Site-Specific DBCO Modification of DEC205 Antibody for Polymer Conjugation


 , and
, and
Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Characterization
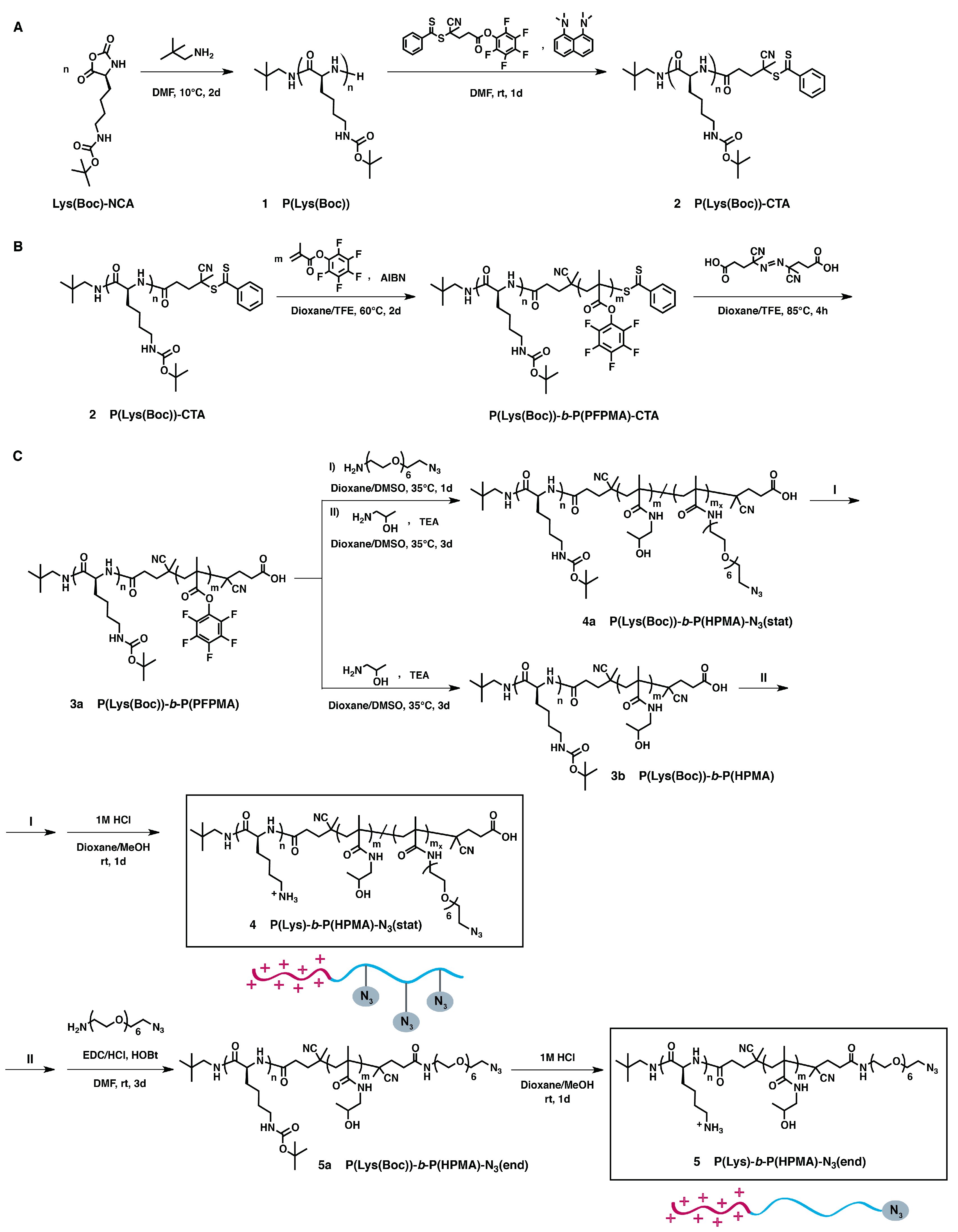
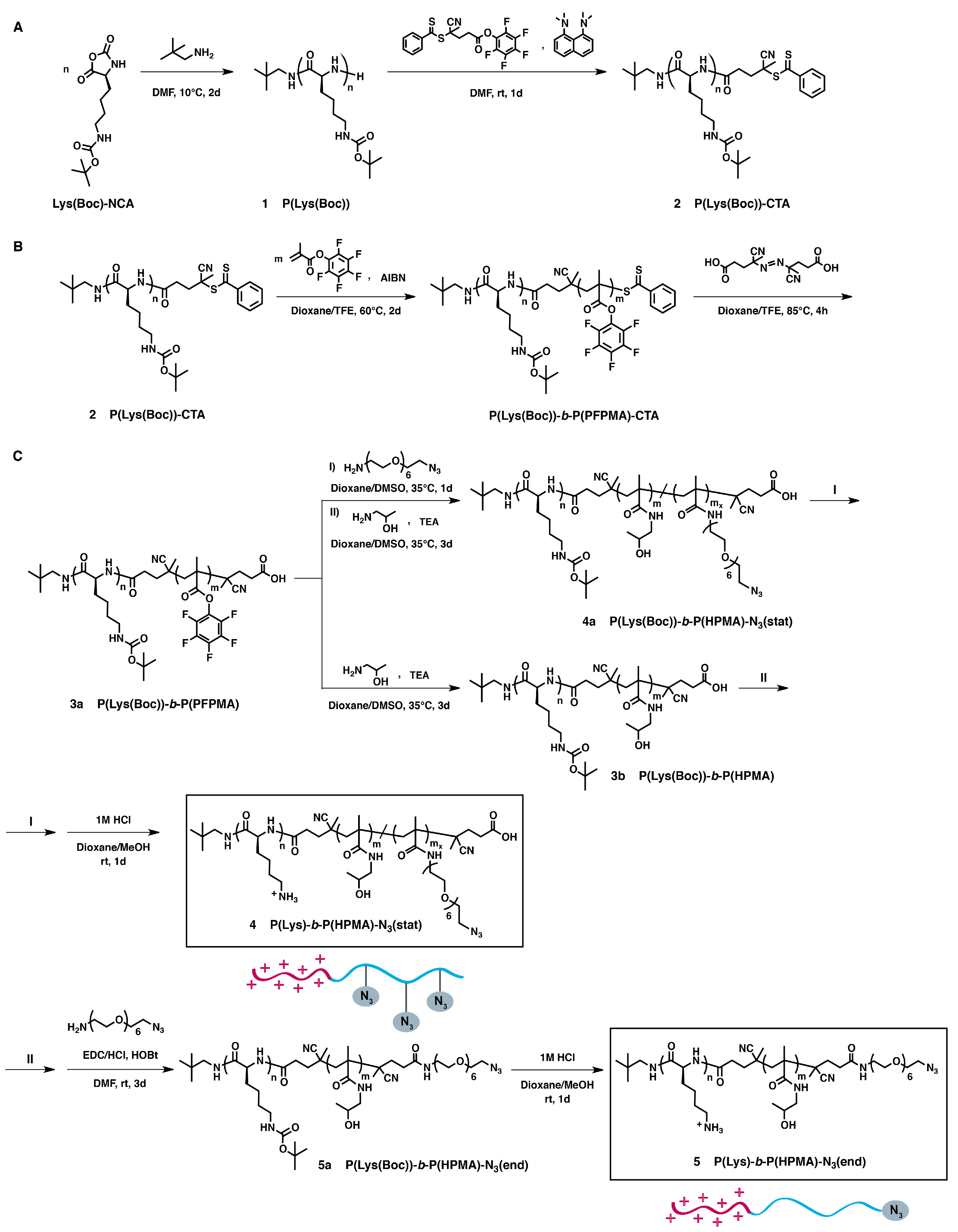
2.3. Synthesis of Azide-Functionalized Block Copolymers: P(Lys)-b-P(HPMA)-N3(stat) and P(Lys)-b-P(HPMA)-N3(end) (SI, Experimental Section)
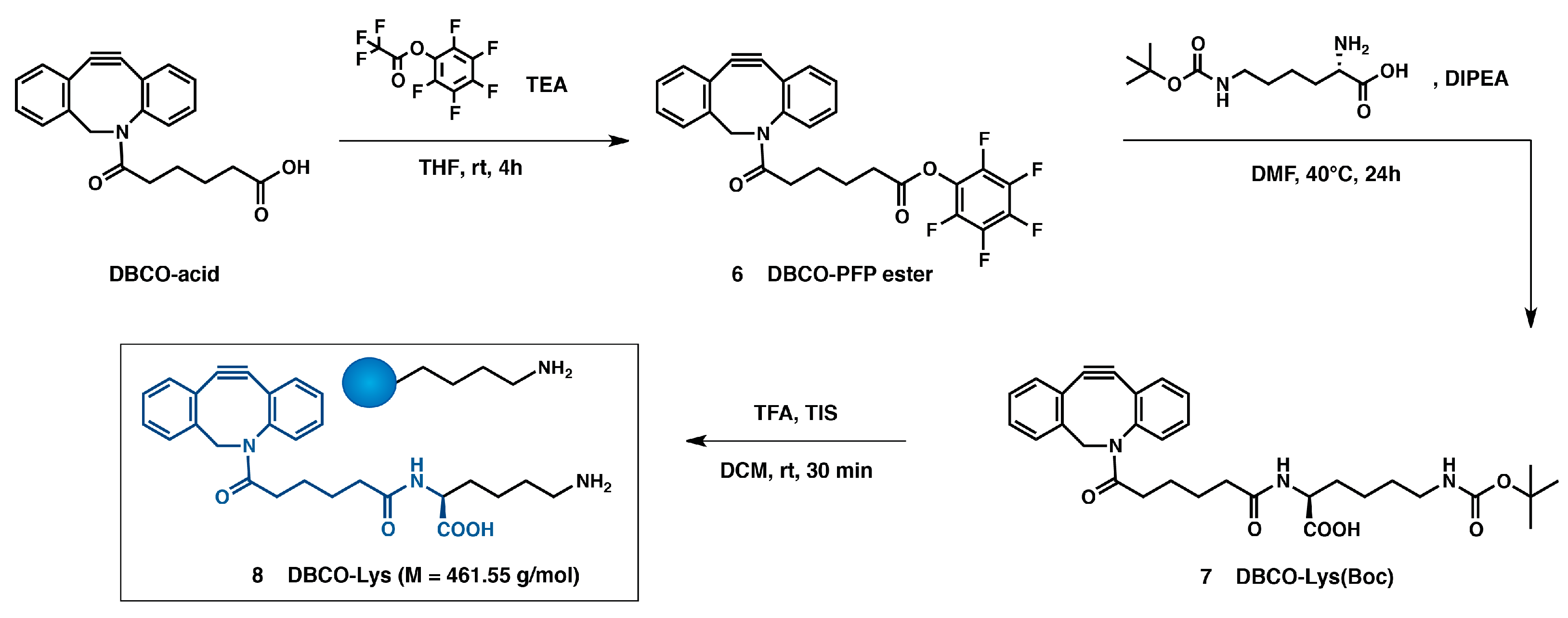
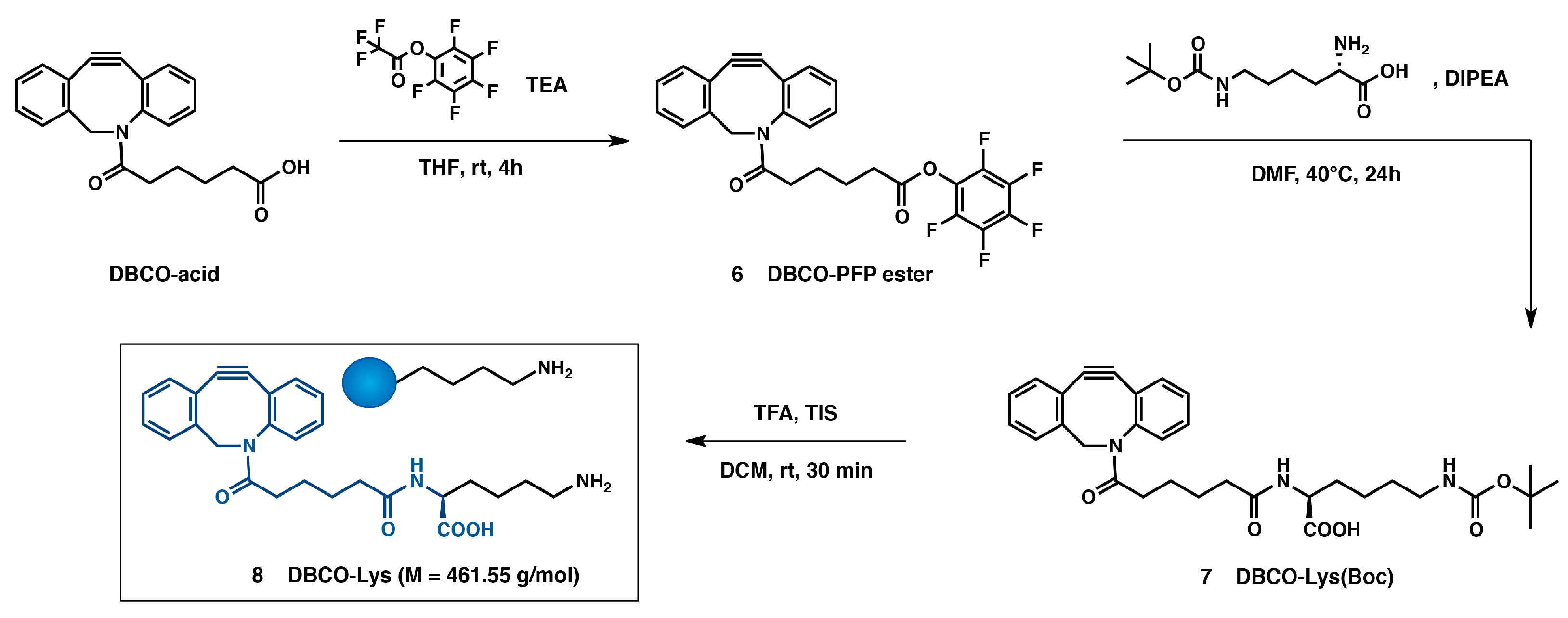
2.4. Synthesis of Dibenzocyclooctyne (DBCO) Linker for DEC205 Antibody Modification (Scheme 4 (8))
2.4.1. Dibenzocyclooctyne-Pentafluorophenylester: DBCO-PFP Ester (Scheme 4 (6))
2.4.2. Dibenzocyclooctyne-N-ε-Boc-l-lysine: DBCO-Lys(Boc) (Scheme 4 (7))
2.4.3. Dibenzocyclooctyne-l-Lysine: DBCO-Lys (Scheme 4 (8))
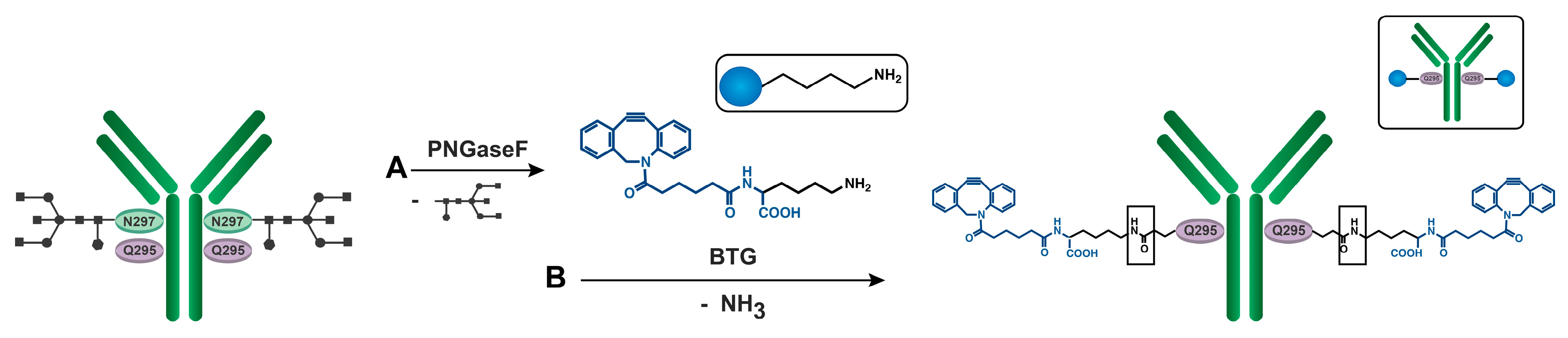
2.5. Enzymatic DBCO Modification of aDEC205
2.5.1. Alexa Fluor 647-Labeling of aDEC205: aDEC205AF647
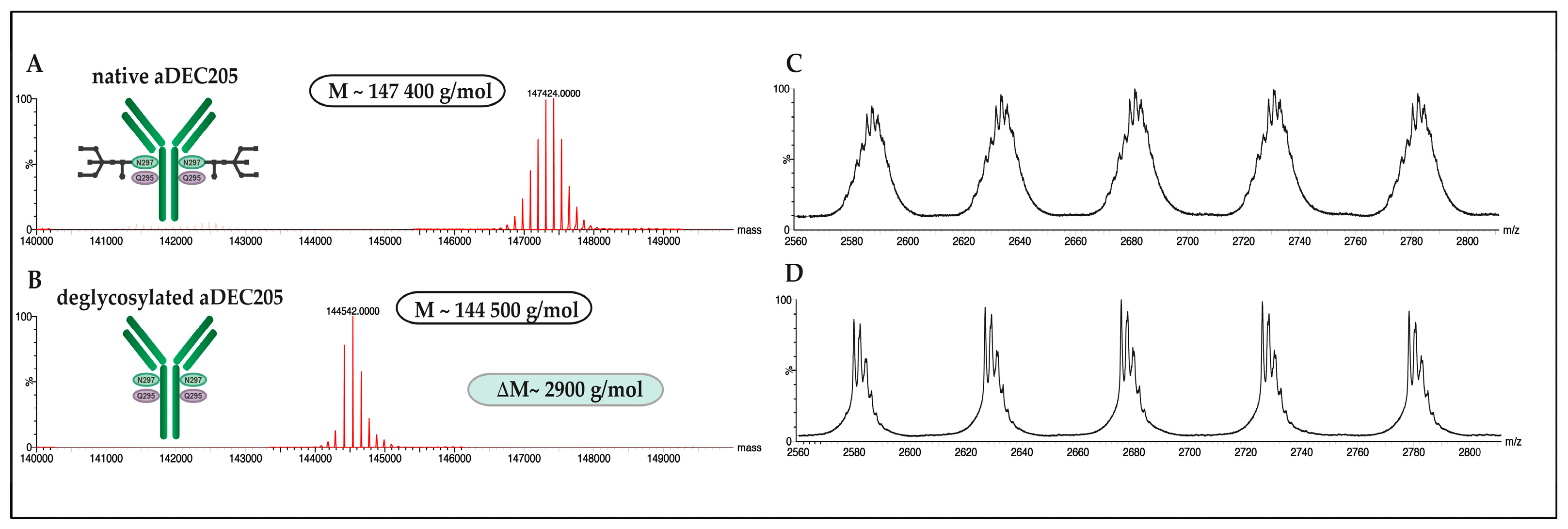
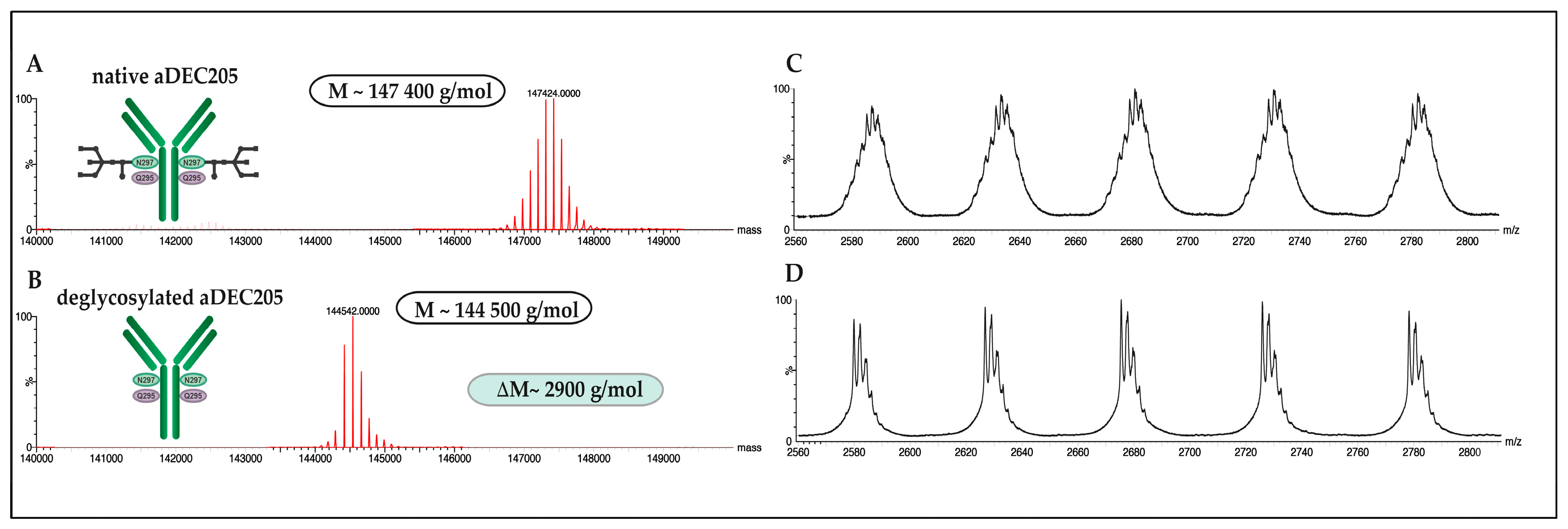
2.5.2. Deglycosylation of aDEC205 at Asp297 by Peptide-N-Glycosidase F (PNGase F): aDEC205(AF647)-dg
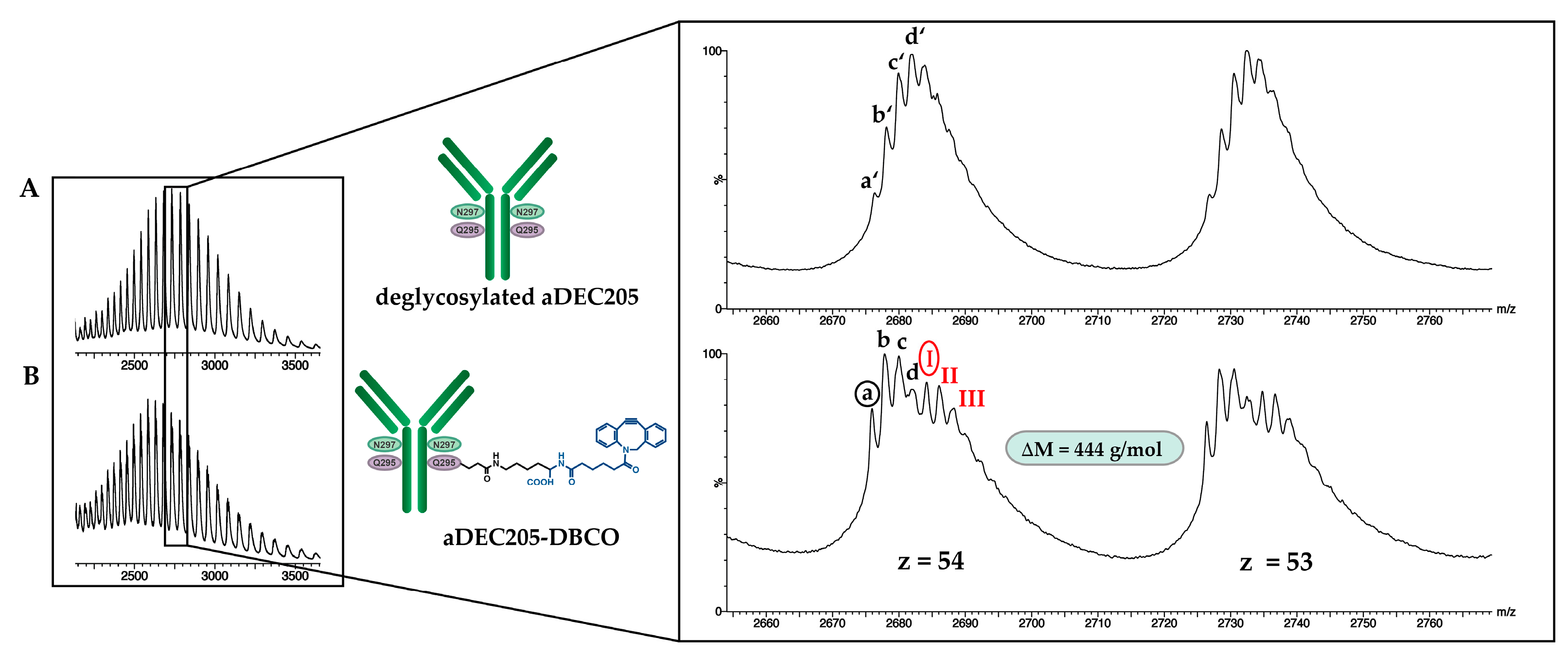
2.5.3. DBCO Modification of aDEC205 at Glu295 by Bacterial Transglutaminase (BTG): aDEC205(AF647)-DBCO
2.6. Strain-Promoted Alkyne-Azide Cycloaddition (SPAAC)
2.6.1. SPAAC of aDEC205-DBCO and 5/6-Carboxyrhodamine 110-PEG3-Azide (CR-110-N3)
2.6.2. SPAAC of aDEC205(AF647)-DBCO and P(Lys)-b-P(HPMA)(OG488)-N3(stat)
2.6.3. SPAAC of aDEC205AF647-DBCO and P(Lys)-b-P(HPMA)-N3(end)
2.6.4. SPAAC of N3-Polyplex and Alexa Fluor 647 DIBO Alkyne (AF647-DBCO)
2.6.5. SPAAC of N3-Polyplex and aDEC205AF647-DBCO
2.7. High Resolution Mass Spectrometry: hr-MS
2.8. Fluorescence Correlation Spectroscopy: FCS
3. Results and Discussion
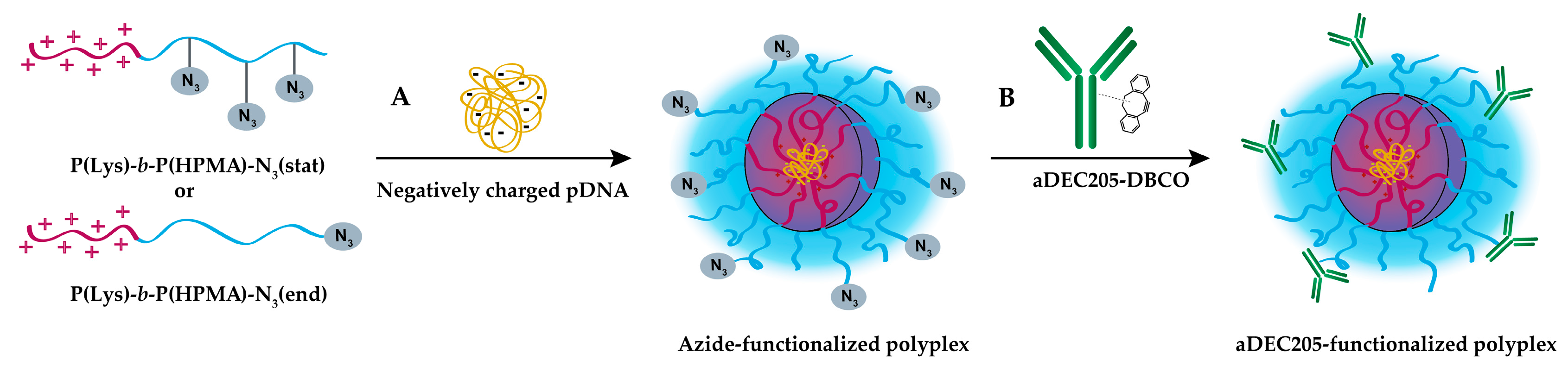
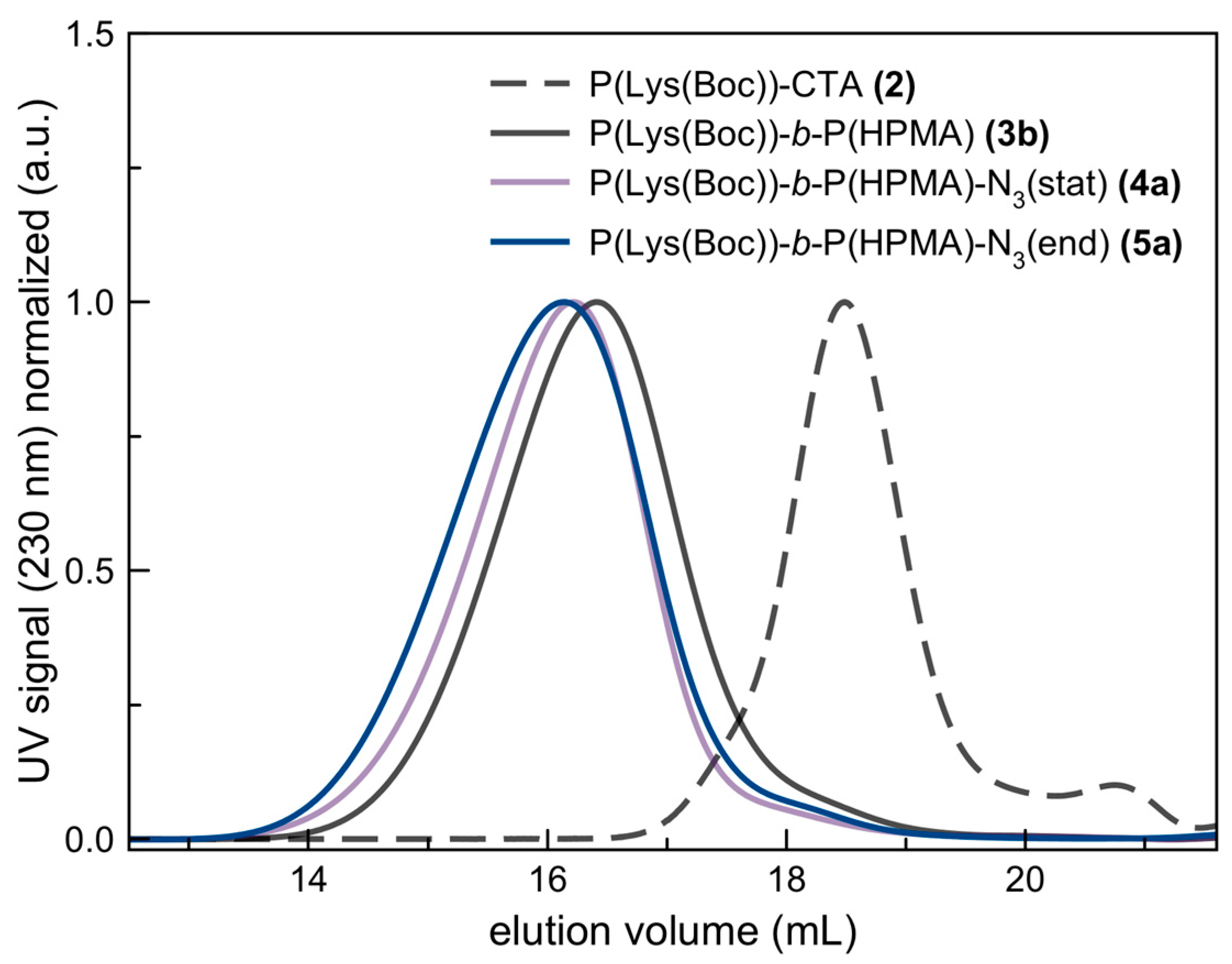
3.1. Synthesis of Azide-Functionalized P(Lys)-b-P(HPMA) Block Copolymers
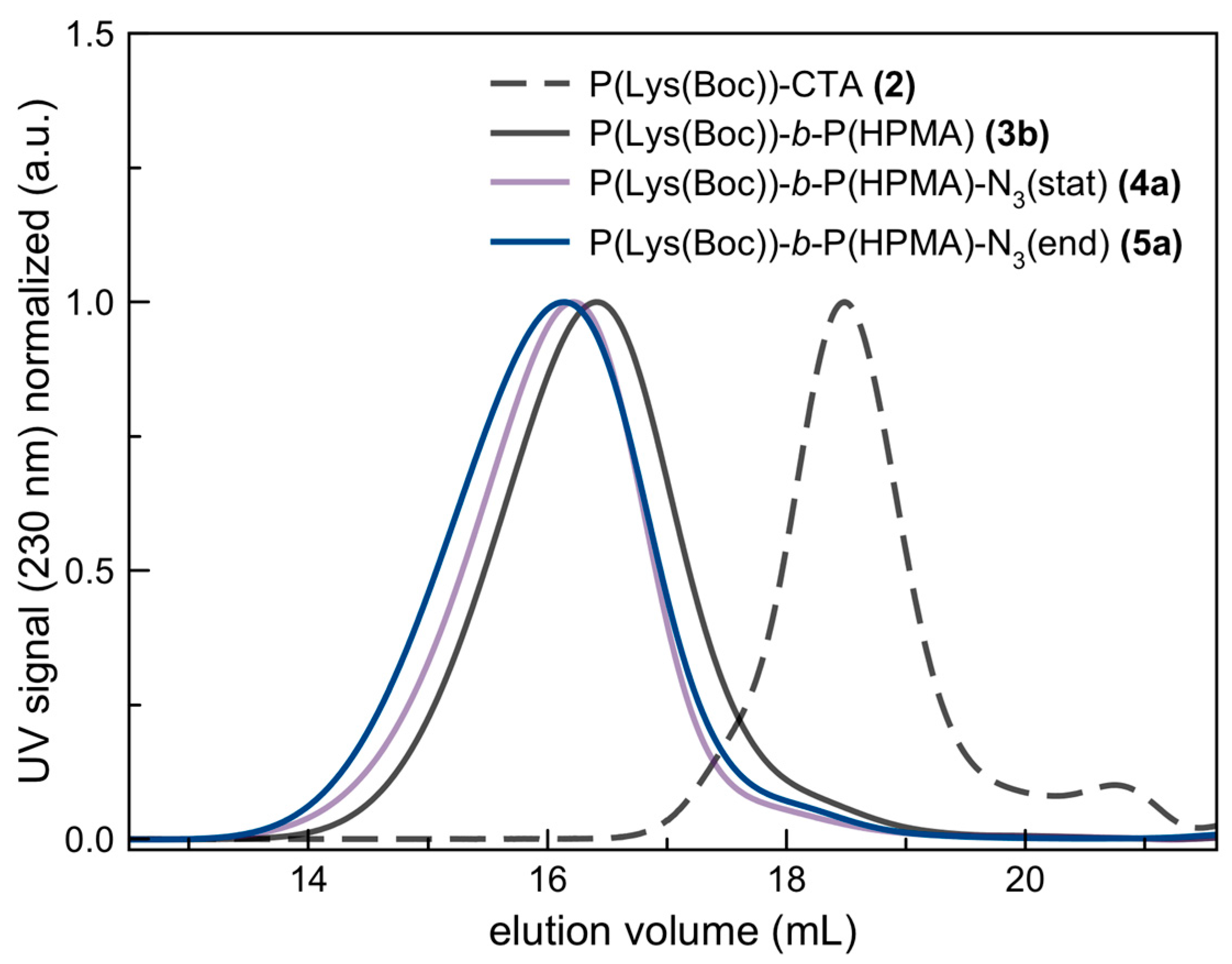
3.2. Characterization of Azide-Modified Block Copolymers: P(Lys)-b-P(HPMA)-N3(stat) and P(Lys)-b-P(HPMA)-N3(end)
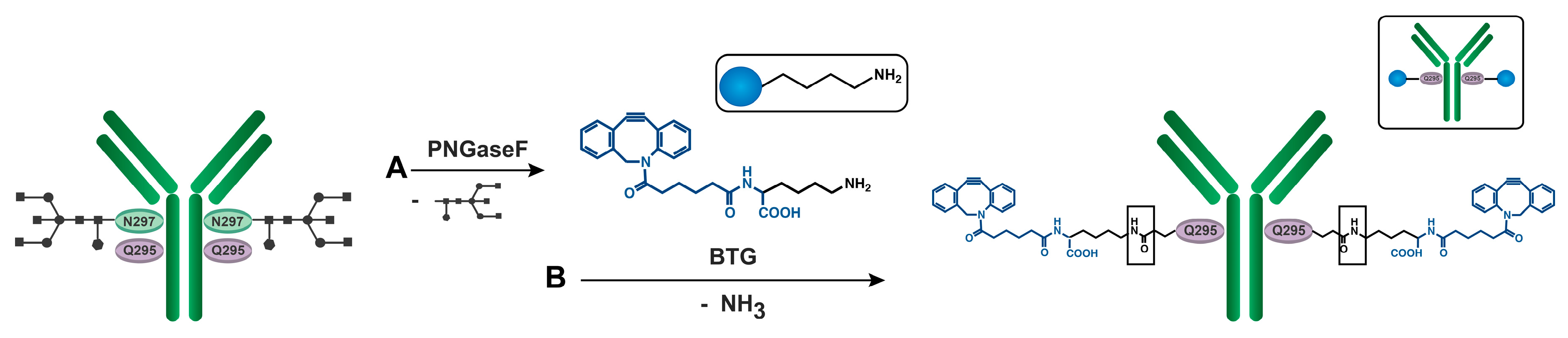
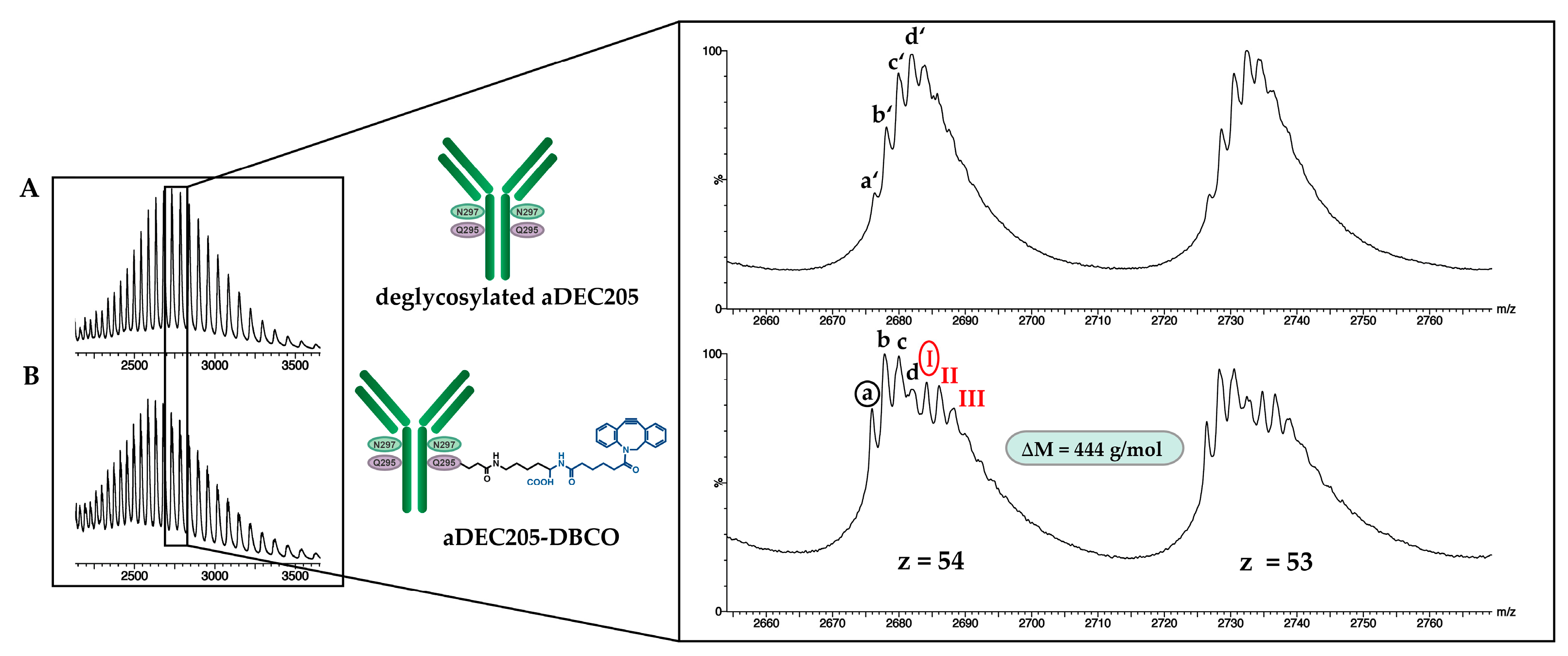
3.3. Site-Specific DBCO Modification of aDEC205
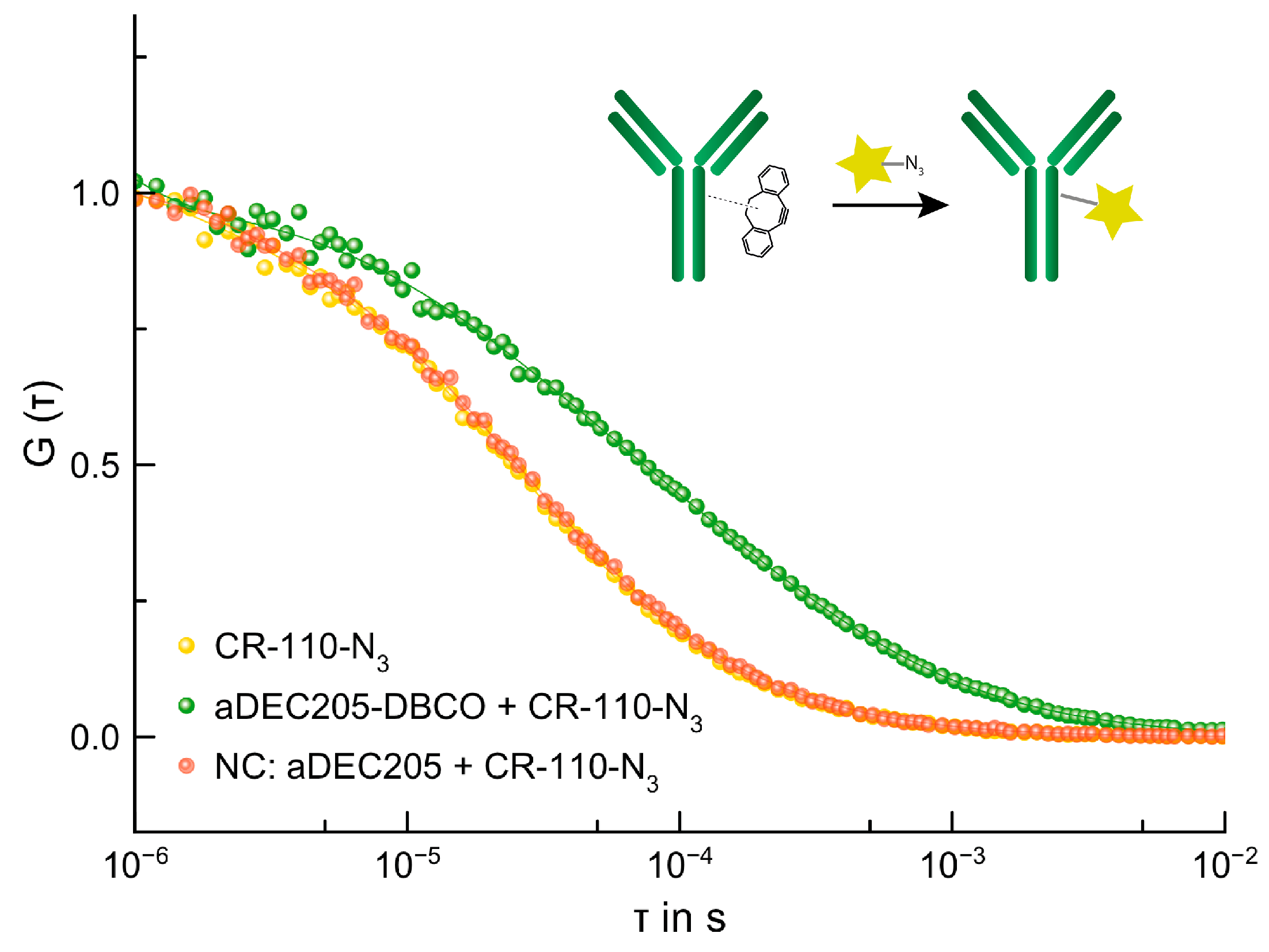
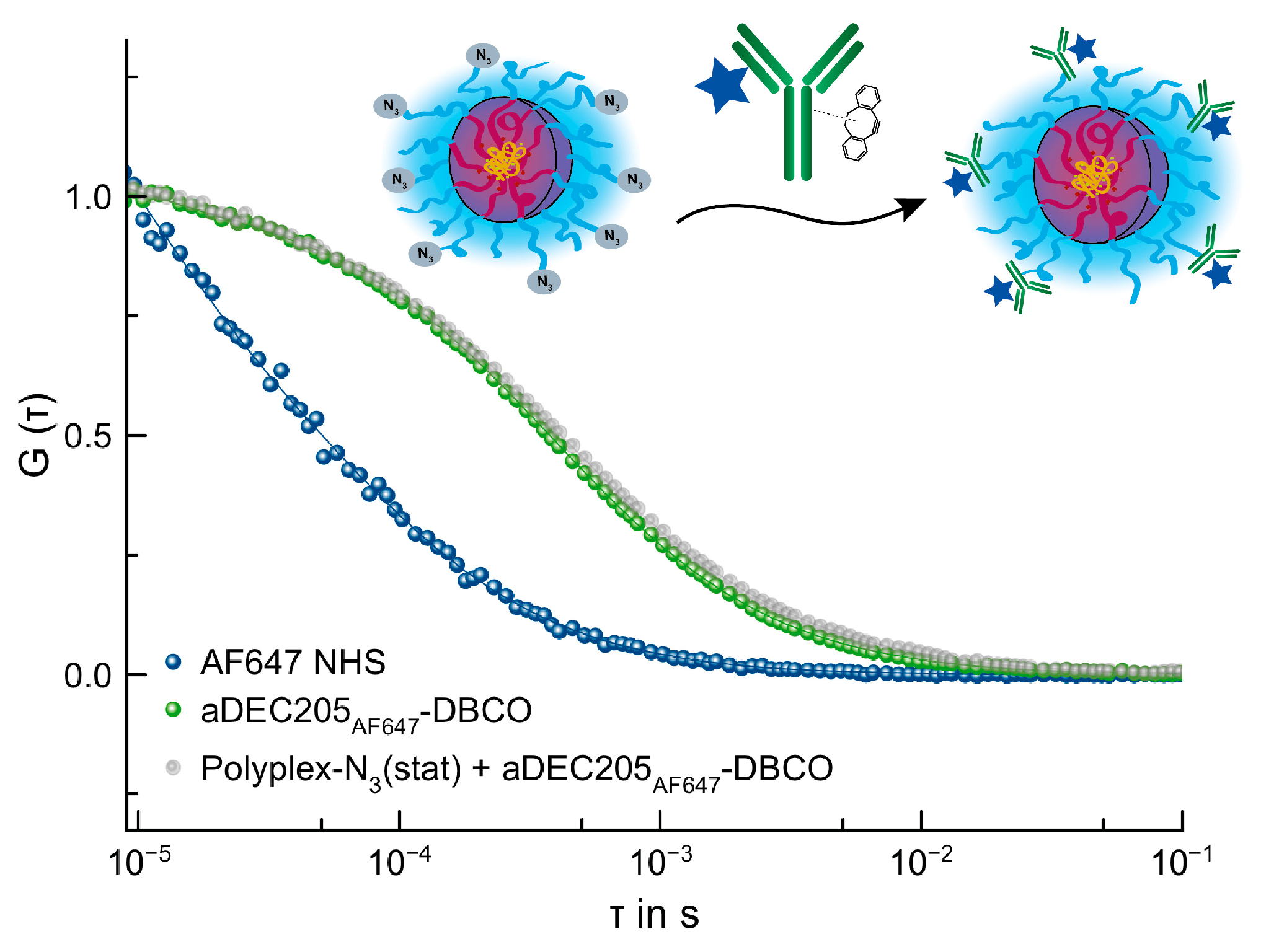
3.4. Conjugation of Polyplex and aDEC205 via Strain-Promoted Alkyne-Azide Cycloaddition (SPAAC)
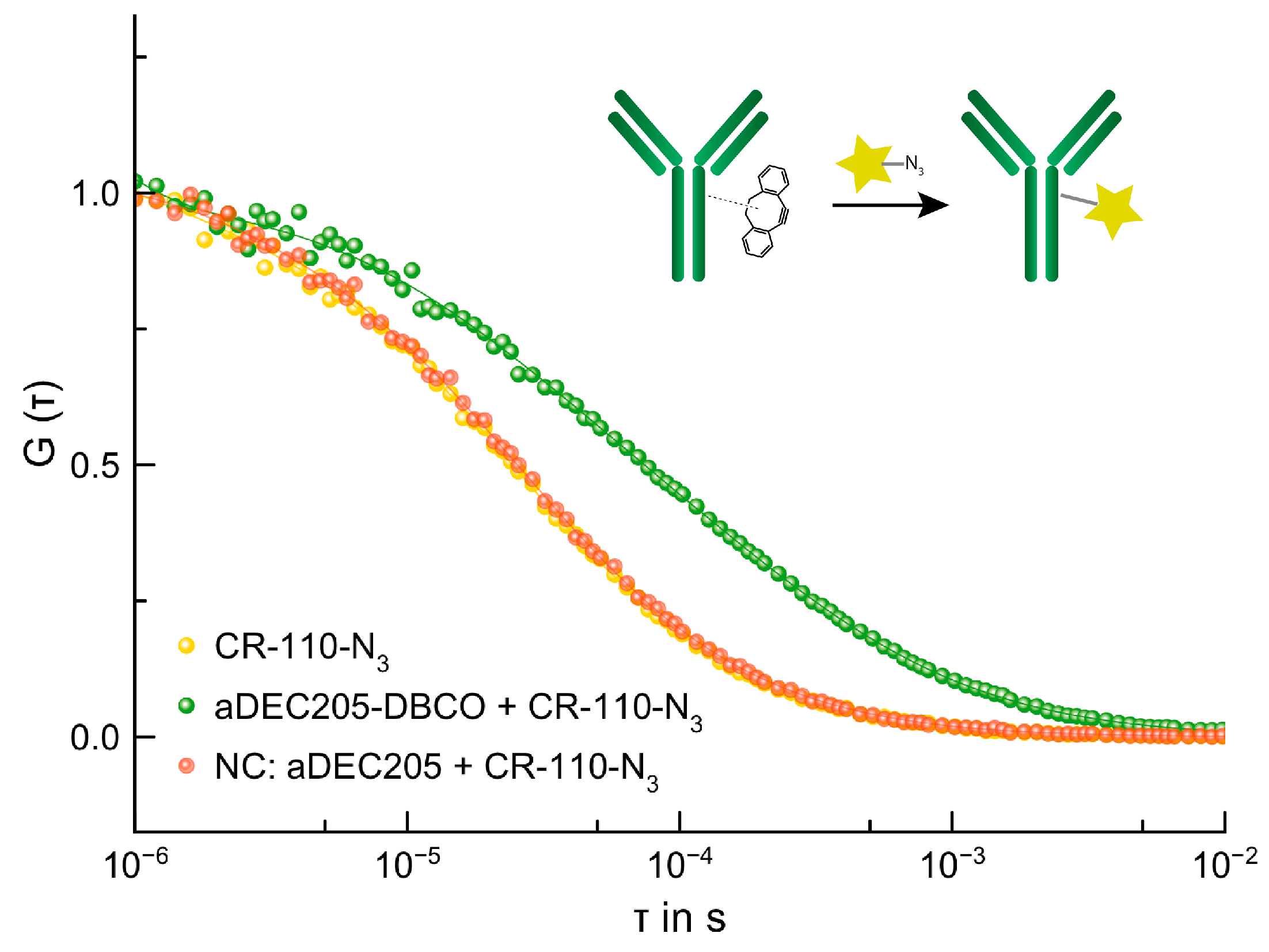
3.4.1. SPAAC of aDEC205-DBCO and 5/6-Carboxyrhodamine 110-PEG3-Azide (CR-110-N3)
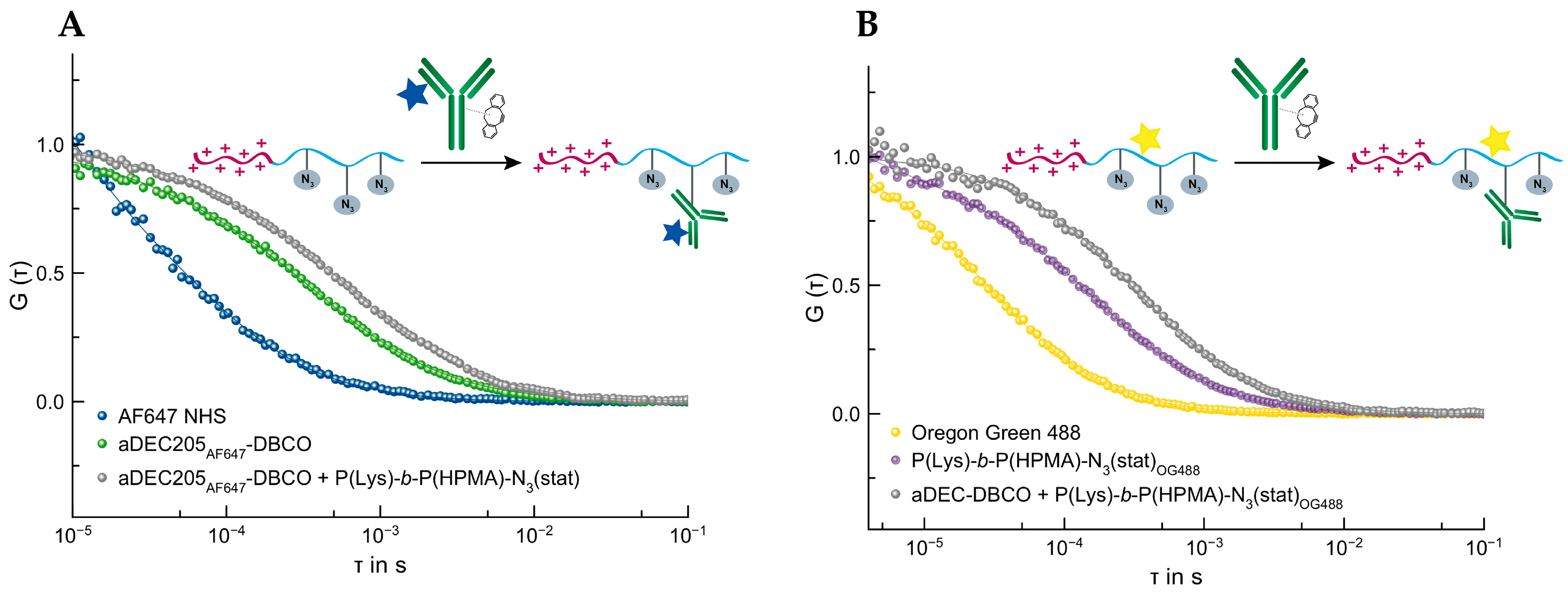
