Intermolecular Interactions in Functional Crystalline Materials: From Data to Knowledge


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
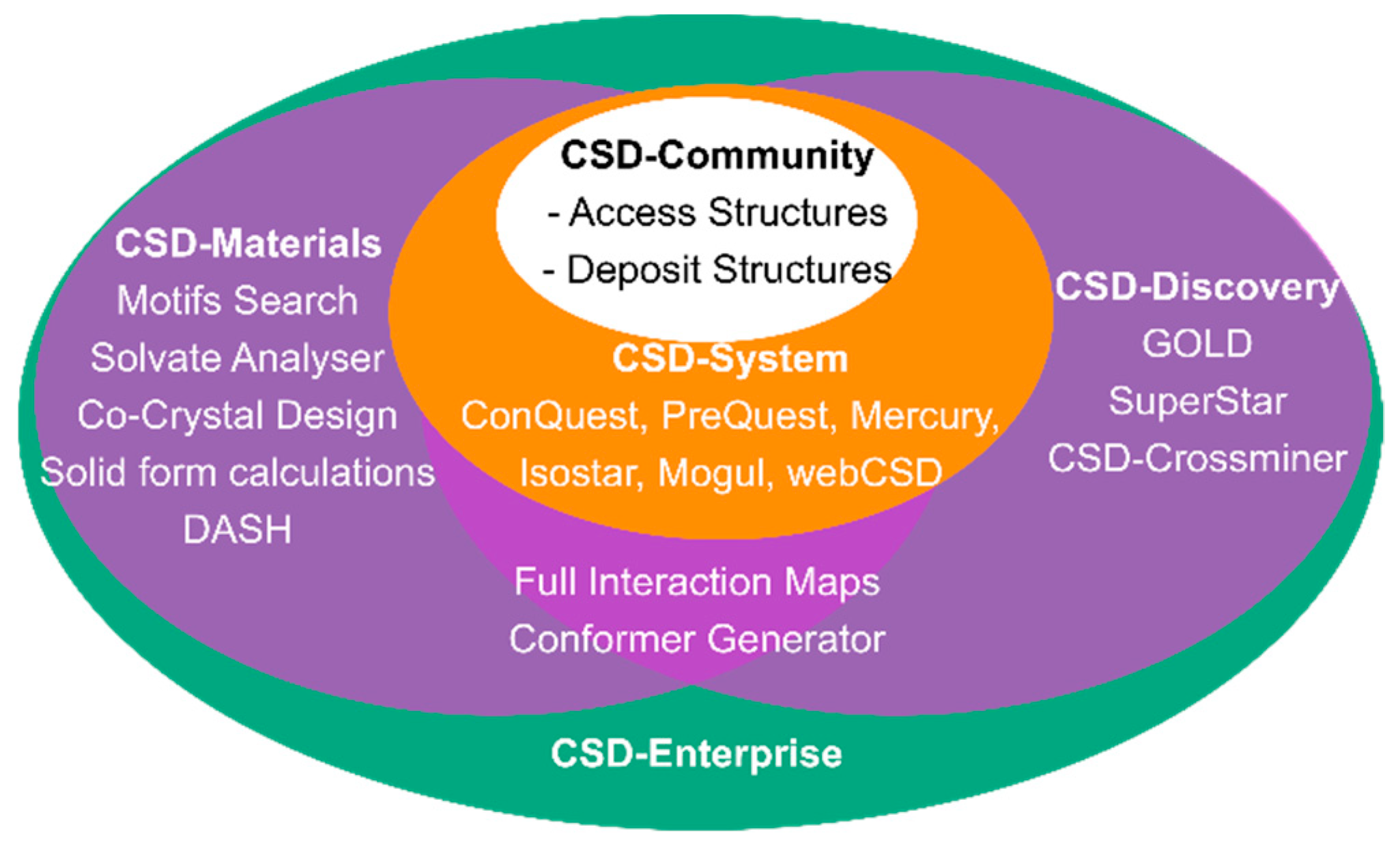
2. The Cambridge Structural Database (CSD) and its’ Libraries and Modules
The CSD-Materials Module
- Analysis of H-bond motifs (searching of motifs and statistics of their occurrences; assessing the risk of polymorphism via H-bond propensities; prediction of co-crystal formation).
- Analysis of crystal packing features (searching on selected motifs, analysis of packing similarity and building of a packing similarity tree diagram).
- Calculation of theoretical crystal morphology, gas phase MOPAC (Molecular Orbital PACkage) calculation and ‘UNI’ (UNIversal) force field intermolecular energy calculations.
- Mapping of interaction preferences around an isolated molecule.
- Analysis of solvate and hydrate crystal structures (searching and classification of H-bond motifs, calculation of the volume occupied in a unit cell with solvent molecules, mapping of interaction preferences around them).
- Generation of conformers based on geometrical statistics from the CSD.
- Solution of crystal structures from powder diffraction data using DASH.
