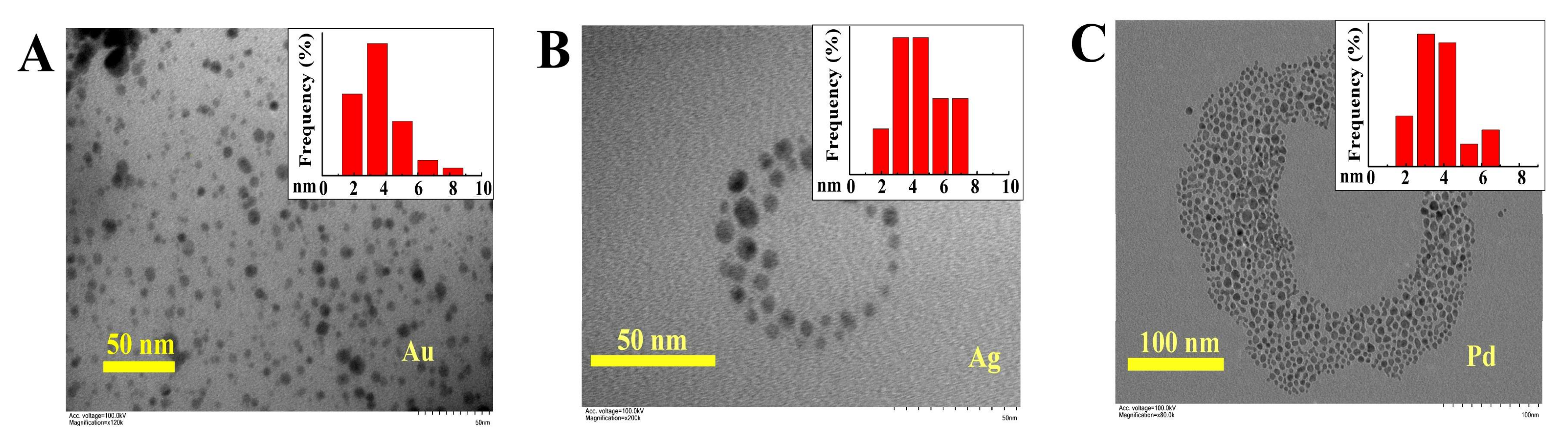
3.3. Functional Group Variety and Application in Encapsulation of Inorganic Nanoparticles
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Scott, D.W. Thermal rearrangement of branched-chain methylpolysiloxanes. J. Am. Chem. Soc. 1946, 68, 356–358. [Google Scholar] [CrossRef]
- Zheng, L.; Kasi, R.M.; Farris, R.J.; Coughlin, E.B. Synthesis and thermal properties of hybrid copolymers of syndiotactic polystyrene and polyhedral oligomeric silsesquioxane. J. Polym. Sci. Part A Polym. Chem. 2002, 40, 885–891. [Google Scholar] [CrossRef]
- Baney, R.H.; Itoh, M.; Sakakibara, A.; Suzuki, T. Silsesquioxanes. Chem. Rev. 1995, 95, 1409–1430. [Google Scholar] [CrossRef]
- Marcolli, C.; Calzaferri, G. Monosubstituted octasilasesquioxanes. Appl. Organomet. Chem. 1999, 13, 213–226. [Google Scholar] [CrossRef]
- Li, G.; Wang, L.; Ni, H.; Pittman, C.U. Polyhedral oligomeric silsesquioxane (POSS) polymers and copolymers: A review. J. Inorg. Organomet. Polym. 2001, 11, 123–154. [Google Scholar] [CrossRef]
- Tereshchenko, T.A. Synthesis and application of polyhedral oligosilsesquioxanes and spherosilicates. Polym. Sci. 2008, 50, 249–262. [Google Scholar] [CrossRef]
- Pielichowski, K.; Njuguna, J.; Janowski, B.; Pielichowski, J. Polyhedral oligomeric silsesquioxanes (POSS)-containing nanohybrid polymers. Adv. Polym. Sci. 2006, 201, 225–296. [Google Scholar] [CrossRef]
- Abe, Y.; Gunji, T. Oligo- and polysiloxanes. Prog. Polym. Chem. 2004, 29, 149–182. [Google Scholar] [CrossRef]
- Blanco, I.; Abate, L.; Bottino, F.A.; Bottino, P. Synthesis, characterization and thermal stability of new dumbbell-shaped isobutyl-substituted POSSs linked by aromatic bridges. J. Therm. Anal. Calorim. 2014, 117, 243–250. [Google Scholar] [CrossRef]
- Fu, B.X.; Hsiao, B.S.; Pagola, S.; Stephens, P.; White, H.; Rafailovich, M.; Sokolov, J.; Mather, P.T.; Jeon, H.G.; Phillips, S. Structural development during deformation of polyurethane containing polyhedral oligomeric silsesquioxanes (POSS) molecules. Polymer 2001, 42, 599–611. [Google Scholar] [CrossRef]
- Lee, A.; Lichtenhan, J.D. Thermal and viscoelastic property of epoxy–clay and hybrid inorganic–organic epoxy nanocomposites. J. Appl. Polym. Sci. 2015, 73, 1993–2001. [Google Scholar] [CrossRef]
- Haddad, T.S.; Lichtenhan, J.D. Hybrid organic−inorganic thermoplastics: Styryl-based polyhedral oligomeric silsesquioxane polymers. Macromolecules 1996, 29, 7302–7304. [Google Scholar] [CrossRef]
- Blanco, I.; Bottino, F.A. Effect of the substituents on the thermal stability of hepta cyclopentyl, phenyl substituted-polyhedral oligomeric silsesquioxane (hcp-POSS)/polystyrene (PS) nanocomposites. AIP Conf. Proc. 2012, 1459, 247–249. [Google Scholar] [CrossRef]