3.4.2. SPAAC of aDEC205-DBCO and P(Lys(Boc))-b-P(HPMA)-N3(stat)
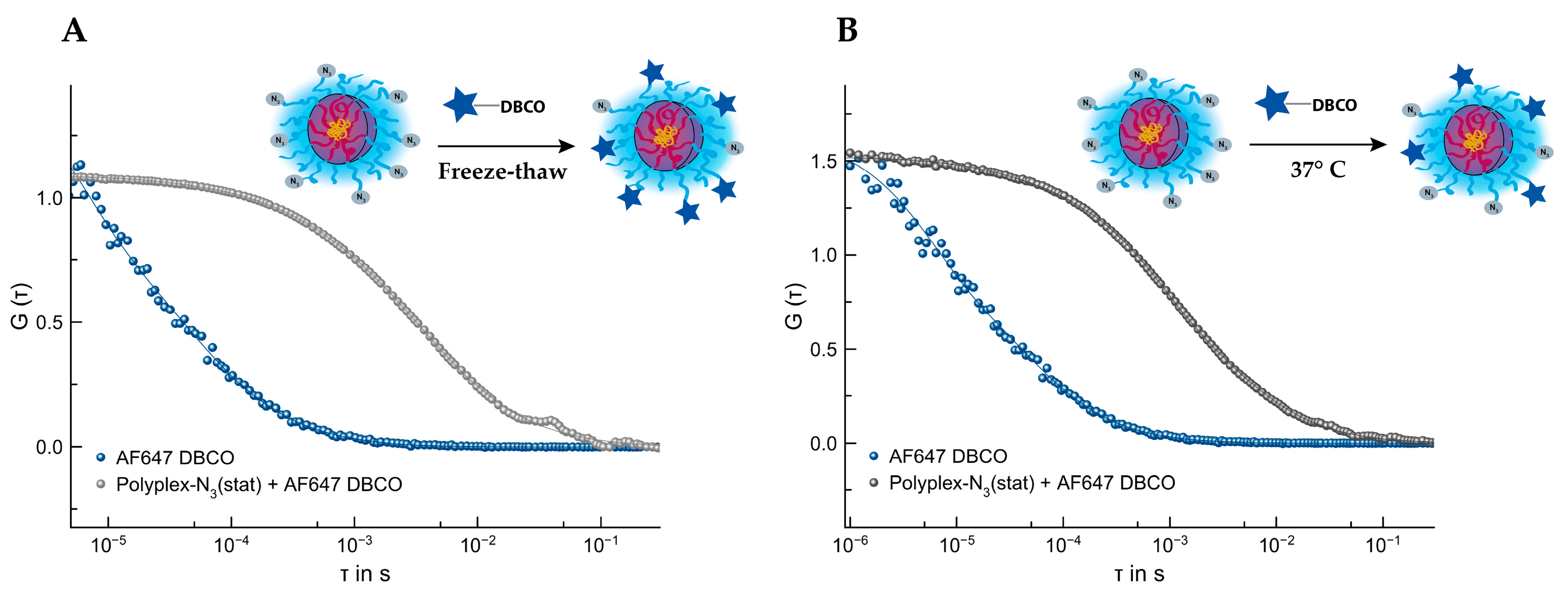
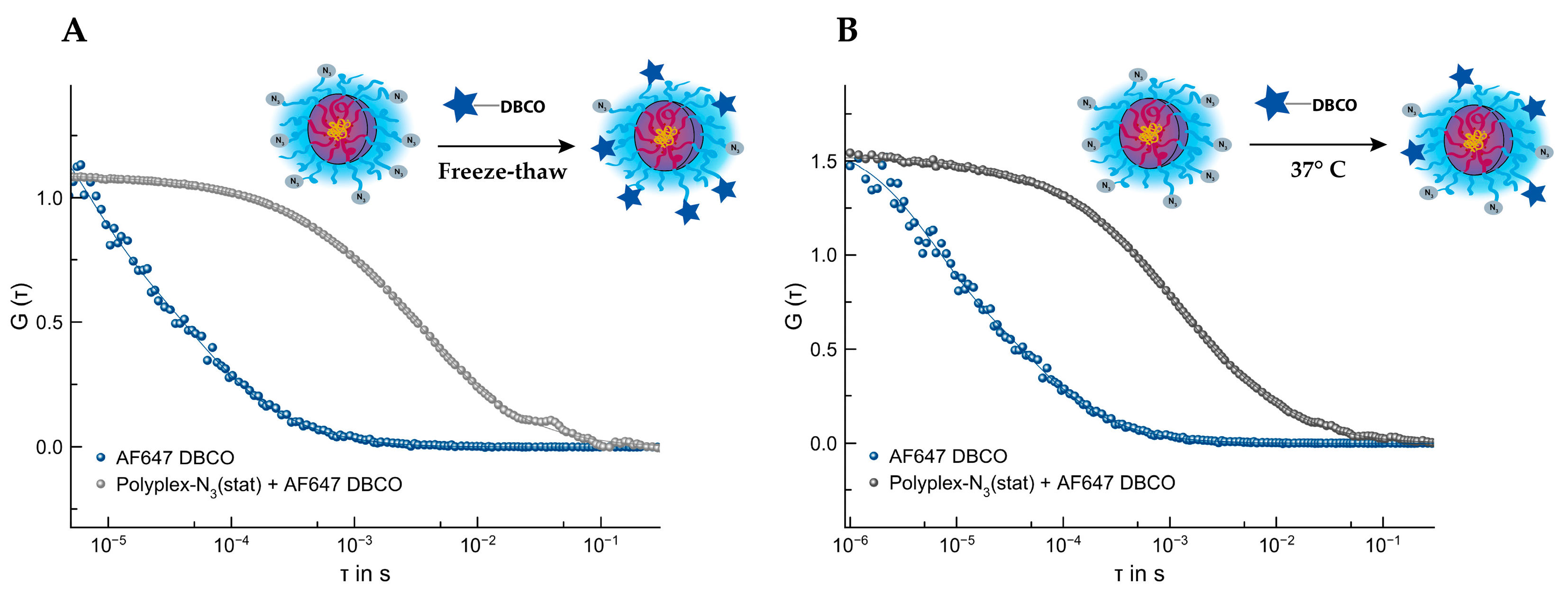
3.4.3. SPAAC of Polyplex by P(Lys)-b-P(HPMA)-N3(stat) and AF647 DBCO
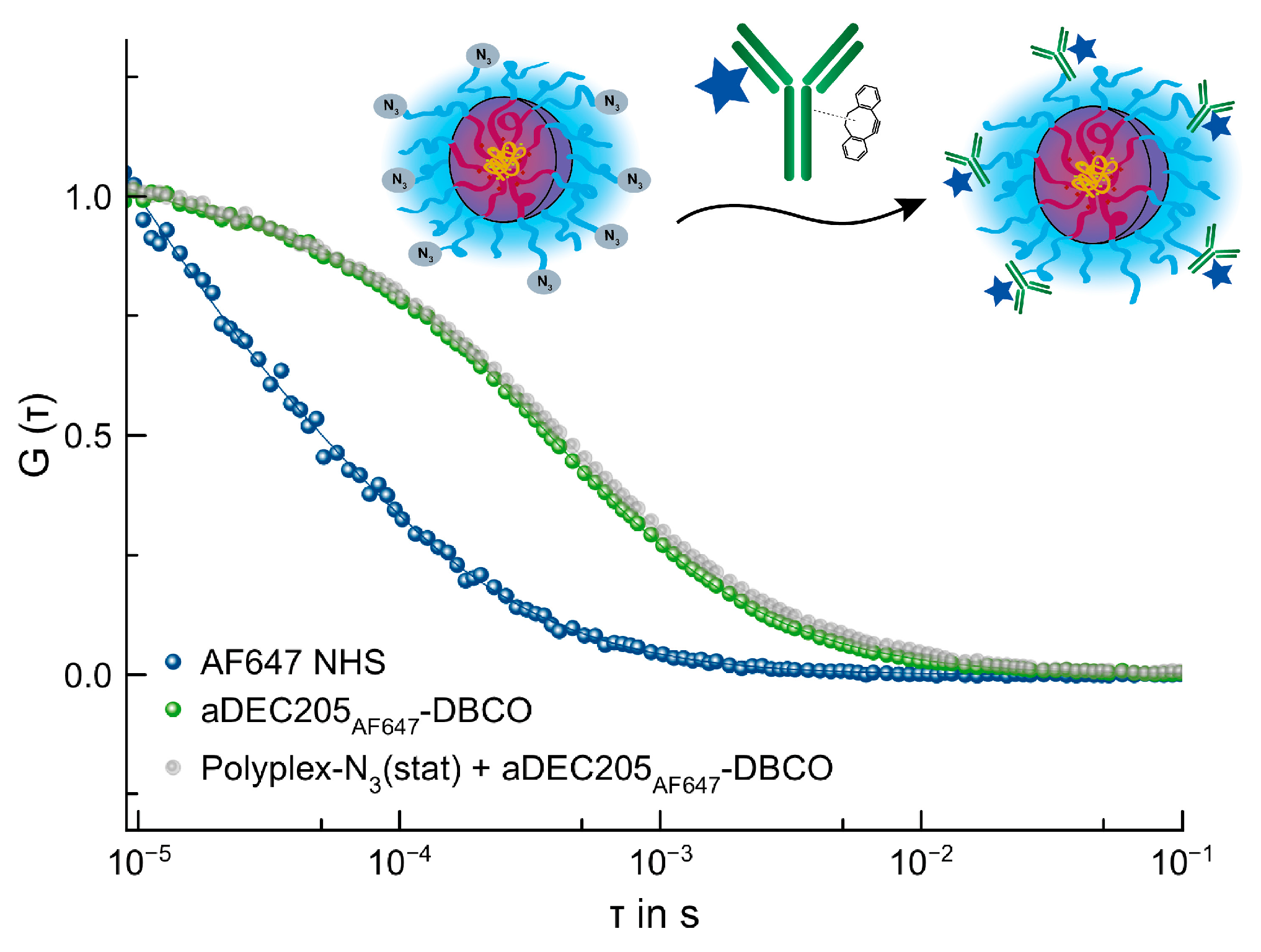
3.4.4. SPAAC of Polyplex by P(Lys)-b-P(HPMA)-N3(stat) and aDEC205AF647-DBCO
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Kearney, C.J.; Mooney, D.J. Macroscale delivery systems for molecular and cellular payloads. Nat. Mater. 2013, 12, 1004–1017. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Votruba, A.R.; Farokhzad, O.C.; Langer, R. Nanotechnology in drug delivery and tissue engineering: From discovery to applications. Nano Lett. 2010, 10, 3223–3230. [Google Scholar] [CrossRef] [PubMed]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, M. Cancer nanotechnology: Opportunities and challenges. Nat. Rev. Cancer 2005, 5, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Maeda, H.; Smancs, A. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accum. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Lammers, T.; Hennink, W.E.; Storm, G. Tumour-targeted nanomedicines: Principles and practice. Br. J. Cancer 2008, 99, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Lammers, T.; Kiessling, F.; Hennink, W.E.; Storm, G. Drug targeting to tumors: Principles, pitfalls and (pre-) clinical progress. J. Control. Release 2012, 161, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R. Polymer conjugates as anticancer nanomedicines. Nat. Rev. Cancer 2006, 6, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Mochida, Y.; Cabral, H.; Miura, Y.; Albertini, F.; Fukushima, S.; Osada, K.; Nishiyama, N.; Kataoka, K. Bundled assembly of helical nanostructures in polymeric micelles loaded with platinum drugs enhancing therapeutic efficiency against pancreatic tumor. ACS Nano 2014, 8, 6724–6738. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Etrych, T.; Chytil, P.; Ohkubo, M.; Fang, J.; Ulbrich, K.; Maeda, H. Two step mechanisms of tumor selective delivery of N-(2-hydroxypropyl) methacrylamide copolymer conjugated with pirarubicin via an acid-cleavable linkage. J. Control. Release 2014, 174, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Xu, L.; Song, G.; Liu, Z. Emerging nanomedicine approaches fighting tumor metastasis: Animal models, metastasis-targeted drug delivery, phototherapy, and immunotherapy. Chem. Soc. Rev. 2016, 45, 6250–6269. [Google Scholar] [CrossRef] [PubMed]