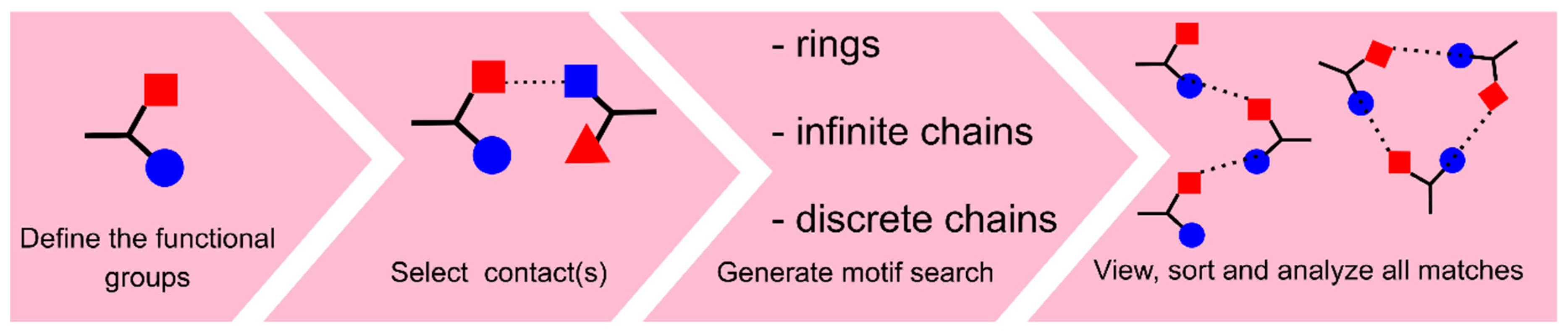
3. Search and Analysis of Supramolecular Associates
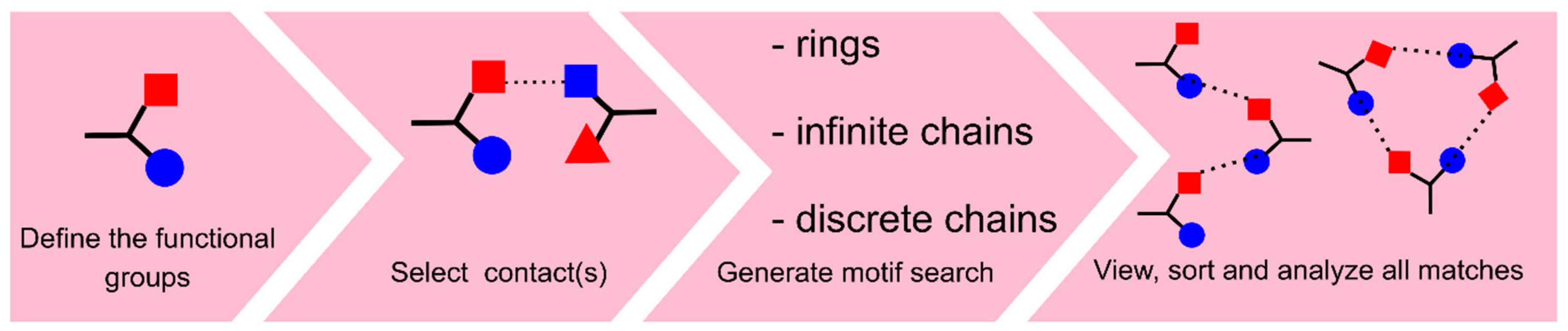
3.1. Search on Hydrogen-Bond Motifs
3.2. StudyingCrystal Packing Features
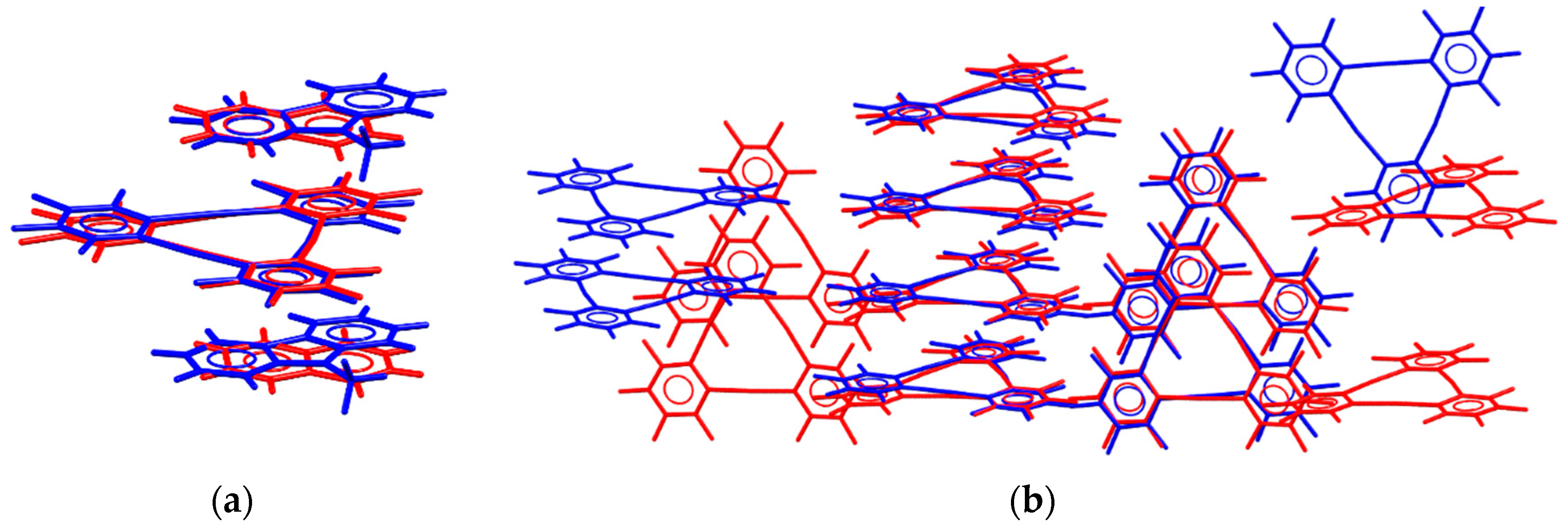
3.3. Crystal Packing Similarity Tool
3.4. CSD-Crossminer
4. Solid Form Calculations
4.1. BFDH Morphology Prediction
5. Knowlede-Based Prediction of Supramolecular Associates and Solid Forms
5.1. Polymorph Assessment
5.2. Co-Crystals Design
5.3. Full Interaction Maps
5.4. CSD-Conformer Generator
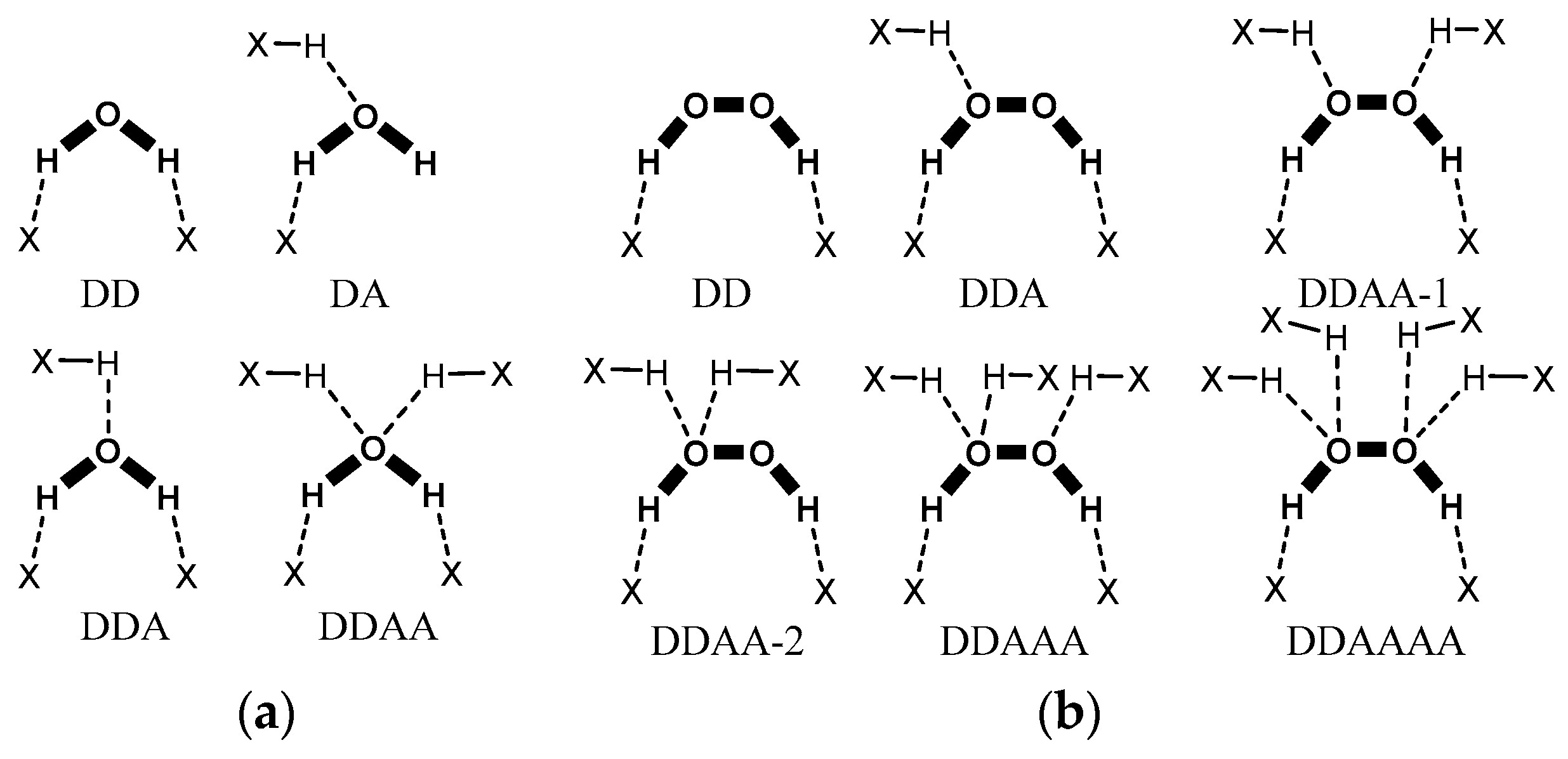
6. Hydrate/Solvate Analyser
6.1. Analysis of the Local Connectivity of a Solvent
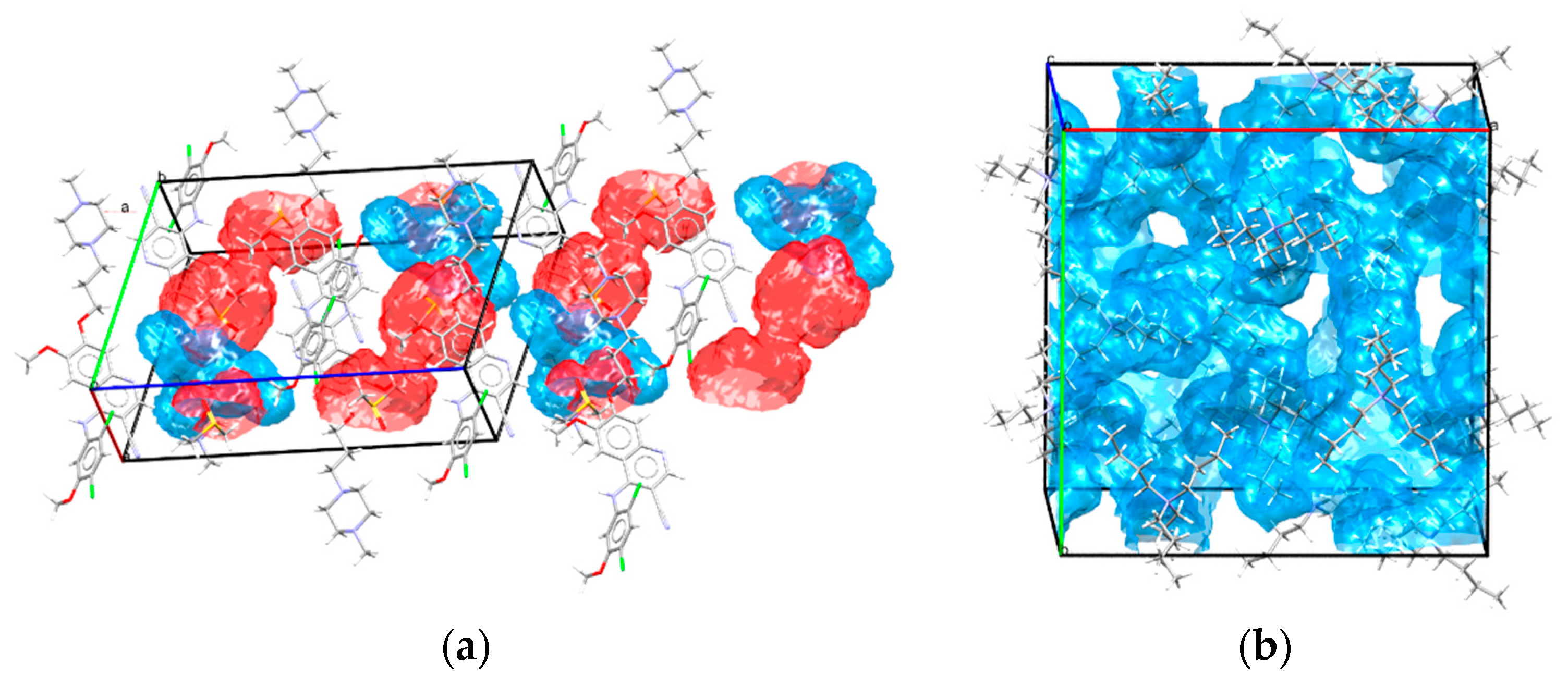
6.2. Analysis of Solvent Associates
7. Additional Software Compatible with the CSD-Materials Analysis of Structure–Property Networks
7.1. CSD Python API
- Analysis (generation of conformers and calculation of their RMSD (conformes_similarity.py) or of all torsion angles (generate_conformers.py) for the loaded structure).
- External (load_in_conquest.py– loading the current structure into the ConQuest sketch window).
- Reports (generating a report in *.html format containing crystallographic details, molecular geometry, and intermolecular hydrogen and halogen bonding (crystal_structure_report.py), or molecular geometry, including Mogul geometry analysis (molecular_geometry_report.py), or crystal packing descriptors (quick_packing_check.py), or basic chemical, crystallographic and publication information about the loaded structure (structure-simple_report.py)).
- Searches within the CSD for entries relevant to the specified chemical name or synonym (chemical_name_search.py) or having similar molecular geometry (molecular_similarity_search.py).