- Ramirez, S.M.; Diaz, Y.J.; Sahagun, C.M.; Duff, M.W.; Lawal, O.B.; Iacono, S.T.; Mabry, J.M. Reversible addition–fragmentation chain transfer (RAFT) copolymerization of fluoroalkyl polyhedral oligomeric silsesquioxane (F-POSS) macromers. Polym. Chem. 2013, 4, 2230–2234. [Google Scholar] [CrossRef]
- Ye, Y.S.; Shen, W.C.; Tseng, C.Y.; Rick, J.; Huang, Y.J.; Chang, F.C.; Hwang, B.J. Versatile grafting approaches to star-shaped POSS-containing hybrid polymers using RAFT polymerization and click chemistry. Chem. Commun. 2011, 47, 10656–10658. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Bernard, J.; Alcouffe, P.; Galy, J.; Dai, L. Nanostructured hybrid polymer networks from in situ self-assembly of RAFT-synthesized POSS-based block copolymers. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 4343–4352. [Google Scholar] [CrossRef]
- Zhang, Z.; Hong, L.; Gao, Y.; Zhang, W. One-pot synthesis of POSS-containing alternating copolymers by RAFT polymerization and their microphase-separated nanostructures. Polym. Chem. 2014, 5, 4534–4541. [Google Scholar] [CrossRef]
- Wang, W.; Ding, W.L.; Yu, J.; Fei, M.; Tang, J.Y. Synthesis and characterization of a novel POSS/PS composite via ATRP of branched functionalized POSS. J. Polym. Res. 2012, 19, 9948–9953. [Google Scholar] [CrossRef]
- Raus, V.; Čadová, E.; Starovoytova, L.; Janata, M. ATRP of POSS monomers revisited: Toward high-molecular weight methacrylate–POSS (co)polymers. Macromolecules 2014, 47, 7311–7320. [Google Scholar] [CrossRef]
- Xu, W.; Kwon, Y.; Chung, C. Synthesis of novel block copolymers containing polyhedral oligomeric silsesquioxane (POSS) pendent groups via ring-opening metathesis polymerization (ROMP). Polymer 2007, 48, 6286–6293. [Google Scholar] [CrossRef]
- Li, L.; Zhang, C.; Zheng, S. Synthesis of POSS-terminated polycyclooctadiene telechelics via ring-opening metathesis polymerization. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 223–233. [Google Scholar] [CrossRef]
- Gnanasekaran, D.; Reddy, B.S.R. Synthesis and characterization of nanocomposites based on copolymers of POSS-ONDI macromonomer and TFONDI: Effect of POSS on thermal, microstructure and morphological properties. Adv. Mater. Res. 2010, 123–125, 775–778. [Google Scholar] [CrossRef]
- Wang, J.; Sun, J.; Zhou, J.; Jin, K.; Fang, Q. Fluorinated and thermo-crosslinked polyhedral oligomeric silsesquioxanes: New organic-inorganic hybrid materials for high performance dielectric application. ACS Appl. Mater. Interfaces 2017, 9, 12782–12790. [Google Scholar] [CrossRef] [PubMed]
- Naka, K.; Masuoka, S.; Shinke, R.; Yamada, M. Synthesis of first- and second-generation imidazole-terminated POSS-core dendrimers and their pH responsive and coordination properties. Polym. J. 2012, 44, 353–359. [Google Scholar] [CrossRef]