- Shao, K.; Singha, S.; Clemente-Casares, X.; Tsai, S.; Yang, Y.; Santamaria, P. Nanoparticle-Based Immunotherapy for Cancer. ACS Nano 2015, 9, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Dömling, A.; Holak, T.A. Programmed death-1: Therapeutic success after more than 100 years of cancer immunotherapy. Angew. Chem. Int. Ed. 2014, 53, 2286–2288. [Google Scholar] [CrossRef] [PubMed]
- Van Parijs, L.; Abbas, A.K. Turning the immune system off: A rticles. Homeostasis and self-tolerance in the immune system: Turning lymphocytes off. Science 1998, 280, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Toth, I.; Skwarczynski, M. The immune system likes nanotechnology. Nanomedicine 2014, 9, 2607–2609. [Google Scholar] [CrossRef] [PubMed]
- Blattman, J.N.; Greenberg, P.D. Cancer immunotherapy: A treatment for the masses. Science 2004, 305, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T. Immunotherapy: Past, present and future. Nat. Med. 2003, 9, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.D.; Unger, W.J.; Storm, G.; van Kooyk, Y.; Mastrobattista, E. Targeting tumor antigens to dendritic cells using particulate carriers. J. Control. Release 2012, 161, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Den Haan, J.M.; Lehar, S.M.; Bevan, M.J. CD8(+) but not CD8(-) dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 2000, 192, 1685–1696. [Google Scholar] [CrossRef] [PubMed]
- Celli, S.; Day, M.; Mu, A.J.; Molina-Paris, C.; Lythe, G.; Bousso, P. Brief report How many dendritic cells are required to initiate a T-cell response? Blood 2012, 120, 3945–3948. [Google Scholar] [CrossRef] [PubMed]
- Herweijer, H.; Zhang, G.; Subbotin, V.M.; Budker, V.; Williams, P.; Wolff, J.A. Time course of gene expression after plasmid DNA gene transfer to the liver. J. Gene Med. 2001, 3, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Höhn, Y.; Sudowe, S.; Reske-kunz, A.B. Biolistic DNA Delivery; Humana Press: New York, NY, USA, 2013; Volume 940, pp. 199–213. [Google Scholar]
- Hanagata, N. Structure-dependent immunostimulatory effect of CpG oligodeoxynucleotides and their delivery system. Int. J. Nanomed. 2012, 7, 2181–2195. [Google Scholar] [CrossRef] [PubMed]
- Caruso, F.; Hyeon, T.; Rotello, V. Nanomedicine themed issue. Chem. Soc. Rev. 2012, 41, 2537–2538. [Google Scholar] [CrossRef] [PubMed]