- One can additionally download from the CSD-Python API Forum [308] the following scripts:
- To generate molecular formula and weight (welcome-and-weight.py), Crystal14 input files (crystal_inputs.py) or Packing Similarity dendrogram (Packing_Similarity_Dendrogram.py);
- To send a Mol2 or CIF files to an external application (send_mol2_to_notepad+.py);
- To generate diagrams for all molecules in a structure file (diagram_to_file.py);
- To find covalently bonded clusters within a structure (dimensionality.py);
- To perform void calculation in a crystal (void_calc.py);
- To merge GOLD docking results (gold_merge.py);
- To filter molecular conformers with unusual torsion angles (conformer_filter_density.py);
- To extract from the CSD unique rings of a specified size (ring_type_count.py);
- To compare protein bound ligand with CSD-Generated conformers (find_binding_conformation.py).
7.2. Combination of Various Tools and Algorythms
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Allen, F.; Kennard, O.; Watson, D.; Brammer, L.; Orpen, A.; Taylor, R. Tables of Bond Lengths Determined by X-Ray and Neutron-Diffraction .1. Bond Lengths in Organic-Compounds. J. Chem. Soc.-Perkin Trans. 2 1987, S1–S19. [Google Scholar] [CrossRef]
- Orpen, A.; Brammer, L.; Allen, F.; Kennard, O.; Watson, D.; Taylor, R. Tables of Bond Lengths\Determined by X-Ray and Neutron-Diffraction .2. Organometallic Compounds and Co-Ordination Complexes of the D-Block and F-Block Metals. J. Chem. Soc.-Dalton Trans. 1989, S1–S83. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Taylor, R. Systematic analysis of structural data as a research technique in organic chemistry. Acc. Chem. Res. 1983, 16, 146–153. [Google Scholar] [CrossRef]
- Allen, F.H.; Kirby, A.J. Bond length and reactivity. Variable length of the carbon-oxygen single bond. J. Am. Chem. Soc. 1984, 106, 6197–6200. [Google Scholar] [CrossRef]
- Kitaigorodskii, A.I. Molecular Crystals and Molecules; Academic Press: New York, NY, USA, 1973. [Google Scholar]
- Desiraju, G.R.; Parshall, G.W. Crystal Engineering: The Design of Organic Solids; Elsevier: Amsterdam, The Netherlands, 1989. [Google Scholar]
- Lehn, J.-M. Supramolecular Chemistry: Concepts and Perspectives; Wiley-VCH: Weinheim, Germany, 1995. [Google Scholar]
- Nangia, A.K.; Desiraju, G.R. Crystal Engineering: An Outlook for the Future. Angew. Chem. Int. Ed. 2019, 58, 4100–4107. [Google Scholar] [CrossRef]
- Corpinot, M.K.; Bučar, D.-K. A Practical Guide to the Design of Molecular Crystals. Cryst. Growth Des. 2019, 19, 1426–1453. [Google Scholar] [CrossRef]
- Duggirala, N.K.; Perry, M.L.; Almarsson, Ö.; Zaworotko, M.J. Pharmaceutical cocrystals: Along the path to improved medicines. Chem. Commun. 2015, 52, 640–655. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, W.; Sheng, P.; Zhao, G.; Zhu, D. Organic Donor–Acceptor Complexes as Novel Organic Semiconductors. Acc. Chem. Res. 2017, 50, 1654–1662. [Google Scholar] [CrossRef]
- Shi, P.-P.; Tang, Y.-Y.; Li, P.-F.; Liao, W.-Q.; Wang, Z.-X.; Ye, Q.; Xiong, R.-G. Symmetry breaking in molecular ferroelectrics. Chem. Soc. Rev. 2016, 45, 3811–3827. [Google Scholar] [CrossRef]
- Turkoglu, G.; Cinar, M.E.; Ozturk, T. Triarylborane-Based Materials for OLED Applications. Molecules 2017, 22, 1522. [Google Scholar] [CrossRef]
- Kidyarov, B.I. Comparative Interrelationship of the Structural, Nonlinear-Optical and Other Acentric Properties for Oxide, Borate and Carbonate Crystals. Crystals 2017, 7, 109. [Google Scholar] [CrossRef]
- Quan, L.N.; Rand, B.P.; Friend, R.H.; Mhaisalkar, S.G.; Lee, T.-W.; Sargent, E.H. Perovskites for Next-Generation Optical Sources. Chem. Rev. 2019, 119, 7444–7477. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Carugo, O.; Blatova, O.A.; Medrish, E.O.; Blatov, V.A.; Proserpio, D.M. Packing topology in crystals of proteins and small molecules: A comparison. Sci. Rep. 2017, 7, 13209. [Google Scholar] [CrossRef]
- Taylor, R.; Wood, P.A. A Million Crystal Structures: The Whole Is Greater than the Sum of Its Parts. Chem. Rev. 2019, 119, 9247–9477. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Allen, F.H. The Cambridge Structural Database in Retrospect and Prospect. Angew. Chem. Int. Ed. 2014, 53, 662–671. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. Sect. B 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Belsky, A.; Hellenbrandt, M.; Karen, V.L.; Luksch, P. New developments in the Inorganic Crystal Structure Database (ICSD): Accessibility in support of materials research and design. Acta Cryst. Sect. B 2002, 58, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Peresypkina, E.V.; Blatov, V.A. Topology of molecular packings in organic crystals. Acta Cryst. Sect. B 2000, 56, 1035–1045. [Google Scholar] [CrossRef] [Green Version]