- Lichtenhan, J.D.; Otonari, Y.A.; Carr, M.J. Linear hybrid polymer building blocks: Methacrylate-functionalized polyhedral oligomeric silsesquioxane monomers and polymers. Macromolecules 1995, 28, 8435–8437. [Google Scholar] [CrossRef]
- Hirai, T.; Leolukman, M.; Hayakawa, T.; Kakimoto, M.; Gopalan, P. Hierarchical nanostructures of organosilicate nanosheets within self-organized block copolymer films. Macromolecules 2008, 41, 4558–4560. [Google Scholar] [CrossRef]
- Yu, C.B.; Ren, L.J.; Wang, W. Synthesis and self-assembly of a series of nPOSS-b-PEO block copolymers with varying shape anisotropy. Macromolecules 2017, 50, 3273–3284. [Google Scholar] [CrossRef]
- Pramudya, I.; Rico, C.G.; Lee, C.; Chung, H. POSS-containing bioinspired adhesives with enhanced mechanical and optical properties for biomedical applications. Biomacromolecules 2016, 17, 3853–3861. [Google Scholar] [CrossRef] [PubMed]
- Chaikittisilp, W.; Kubo, M.; Moteki, T.; Sugawaranarutaki, A.; Shimojima, A.; Okubo, T. Porous siloxaneorganic hybrid with ultrahigh surface area through simultaneous polymerizationdestruction of functionalized cubic siloxane cages. J. Am. Chem. Soc. 2011, 133, 13832–13835. [Google Scholar] [CrossRef] [PubMed]
- Nischang, I.; Brüggemann, O.; Teasdale, I. Facile, Single-step preparation of versatile, high-surface-area, hierarchically structured hybrid materials. Angew. Chem. Int. Ed. 2011, 50, 4592–4596. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Xue, L.; Li, L.; Deng, B.; Feng, S.; Liu, H.; Zhao, X. Rational design and synthesis of hybrid porous polymers derived from polyhedral oligomeric silsesquioxanes via heck coupling reactions. Macromol. Rapid Commun. 2013, 34, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Naka, K.; Irie, Y. Synthesis of single component element-block materials based on siloxane-based cage frameworks. Polym. Int. 2017, 66, 187–194. [Google Scholar] [CrossRef]
- Shioda, T.; Gunji, T.; Abe, N.; Abe, Y. Preparation and properties of polyhedral oligomeric silsesquioxane polymers. Appl. Organomet. Chem. 2011, 25, 661–664. [Google Scholar] [CrossRef]
- Gunji, T.; Igarashi, T.; Tsukada, S.; Abe, Y. Syntheses of cage octasilicate polymers. J. Sol-Gel Sci. Technol. 2017, 81, 21–26. [Google Scholar] [CrossRef]
- Lichtenhan, J.D.; Vu, N.Q.; Carter, J.A.; Gilman, J.W.; Feher, F.J. Silsesquioxane–siloxane copolymers from polyhedral silsesquioxane. Macromolecules 1993, 26, 2141–2142. [Google Scholar] [CrossRef]
- Wu, C.Y.; Liu, Y.Z. Hyperbranched polysiloxane with highly constrained rings and the effect of the attached arms on the assembly behavior. Polym. Chem. 2017, 8, 6490–6495. [Google Scholar] [CrossRef]
- Rubinsztajn, S.; Cella, J.A. A new polycondensation process for the preparation of polysiloxane copolymers. Macromolecules 2005, 38, 1061–1063. [Google Scholar] [CrossRef]