- Katayose, S.; Kataoka, K. Water-soluble polyion complex associates of DNA and poly(ethylene glycol)-poly(l-lysine) block copolymer. Bioconjug. Chem. 1997, 8, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Kakizawa, Y.; Harada, A.; Kataoka, K. Glutathione-sensitive stabilization of block copolymer micelles composed of antisense DNA and thiolated poly(ethylene glycol)-block-poly(l-lysine): A potential carrier for systemic delivery of antisense DNA. Biomacromolecules 2001, 2, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Efthimiadou, E.K.; Tapeinos, C.; Bilalis, P.; Kordas, G. New approach in synthesis, characterization and release study of pH-sensitive polymeric micelles, based on PLA-Lys-b-PEGm, conjugated with doxorubicin. J. Nanopart. Res. 2011, 13, 6725–6736. [Google Scholar] [CrossRef]
- Heller, P.; Birke, A.; Huesmann, D.; Weber, B.; Fischer, K.; Reske-Kunz, A.; Bros, M.; Barz, M. Introducing PeptoPlexes: Polylysine-block-polysarcosine based polyplexes for transfection of HEK 293T cells. Macromol. Biosci. 2014, 14, 1380–1395. [Google Scholar] [CrossRef] [PubMed]
- Heller, P.; Hobernik, D.; Lächelt, U.; Schinnerer, M.; Weber, B.; Schmidt, M.; Wagner, E.; Bros, M.; Barz, M. Combining reactive triblock copolymers with functional cross-linkers: A versatile pathway to disulfide stabilized-polyplex libraries and their application as pDNA vaccines. J. Control. Release 2017, 258, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Tappertzhofen, K.; Weiser, F.; Montermann, E.; Reske-Kunz, A.; Bros, M.; Zentel, R. Poly-l-Lysine-Poly[HPMA] block copolymers obtained by RAFT polymerization as polyplex-transfection reagents with minimal toxicity. Macromol. Biosci. 2015, 15, 1159–1173. [Google Scholar] [CrossRef] [PubMed]
- Tappertzhofen, K.; Beck, S.; Montermann, E.; Huesmann, D.; Barz, M.; Koynov, K.; Bros, M.; Zentel, R. Bioreducible Poly-l-Lysine-Poly[HPMA] block copolymers obtained by RAFT-polymerization as efficient polyplex-transfection reagents. Macromol. Biosci. 2015, 16, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Raghuwanshi, D.; Mishra, V.; Suresh, M.R.; Kaur, K. A simple approach for enhanced immune response using engineered dendritic cell targeted nanoparticles. Vaccine 2012, 30, 7292–7299. [Google Scholar] [CrossRef] [PubMed]
- Heller, P.; Mohr, N.; Birke, A.; Weber, B.; Reske-Kunz, A.; Bros, M.; Barz, M. Directed interactions of block copolypept(o) ides with mannose-binding receptors: Peptomicelles targeted to cells of the innate immune system. Macromol. Biosci. 2015, 15, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Mintern, J.D.; Percival, C.; Kamphuis, M.M.J.; Chin, W.J.; Caruso, F.; Johnston, A.P.R. Targeting dendritic cells: The role of specific receptors in the internalization of polymer capsules. Adv. Healthc. Mater. 2013, 2, 940–944. [Google Scholar] [CrossRef] [PubMed]