- Peresypkina, E.V.; Blatov, V.A. Molecular coordination numbers in crystal structures of organic compounds. Acta Cryst. Sect. B 2000, 56, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Pathaneni, S.S.; Desiraju, G.R. Database analysis of Au⋯Au interactions. J. Chem. Soc. Dalton Trans. 1993, 319–322. [Google Scholar] [CrossRef]
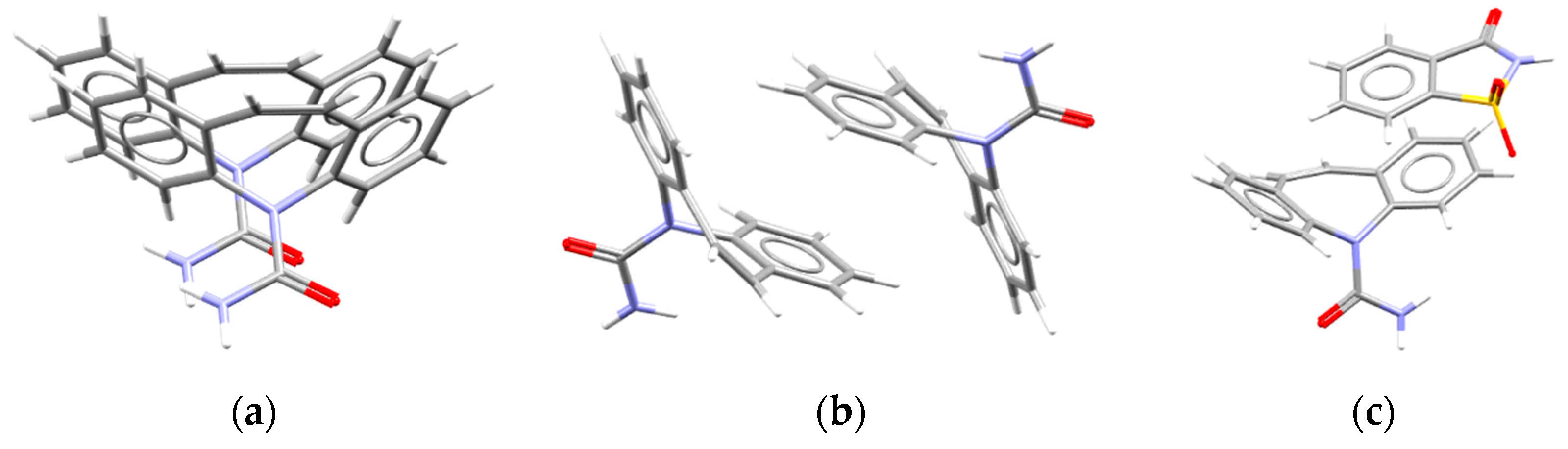
- Terrasson, V.; Planas, J.G.; Prim, D.; Teixidor, F.; Viñas, C.; Light, M.E.; Hursthouse, M.B. General Access to Aminobenzyl-o-carboranes as a New Class of Carborane Derivatives: Entry to Enantiopure Carborane–Amine Combinations. Chem.—A Eur. J. 2009, 15, 12030–12042. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.P.; Pavan, M.S.; Row, T.N.G. Experimental evidence for ‘carbon bonding’ in the solid state from charge density analysis. Chem. Commun. 2013, 50, 49–51. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Del Bene, J.E.; Elguero, J. H2XP:OH2 Complexes: Hydrogen vs. Pnicogen Bonds. Crystals 2016, 6, 19. [Google Scholar] [CrossRef]
- Lodochnikova, O.A.; Latypova, L.Z.; Madzhidov, T.I.; Chmutova, G.A.; Voronina, J.K.; Gubaidullin, A.T.; Kurbangalieva, A.R. “Lp⋯synthon” interaction as a reason for the strong amplification of synthon-forming hydrogen bonds. CrystEngComm 2019, 21, 1499–1511. [Google Scholar] [CrossRef]
- Taylor, R. Which intermolecular interactions have a significant influence on crystal packing? CrystEngComm 2014, 16, 6852–6865. [Google Scholar] [CrossRef]
- Taylor, R. It Isn’t, It Is: The C–H···X (X = O, N, F, Cl) Interaction Really Is Significant in Crystal Packing. Cryst. Growth Des. 2016, 16, 4165–4168. [Google Scholar] [CrossRef]
- Jelsch, C.; Ejsmont, K.; Huder, L. The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis. IUCrJ 2014, 1, 119–128. [Google Scholar] [CrossRef]
- Jelsch, C.; Bibila Mayaya Bisseyou, Y. Atom interaction propensities of oxygenated chemical functions in crystal packings. IUCrJ 2017, 4, 158–174. [Google Scholar] [CrossRef]
- Buergi, H.B.; Dunitz, J.D. From crystal statics to chemical dynamics. Acc. Chem. Res. 1983, 16, 153–161. [Google Scholar] [CrossRef]
- Bürgi, H.-B.; Dunitz, J.D. Structure Correlation; Wiley-VCH: Weinheim, Germany, 1994. [Google Scholar]
- Vologzhanina, A.V.; Korlyukov, A.A.; Antipin, M.Y. Special features of intermolecular bonding A⋯D (A = Si, Ge and D = nucleophile) in crystal structures. Acta Cryst. Sect. B 2008, 64, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Bruno, I.J.; Cole, J.C.; Kessler, M.; Luo, J.; Motherwell, W.D.S.; Purkis, L.H.; Smith, B.R.; Taylor, R.; Cooper, R.I.; Harris, S.E.; et al. Retrieval of Crystallographically-Derived Molecular Geometry Information. J. Chem. Inf. Comput. Sci. 2004, 44, 2133–2144. [Google Scholar] [CrossRef] [PubMed]
- Bruno, I.J.; Cole, J.C.; Lommerse, J.P.M.; Rowland, R.S.; Taylor, R.; Verdonk, M.L. IsoStar: A library of information about nonbonded interactions. J. Comput. Aided Mol. Des. 1997, 11, 525–537. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, J.; Feeder, N.; Hunter, C.A. H-bond competition experiments in solution and the solid state. CrystEngComm 2016, 18, 394–397. [Google Scholar] [CrossRef]
- Mascal, M.; Infantes, L.; Chisholm, J. Water Oligomers in Crystal Hydrates—What’s News and What Isn’t? Angew. Chem. Int. Ed. 2006, 45, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Manna, U.; Halder, S.; Das, G. Ice-like Cyclic Water Hexamer Trapped within a Halide Encapsulated Hexameric Neutral Receptor Core: First Crystallographic Evidence of a Water Cluster Confined within a Receptor-Anion Capsular Assembly. Cryst. Growth Des. 2018, 18, 1818–1825. [Google Scholar] [CrossRef]
- Desiraju, G.R. Supramolecular Synthons in Crystal Engineering—A New Organic Synthesis. Angew. Chem. Int. Ed. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Aakeröy, C.B. Crystal Engineering: Strategies and Architectures. Acta Cryst. Sect. B 1997, 53, 569–586. [Google Scholar] [CrossRef]
- Hollingsworth, M.D. Crystal Engineering: From Structure to Function. Science 2002, 295, 2410–2413. [Google Scholar]
- Nagarathinam, M.; Peedikakkal, A.M.P.; Vittal, J.J. Stacking of double bonds for photochemical [2+2] cycloaddition reactions in the solid state. Chem. Commun. 2008, 5277–5288. [Google Scholar] [CrossRef] [PubMed]
- Ramamurthy, V.; Sivaguru, J. Supramolecular Photochemistry as a Potential Synthetic Tool: Photocycloaddition. Chem. Rev. 2016, 116, 9914–9993. [Google Scholar] [CrossRef] [PubMed]