- Yu, J.Y.; Liu, Y.Z. Cyclic polysiloxanes with linked cyclotetrasiloxane subunits. Angew. Chem. Int. Ed. 2017, 56, 8706–8871. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.; Chen, T.; Hayes, R.; Dickie, T.; Urlicha, T.; Brook, M.A. Facile synthesis of dendron branched silicone polymers. Polym. Chem. 2017, 8, 2743–2746. [Google Scholar] [CrossRef]
- Park, S.H.; Liu, H.M.; Kleinsorge, M.; Grey, C.P.; Toby, B.H.; Parise, J.B. [Li–Si–O]-MFI: A new microporous lithosilicate with the MFI topology. Chem. Mater. 2004, 16, 2605–2614. [Google Scholar] [CrossRef]
- Mintcheva, N.; Tanabe, M.; Osakada, K.; Georgieva, I.; Mihailov, T.; Trendafilova, N. Synthesis and characterization of a dinuclear platinum complex with silsesquioxanate ligand. J. Organomet. Chem. 2010, 695, 1738–1743. [Google Scholar] [CrossRef]
- Malinovskii, S.T.; Tesuro Vallina, A.; Stoeckli-Evans, H. X-ray diffraction investigation of siloxanes. III. Structure and configuration of cyclic tetra- and pentasiloxanes bearing different organic substituents at silicon atoms. J. Struct. Chem. 2007, 48, 128–136. [Google Scholar] [CrossRef]
- Bassindale, A.R.; Liu, Z.H.; Parker, D.J.; Taylor, P.G.; Horton, P.N.; Hursthouse, M.B.; Light, M.E. The reactions of dialkyl and diarylethoxysilanes with T6 silsesquioxane cages: X-ray crystallographic studies of the mono-T6D1 and bis-T6D2 insertion ring expansion products. J. Organomet. Chem. 2003, 687, 1–11. [Google Scholar] [CrossRef]
- Janaa, R.N.; Mukundab, P.G.; Nandoa, G.B. Thermogravimetric analysis of compatibilized blends of low density polyethylene and poly(dimethyl siloxane) rubber. Polym. Degrad. Stab. 2003, 80, 75–82. [Google Scholar] [CrossRef]
- Fina, A.; Tabuani, D.; Carniato, F.; Frache, A.; Boccaleri, E.; Camino, G. Polyhedral oligomeric silsesquioxanes (POSS) thermal degradation. Thermochim. Acta 2006, 440, 36–42. [Google Scholar] [CrossRef]
- Blanco, I.; Abate, L.; Bottino, F.A. Mono substituted octaphenyl POSSs: The effects of substituents on thermal properties and solubility. Thermochim. Acta 2017, 655, 117–123. [Google Scholar] [CrossRef]
- Chiang, C.L.; Ma, C.C.M. Synthesis, characterization, and properties of novel ladderlike phosphorus-containing polysilsesquioxanes. J. Polym. Sci. Part A Polym. Chem. 2003, 41, 1371–1379. [Google Scholar] [CrossRef]
- Li, Z.Q.; Yang, R.J. Flame retardancy, thermal and mechanical properties of sulfonate-containing polyhedral oligomeric silsesquioxane (S-POSS)/polycarbonate composites. Polym. Degrad. Stab. 2015, 116, 81–87. [Google Scholar] [CrossRef]
- Sheen, Y.C.; Lu, C.H.; Huang, C.F.; Kuo, S.W.; Chang, F.C. Synthesis and characterization of amorphous octakis-functionalized polyhedral oligomeric silsesquioxanes for polymer nanocomposites. Polymer 2008, 49, 4017–4024. [Google Scholar] [CrossRef]