- Tappertzhofen, K.; Bednarczyk, M.; Koynov, K.; Bros, M.; Grabbe, S.; Zentel, R. Toward anticancer immunotherapeutics: Well-defined polymer-antibody conjugates for selective dendritic cell targeting. Macromol. Biosci. 2014, 14, 1444–1457. [Google Scholar] [CrossRef] [PubMed]
- Bühler, J.; Gietzen, S.; Reuter, A.; Kappel, C.; Fischer, K.; Decker, S.; Schäffel, D.; Koynov, K.; Bros, M.; Tubbe, I.; et al. Selective uptake of cylindrical poly(2-oxazoline) brush-antiDEC205 antibody-OVA antigen conjugates into DEC-positive dendritic cells and subsequent T-cell activation. Chemistry 2014, 20, 12405–12410. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R. Polymer therapeutics as nanomedicines: New perspectives. Curr. Opin. Biotechnol. 2011, 22, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Barz, M.; Luxenhofer, R.; Zentel, R.; Vicent, M.J. Overcoming the PEG-addiction: Well-defined alternatives to PEG, from structure–property relationships to better defined therapeutics. Polym. Chem. 2011, 2, 1900–1918. [Google Scholar] [CrossRef]
- Aghemo, A.; Rumi, M.G.; Colombo, M. Pegylated interferons α2a and α2b in the treatment of chronic hepatitis C. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, M.; Mruk, R.; Zentel, R.; Théato, P. Synthesis of pentafluorophenyl(meth)acrylate polymers: New precursor polymers for the synthesis of multifunctional materials. Eur. Polym. J. 2005, 41, 1569–1575. [Google Scholar] [CrossRef]
- Mohr, N.; Barz, M.; Forst, R.; Zentel, R. A Deeper insight into the postpolymerization modifi cation of polypenta fluorophenyl methacrylates to poly (N-(2-hydroxypropyl) methacrylamide). Macromol. Rapid Commun. 2014, 35, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Nuhn, L.; Barz, M.; Zentel, R. New perspectives of HPMA-based copolymers derived by post-polymerization modification. Macromol. Biosci. 2014, 14, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. Engl. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Agard, N.J.; Prescher, J.A.; Bertozzi, C.R. A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc. 2004, 126, 15046–15047. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, M.A.; Klok, H.-A. Peptide/protein-polymer conjugates: Synthetic strategies and design concepts. Chem. Commun. (Camb.) 2008, 2591–2611. [Google Scholar] [CrossRef] [PubMed]
- Broyer, R.M.; Grover, G.N.; Maynard, H.D. Emerging synthetic approaches for protein-polymer conjugations. Chem. Commun. (Camb.) 2011, 47, 2212–2226. [Google Scholar] [CrossRef] [PubMed]