- Kreuer, K.-D. Proton Conductivity: Materials and Applications. Chem. Mater. 1996, 8, 610–641. [Google Scholar] [CrossRef]
- Horiuchi, S.; Kumai, R.; Tokura, Y. Hydrogen-Bonding Molecular Chains for High-Temperature Ferroelectricity. Adv. Mater. 2011, 23, 2098–2103. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, S.; Noda, Y.; Hasegawa, T.; Kagawa, F.; Ishibashi, S. Correlated Proton Transfer and Ferroelectricity along Alternating Zwitterionic and Nonzwitterionic Anthranilic Acid Molecules. Chem. Mater. 2015, 27, 6193–6197. [Google Scholar] [CrossRef]
- Horiuchi, S.; Kagawa, F.; Hatahara, K.; Kobayashi, K.; Kumai, R.; Murakami, Y.; Tokura, Y. Above-room-temperature ferroelectricity and antiferroelectricity in benzimidazoles. Nat. Commun. 2012, 3, 1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owczarek, M.; Hujsak, K.A.; Ferris, D.P.; Prokofjevs, A.; Majerz, I.; Szklarz, P.; Zhang, H.; Sarjeant, A.A.; Stern, C.L.; Jakubas, R.; et al. Flexible ferroelectric organic crystals. Nat. Commun. 2016, 7, 13108. [Google Scholar] [CrossRef] [PubMed]
- Borel, C.; Larsson, K.; Håkansson, M.; Olsson, B.E.; Bond, A.D.; Öhrström, L. Oxalate- and Squarate-Biimidazole Supramolecular Synthons: Hydrogen-Bonded Networks Based on [Co(H2biimidazole)3]3+. Cryst. Growth Des. 2009, 9, 2821–2827. [Google Scholar] [CrossRef]
- Pallipurath, A.R.; Civati, F.; Eziashi, M.; Omar, E.; McArdle, P.; Erxleben, A. Tailoring Cocrystal and Salt Formation and Controlling the Crystal Habit of Diflunisal. Cryst. Growth Des. 2016, 16, 6468–6478. [Google Scholar] [CrossRef] [Green Version]
- George, F.; Norberg, B.; Wouters, J.; Leyssens, T. Structural Investigation of Substituent Effect on Hydrogen Bonding in (S)-Phenylglycine Amide Benzaldimines. Cryst. Growth Des. 2015, 15, 4005–4019. [Google Scholar] [CrossRef]
- Bernstein, J. Polymorphism in Molecular Crystals; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Bauer, J.; Spanton, S.; Henry, R.; Quick, J.; Dziki, W.; Porter, W.; Morris, J. Ritonavir: An Extraordinary Example of Conformational Polymorphism. Pharm. Res. 2001, 18, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Dalinger, I.L.; Vatsadze, I.A.; Shkineva, T.K.; Kormanov, A.V.; Struchkova, M.I.; Suponitsky, K.Yu.; Bragin, A.A.; Monogarov, K.A.; Sinditskii, V.P.; Sheremetev, A.B. Novel Highly Energetic Pyrazoles: N-Trinitromethyl-Substituted Nitropyrazoles. Chem.—Asian J. 2015, 10, 1987–1996. [Google Scholar] [CrossRef] [PubMed]
- Dalinger, I.L.; Kormanov, A.V.; Suponitsky, K.Y.; Muravyev, N.V.; Sheremetev, A.B. Pyrazole-Tetrazole Hybrid with Trinitromethyl, Fluorodinitromethyl, or (Difluoroamino)dinitromethyl Groups: High-Performance Energetic Materials. Chem.—Asian J. 2018, 13, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Dalinger, I.L.; Serushkina, O.V.; Muravyev, N.V.; Meerov, D.B.; Miroshnichenko, E.A.; Kon’kova, T.S.; Suponitsky, K.Y.; Vener, M.V.; Sheremetev, A.B. Azasydnone—Novel “green” building block for designing high energetic compounds. J. Mater. Chem. A 2018, 6, 18669–18676. [Google Scholar] [CrossRef]
- Bredikhin, A.A.; Bredikhina, Z.A.; Zakharychev, D.V. Crystallization of chiral compounds: Thermodynamical, structural and practical aspects. Mendeleev Commun. 2012, 22, 171–180. [Google Scholar] [CrossRef]
- Bredikhin, A.A.; Zakharychev, D.V.; Bredikhina, Z.A.; Kurenkov, A.V.; Krivolapov, D.B.; Gubaidullin, A.T. Spontaneous Resolution of Chiral 3-(2,3-Dimethylphenoxy)propane-1,2-diol under the Circumstances of an Unusual Diversity of Racemic Crystalline Modifications. Cryst. Growth Des. 2017, 17, 4196–4206. [Google Scholar] [CrossRef]
- Kinbara, K.; Hashimoto, Y.; Sukegawa, M.; Nohira, H.; Saigo, K. Crystal Structures of the Salts of Chiral Primary Amines with Achiral Carboxylic Acids: Recognition of the Commonly-Occurring Supramolecular Assemblies of Hydrogen-Bond Networks and Their Role in the Formation of Conglomerates. J. Am. Chem. Soc. 1996, 118, 3441–3449. [Google Scholar] [CrossRef]
- Haynes, D.A.; Chisholm, J.A.; Jones, W.; Motherwell, W.D.S. Supramolecular synthon competition in organic sulfonates: A CSD survey. CrystEngComm 2004, 6, 584–588. [Google Scholar] [CrossRef]
- da Silva, C.C.P.; de Oliveira, R.; Tenorio, J.C.; Honorato, S.B.; Ayala, A.P.; Ellena, J. The Continuum in 5-Fluorocytosine. Toward Salt Formation. Cryst. Growth Des. 2013, 13, 4315–4322. [Google Scholar] [CrossRef]
- Chisholm, J.A.; Motherwell, S. COMPACK: A program for identifying crystal structure similarity using distances. J. Appl. Cryst. 2005, 38, 228–231. [Google Scholar] [CrossRef]