- Aryabadie, S.; Sadeghi-kiakhani, M.; Arami, M. Antimicrobial and dyeing studies of treated cotton fabrics by prepared chitosan-PAMAM dendrimer/Ag nanoemulsion. Fibers Polym. 2016, 16, 2529–2537. [Google Scholar] [CrossRef]
- Gao, L.; Kojima, K.; Nagashima, H. Transition metal nanoparticles stabilized by ammonium salts of hyperbranched polystyrene: Effect of metals on catalysis of the biphasic hydrogenation of alkenes and arenes. Tetrahedron 2015, 71, 6414–6423. [Google Scholar] [CrossRef]
- Tang, Q.; Cheng, F.; Lou, X.L.; Liu, H.J.; Chen, Y. Comparative study of thiol-free amphiphilic hyperbranched and linear polymers for the stabilization of large gold nanoparticles in organic solvent. J. Colloid Interface Sci. 2009, 337, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Jiang, X.S.; Yin, J. Responsive hybrid nanosheets of hyperbranched poly(ether amine) as a 2D-platform for metal nanoparticles. Chem. Commun. 2013, 49, 603–605. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P.L.; Chen, C.C.; Yuen, S.M. Protection effects of hydrophile-grafted silicone copolymers on the formation of colloidal silver nanoparticles. Macromolecules 2009, 42, 4937–4940. [Google Scholar] [CrossRef]
- Shankar, R.; Jangira, B.; Sharmaa, A. A novel synthetic approach to poly(hydrosiloxane)s via hydrolytic oxidation of primary organosilanes with a AuNPs-stabilized Pickering interfacial catalyst. RSC Adv. 2017, 7, 344–351. [Google Scholar] [CrossRef]
- Stochma, E.; Strzezik, J.; Krowiak, A. Preparation and characterization of polysiloxane networks containing metallic platinum particles. J. Appl. Polym. Sci. 2016, 133, 43096. [Google Scholar] [CrossRef]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, N.; Yu, J.; Meng, Y.; Liu, Y. Hyperbranched Polysiloxanes Based on Polyhedral Oligomeric Silsesquioxane Cages with Ultra-High Molecular Weight and Structural Tuneability. Polymers 2018, 10, 496. https://doi.org/10.3390/polym10050496
Liu N, Yu J, Meng Y, Liu Y. Hyperbranched Polysiloxanes Based on Polyhedral Oligomeric Silsesquioxane Cages with Ultra-High Molecular Weight and Structural Tuneability. Polymers. 2018; 10(5):496. https://doi.org/10.3390/polym10050496
Chicago/Turabian StyleLiu, Ning, Jianyi Yu, Yaoyong Meng, and Yuzhou Liu. 2018. "Hyperbranched Polysiloxanes Based on Polyhedral Oligomeric Silsesquioxane Cages with Ultra-High Molecular Weight and Structural Tuneability" Polymers 10, no. 5: 496. https://doi.org/10.3390/polym10050496
APA StyleLiu, N., Yu, J., Meng, Y., & Liu, Y. (2018). Hyperbranched Polysiloxanes Based on Polyhedral Oligomeric Silsesquioxane Cages with Ultra-High Molecular Weight and Structural Tuneability. Polymers, 10(5), 496. https://doi.org/10.3390/polym10050496