- Gerngross, T.U. Advances in the production of human therapeutic proteins in yeasts and filamentous fungi. Nat. Biotechnol. 2004, 22, 1409–1414. [Google Scholar] [CrossRef] [PubMed]
- Strop, P.; Liu, S.H.; Dorywalska, M.; Delaria, K.; Dushin, R.G.; Tran, T.T.; Ho, W.H.; Farias, S.; Casas, M.G.; Abdiche, Y.; et al. Location matters: Site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem. Biol. 2013, 20, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Jeger, S.; Zimmermann, K.; Blanc, A.; Grünberg, J.; Honer, M.; Hunziker, P.; Struthers, H.; Schibli, R. Site-specific and stoichiometric modification of antibodies by bacterial transglutaminase. Angew. Chem. Int. Ed. 2010, 9995–9997. [Google Scholar] [CrossRef] [PubMed]
- Dennler, P.; Chiotellis, A.; Fischer, E.; Brégeon, D.; Belmant, C.; Gauthier, L.; Lhospice, F.; Romagne, F.; Schibli, R. Transglutaminase-based chemo-enzymatic conjugation approach yields homogeneous antibody-drug conjugates. Bioconjug. Chem. 2014, 25, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Heller, P.; Zhou, J.; Weber, B.; Hobernik, D.; Bros, M.; Schmid, F.; Barz, M. The influence of block ionomer microstructure on polyplex properties: Can simulations help to understand differences in transfection efficiency? Small 2017, 13, 1603694. [Google Scholar] [CrossRef] [PubMed]
- Rigler, R.; Wennmalm, S.; Edman, L. FCS in Single Molecule Analysis. In Fluorescence Correlation Spectroscopy; Springer Series in Chemical Physics; Springer: Berlin/Heidelberg, Germany, 2001; Volume 65, pp. 459–476. ISBN 978-3-642-59542-4. [Google Scholar]
- Heller, P.; Weber, B.; Birke, A.; Barz, M. Synthesis and sequential deprotection of triblock copolypept(o)ides using orthogonal protective group chemistry. Macromol. Rapid Commun. 2015, 36, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Pissuwan, D.; Boyer, C.; Gunasekaran, K.; Davis, T.P.; Bulmus, V. In vitro cytotoxicity of RAFT polymers. Biomacromolecules 2010, 11, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Huesmann, D.; Birke, A.; Klinker, K.; Türk, S.; Räder, H.J.; Barz, M. Revisiting secondary structures in NCA polymerization: Influences on the analysis of protected polylysines. Macromolecules 2014, 47, 928–936. [Google Scholar] [CrossRef]
- Hesse, M.; Meier, H.; Bienz, S.; Bigler, L.; Fox, T. Spektroskopische Methoden in der Organischen Chemie; 8 Überarb Auflage 2011; Stuttgart Thieme: Verlag, Germany, 2011; ISBN 313576107X. [Google Scholar]
- Maley, F.; Trimble, R.B.; Tarentino, A.L.; Plummer, T.H. Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal. Biochem. 1989, 180, 195–204. [Google Scholar] [CrossRef]