- Wanat, M.; Malinska, M.; Kutner, A.; Wozniak, K. Effect of Vitamin D Conformation on Interactions and Packing in the Crystal Lattice. Cryst. Growth Des. 2018, 18, 3385–3396. [Google Scholar] [CrossRef]
- Infantes, L.; Motherwell, S. Water clusters in organic molecular crystals. CrystEngComm 2002, 4, 454–461. [Google Scholar] [CrossRef]
- Blatov, V.A.; O’Keeffe, M.; Proserpio, D.M. Vertex-, face-, point-, Schläfli-, and Delaney-symbols in nets, polyhedra and tilings: Recommended terminology. CrystEngComm 2009, 12, 44–48. [Google Scholar] [CrossRef]
- Batten, S.R.; Champness, N.R.; Chen, X.-M.; Garcia-Martinez, J.; Kitagawa, S.; Ohrstrom, L.; O’Keeffe, M.; Suh, M.P.; Reedijk, J. Terminology of metal–organic frameworks and coordination polymers (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1715–1724. [Google Scholar] [CrossRef]
- Ohrstrom, L. Designing, Describing and Disseminating New Materials by using the Network Topology Approach. Chem.—Eur. J. 2016, 22, 13758–13763. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, C.; O’Keeffe, M.; Proserpio, D.M.; Blatov, V.A.; Batten, S.R.; Bourne, S.A.; Lah, M.S.; Eon, J.-G.; Hyde, S.T.; Wiggin, S.B.; et al. Deconstruction of Crystalline Networks into Underlying Nets: Relevance for Terminology Guidelines and Crystallographic Databases. Cryst. Growth Des. 2018, 18, 3411–3418. [Google Scholar] [CrossRef]
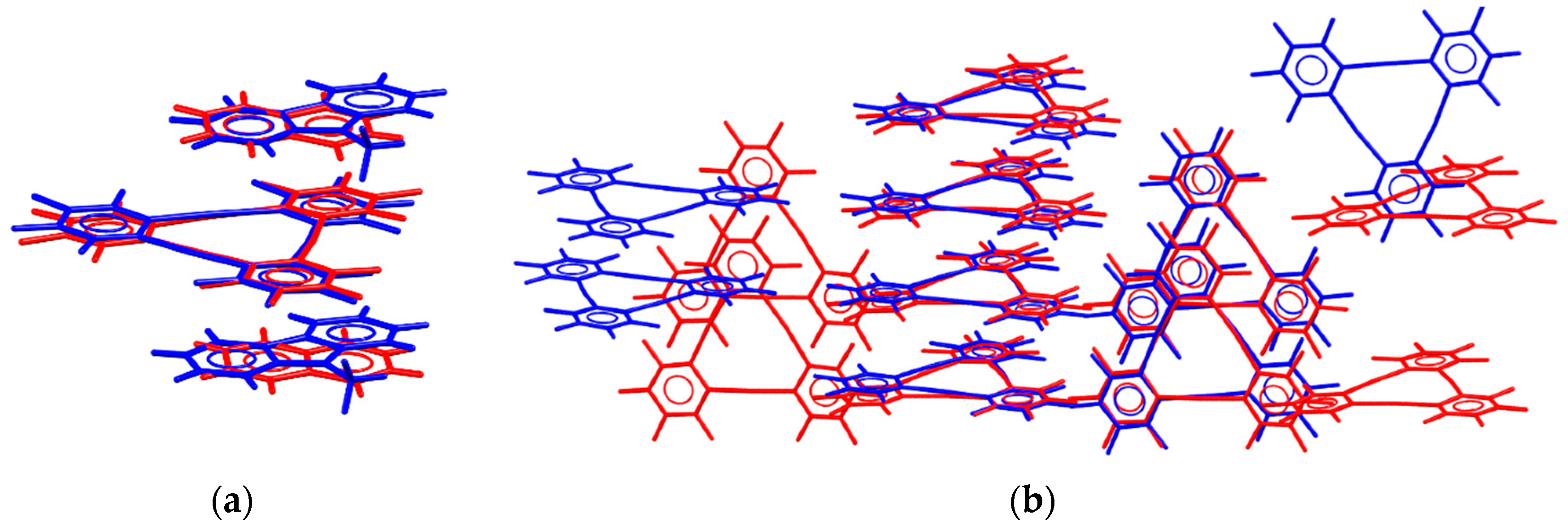
- Haneline, M.R.; Tsunoda, M.; Gabbaï, F.P. π-Complexation of Biphenyl, Naphthalene, and Triphenylene to Trimeric Perfluoro-ortho-phenylene Mercury. Formation of Extended Binary Stacks with Unusual Luminescent Properties. J. Am. Chem. Soc. 2002, 124, 3737–3742. [Google Scholar] [CrossRef] [PubMed]
- Burress, C.; Elbjeirami, O.; Omary, M.A.; Gabbaï, F.P. Five-Order-of-Magnitude Reduction of the Triplet Lifetimes of N-Heterocycles by Complexation to a Trinuclear Mercury Complex. J. Am. Chem. Soc. 2005, 127, 12166–12167. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.J.; Burress, C.N.; Gabbaï, F.P. Lewis Acidic Behavior of Fluorinated Organomercurials. Organometallics 2007, 26, 5252–5263. [Google Scholar] [CrossRef]
- Ferraris, J.; Cowan, D.O.; Walatka, V.; Perlstein, J.H. Electron transfer in a new highly conducting donor-acceptor complex. J. Am. Chem. Soc. 1973, 95, 948–949. [Google Scholar] [CrossRef]
- Giri, G.; Verploegen, E.; Mannsfeld, S.C.B.; Atahan-Evrenk, S.; Kim, D.H.; Lee, S.Y.; Becerril, H.A.; Aspuru-Guzik, A.; Toney, M.F.; Bao, Z. Tuning charge transport in solution-sheared organic semiconductors using lattice strain. Nature 2011, 480, 504–508. [Google Scholar] [CrossRef] [PubMed]
- Varghese, S.; Das, S. Role of Molecular Packing in Determining Solid-State Optical Properties of π-Conjugated Materials. J. Phys. Chem. Lett. 2011, 2, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.; Wang, X.; Ju, H.; Zhao, L.; Guo, T.; Wu, W.; Wang, H. Controllable Molecular Packing Motif and Overlap Type in Organic Nanomaterials for Advanced Optical Properties. Crystals 2018, 8, 22. [Google Scholar] [CrossRef]
- Larin, A.A.; Muravyev, N.V.; Pivkina, A.N.; Suponitsky, K.Y.; Ananyev, I.V.; Khakimov, D.V.; Fershtat, L.L.; Makhova, N.N. Assembly of Tetrazolylfuroxan Organic Salts: Multipurpose Green Energetic Materials with High Enthalpies of Formation and Excellent Detonation Performance. Chem.—Eur. J. 2019, 25, 4225–4233. [Google Scholar] [CrossRef] [PubMed]
- Dolgushin, F.M.; Smol’yakov, A.F.; Suponitsky, K.Y.; Vologzhanina, A.V.; Fedyanin, I.V.; Shishkina, S.V. Intermolecular interactions in polymorphs of the cyclic trimeric perfluoro-ortho-phenylene mercury from geometric, energetic and AIM viewpoints: DFT study and Hirshfeld surface analysis. Struct. Chem. 2016, 27, 37–49. [Google Scholar] [CrossRef]
- Torubaev, Y.V.; Rai, D.K.; Skabitsky, I.V.; Pakhira, S.; Dmitrienko, A. Energy framework approach to the supramolecular reactions: Interplay of the secondary bonding interaction in Ph2E2 (E = Se, Te)/p-I-C6F4-I co-crystals. New J. Chem. 2019, 43, 7941–7949. [Google Scholar] [CrossRef]
- Martins, F.T.; Paparidis, N.; Doriguetto, A.C.; Ellena, J. Crystal Engineering of an Anti-HIV Drug Based on the Recognition of Assembling Molecular Frameworks. Cryst. Growth Des. 2009, 9, 5283–5292. [Google Scholar] [CrossRef]
- Pogoda, D.; Janczak, J.; Videnova-Adrabinska, V. New polymorphs of an old drug: Conformational and synthon polymorphism of 5-nitrofurazone. Acta Cryst. Sect. B 2016, 72, 263–273. [Google Scholar] [CrossRef]
- Reilly, A.M.; Cooper, R.I.; Adjiman, C.S.; Bhattacharya, S.; Boese, A.D.; Brandenburg, J.G.; Bygrave, P.J.; Bylsma, R.; Campbell, J.E.; Car, R.; et al. Report on the sixth blind test of organic crystal structure prediction methods. Acta Cryst. Sect. B 2016, 72, 439–459. [Google Scholar] [CrossRef]