- Tretter, V.; Altmann, F.; März, L. Peptide-N4-(N-acetyl-β-glucosaminyl)asparagine amidase F cannot release glycans with fucose attached α1 → 3 to the asparagine-linked N-acetylglucosamine residue. Eur. J. Biochem. 1991, 199, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Plummer, T.H.; Tarentino, A.L. Purification of the oligosaccharide-cleaving enzymes of Flavobacterium meningosepticum. Glycobiology 1991, 1, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Krapp, S.; Mimura, Y.; Jefferis, R.; Huber, R.; Sondermann, P. Structural analysis of human IgG-Fc glycoforms reveals a correlation between glycosylation and structural integrity. J. Mol. Biol. 2003, 325, 979–989. [Google Scholar] [CrossRef]
- Matsumiya, S.; Yamaguchi, Y.; Saito, J.; Nagano, M.; Sasakawa, H.; Otaki, S.; Satoh, M.; Shitara, K.; Kato, K. Structural comparison of fucosylated and nonfucosylated Fc fragments of human immunoglobulin G1. J. Mol. Biol. 2007, 368, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Feige, M.J.; Nath, S.; Catharino, S.R.; Weinfurtner, D.; Steinbacher, S.; Buchner, J. Structure of the murine unglycosylated IgG1 Fc fragment. J. Mol. Biol. 2009, 391, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Fontana, A.; Spolaore, B.; Mero, A.; Veronese, F.M. Site-specific modification and PEGylation of pharmaceutical proteins mediated by transglutaminase. Adv. Drug Deliv. Rev. 2008, 60, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Pearson, D.A.; Blanchette, M.; Baker, M.; Guindon, C.A. Trialkylsilanes as scavengers for the trifluoroacetic acid deblocking of protecting groups in peptide synthesis. Tetrahedron Lett. 1989, 30, 2739–2742. [Google Scholar] [CrossRef]
- Wagner-Rousset, E.; Bednarczyk, A.; Bussat, M.C.; Colas, O.; Corvaia, N.; Schaeffer, C.; Van Dorsselaer, A.; Beck, A. The way forward, enhanced characterization of therapeutic antibody glycosylation: Comparison of three level mass spectrometry-based strategies. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 872, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Schur, P.H.; Becker, E.L. Pepsin digestion of rabbit and sheep antibodies. J. Exp. Med. 1963, 891–904. [Google Scholar] [CrossRef]
- Adamczyk, M.; Gebler, J.C.; Wu, J. Papain digestion of different mouse IgG subclasses as studied by electrospray mass spectrometry. J. Immunol. Methods 2000, 237, 95–104. [Google Scholar] [CrossRef]
- Debets, M.F.; van Berkel, S.S.; Schoffelen, S.; Rutjes, F.P.J.T.; van Hest, J.C.M.; van Delft, F.L. Aza-dibenzocyclooctynes for fast and efficient enzyme PEGylation via copper-free (3 + 2) cycloaddition. Chem. Commun. 2010, 46, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, S.; Chin, J.; Schirrmacher, R.; Popik, V.V.; Kostikov, A.P. [18F]Azadibenzocyclooctyne ([18F]ADIBO): A biocompatible radioactive labeling synthon for peptides using catalyst free [3 + 2] cycloaddition. Bioorg. Med. Chem. Lett. 2011, 21, 6987–6991. [Google Scholar] [CrossRef] [PubMed]
- The Alexa Fluor Dye Series—Note 1.1. Available online: http://www.thermofisher.com/de/de/home/references/molecular-probes-the-handbook/technical-notes-and-product-highlights/the-alexa-fluor-dye-series.html (accessed on 15 May 2016).
- Koynov, K.; Butt, H.-J. Fluorescence correlation spectroscopy in colloid and interface science. Curr. Opin. Colloid Interface Sci. 2012, 17, 377–387. [Google Scholar] [CrossRef]
- Takemoto, H.; Miyata, K.; Ishii, T.; Hattori, S.; Osawa, S.; Nishiyama, N.; Kataoka, K. Accelerated polymer-polymer click conjugation by freeze-thaw treatment. Bioconjug. Chem. 2012, 23, 1503–1506. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Xn | Block Ratio P(Lys):P(HPMA) | Đ (HFIP GPC) | Mn (1H/19F NMR) [g/mol] |
|---|---|---|---|---|
| P(Lys(Boc))-CTA (2) | 32 | - | 1.26 | 7687 |
| P(Lys(Boc))-b-P(HPMA) (3b) | 32:162 | 1:5 | 1.41 | 30,857 * |
| P(Lys(Boc))-b-P(HPMA)-N3(stat) (4a) | 32:162 | 1:5 | 1.53 | 35,537 */** |
| P(Lys(Boc))-b-P(HPMA)-N3(end) (5a) | 32:162 | 1:5 | 1.58 | 30,880 */** |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beck, S.; Schultze, J.; Räder, H.-J.; Holm, R.; Schinnerer, M.; Barz, M.; Koynov, K.; Zentel, R. Site-Specific DBCO Modification of DEC205 Antibody for Polymer Conjugation. Polymers 2018, 10, 141. https://doi.org/10.3390/polym10020141
Beck S, Schultze J, Räder H-J, Holm R, Schinnerer M, Barz M, Koynov K, Zentel R. Site-Specific DBCO Modification of DEC205 Antibody for Polymer Conjugation. Polymers. 2018; 10(2):141. https://doi.org/10.3390/polym10020141
Chicago/Turabian StyleBeck, Simone, Jennifer Schultze, Hans-Joachim Räder, Regina Holm, Meike Schinnerer, Matthias Barz, Kaloian Koynov, and Rudolf Zentel. 2018. "Site-Specific DBCO Modification of DEC205 Antibody for Polymer Conjugation" Polymers 10, no. 2: 141. https://doi.org/10.3390/polym10020141
APA StyleBeck, S., Schultze, J., Räder, H.-J., Holm, R., Schinnerer, M., Barz, M., Koynov, K., & Zentel, R. (2018). Site-Specific DBCO Modification of DEC205 Antibody for Polymer Conjugation. Polymers, 10(2), 141. https://doi.org/10.3390/polym10020141