- Giangreco, I.; Cole, J.C.; Thomas, E. Mining the Cambridge Structural Database for Matched Molecular Crystal Structures: A Systematic Exploration of Isostructurality. Cryst. Growth Des. 2017, 17, 3192–3203. [Google Scholar] [CrossRef]
- Carletta, A.; Colaço, M.; Mouchet, S.R.; Plas, A.; Tumanov, N.; Fusaro, L.; Champagne, B.; Lanners, S.; Wouters, J. Tetraphenylborate Anion Induces Photochromism in N-Salicylideneamino-1-alkylpyridinium Derivatives Through Formation of Tetra-Aryl Boxes. J. Phys. Chem. C 2018, 122, 10999–11007. [Google Scholar] [CrossRef]
- Nath, N.K.; Saha, B.K.; Nangia, A. Isostructural polymorphs of triiodophloroglucinol and triiodoresorcinol. New J. Chem. 2008, 32, 1693–1701. [Google Scholar] [CrossRef]
- Moorthy, J.N.; Mandal, S.; Venugopalan, P. Hydrogen-Bonded Helical Self-Assembly of Sterically-Hindered Benzyl Alcohols: Rare Isostructurality and Synthon Equivalence Between Alcohols and Acids. Cryst. Growth Des. 2012, 12, 2942–2947. [Google Scholar] [CrossRef]
- Reddy, C.M.; Kirchner, M.T.; Gundakaram, R.C.; Padmanabhan, K.A.; Desiraju, G.R. Isostructurality, Polymorphism and Mechanical Properties of Some Hexahalogenated Benzenes: The Nature of Halogen⋅⋅⋅Halogen Interactions. Chem.—Eur. J. 2006, 12, 2222–2234. [Google Scholar] [CrossRef] [PubMed]
- Asmadi, A.; Kendrick, J.; Leusen, F.J.J. Crystal Structure Prediction and Isostructurality of Three Small Molecules. Chem.—Eur. J. 2010, 16, 12701–12709. [Google Scholar] [CrossRef]
- Owczarzak, A.M.; Kourkoumelis, N.; Hadjikakou, S.K.; Kubicki, M. The impact of the anion size on the crystal packing in 2-mercaptopyrimidine halides; isostructurality and polymorphism. CrystEngComm 2013, 15, 3607–3614. [Google Scholar] [CrossRef]
- Ravat, P.; SeethaLekshmi, S.; Biswas, S.N.; Nandy, P.; Varughese, S. Equivalence of Ethylene and Azo-Bridges in the Modular Design of Molecular Complexes: Role of Weak Interactions. Cryst. Growth Des. 2015, 15, 2389–2401. [Google Scholar] [CrossRef]
- Nath, N.K.; Nangia, A. Isomorphous Crystals by Chloro–Methyl Exchange in Polymorphic Fuchsones. Cryst. Growth Des. 2012, 12, 5411–5425. [Google Scholar] [CrossRef]
- Gonnade, R.G.; Bhadbhade, M.M.; Shashidhar, M.S. Crystal-to-crystal thermal phase transition amongst dimorphs of hexa-O-p-toluoyl-myo-inositol conserving two-dimensional isostructurality. CrystEngComm 2010, 12, 478–484. [Google Scholar] [CrossRef]
- Gelbrich, T.; Hughes, D.S.; Hursthouse, M.B.; Threlfall, T.L. Packing similarity in polymorphs of sulfathiazole. CrystEngComm 2008, 10, 1328–1334. [Google Scholar] [CrossRef]
- Panini, P.; Mohan, T.P.; Gangwar, U.; Sankolli, R.; Chopra, D. Quantitative crystal structure analysis of 1,3,4-thiadiazole derivatives. CrystEngComm 2013, 15, 4549–4564. [Google Scholar] [CrossRef]
- Chopra, D.; Row, T.N.G. Evaluation of the interchangeability of C–H and C–F groups: Insights from crystal packing in a series of isomeric fluorinated benzanilides. CrystEngComm 2007, 10, 54–67. [Google Scholar] [CrossRef]
- Thakuria, R.; Nath, N.K.; Roy, S.; Nangia, A. Polymorphism and isostructurality in sulfonylhydrazones. CrystEngComm 2014, 16, 4681–4690. [Google Scholar] [CrossRef]
- Gelbrich, T.; Hursthouse, M.B. A versatile procedure for the identification, description and quantification of structural similarity in molecular crystals. CrystEngComm 2005, 7, 324–336. [Google Scholar] [CrossRef]
- Dzyabchenko, A.V. Method of crystal-structure similarity searching. Acta Cryst. Sect. B 1994, 50, 414–425. [Google Scholar]
- Suponitsky, K.Y.; Lyssenko, K.A.; Ananyev, I.V.; Kozeev, A.M.; Sheremetev, A.B. Role of Weak Intermolecular Interactions in the Crystal Structure of Tetrakis-furazano[3,4-c:3′,4′-g:3″,4″-k:3‴,4‴-o][1,2,5,6,9,10,13,14]octaazacyclohexadecine and Its Solvates. Cryst. Growth Des. 2014, 14, 4439–4449. [Google Scholar] [CrossRef]
- Childs, S.L.; Wood, P.A.; Rodríguez-Hornedo, N.; Reddy, L.S.; Hardcastle, K.I. Analysis of 50 Crystal Structures Containing Carbamazepine Using the Materials Module of Mercury CSD. Cryst. Growth Des. 2009, 9, 1869–1888. [Google Scholar] [CrossRef]
- Gelbrich, T.; Hursthouse, M.B. Systematic investigation of the relationships between 25 crystal structures containing the carbamazepine molecule or a close analogue: A case study of the XPac method. CrystEngComm 2006, 8, 448–460. [Google Scholar] [CrossRef]
- Prohens, R.; Font-Bardia, M.; Barbas, R. Water wires in the nanoporous form II of carbamazepine: A single-crystal X-ray diffraction analysis. CrystEngComm 2013, 15, 845–847. [Google Scholar] [CrossRef]
- Zhong, Z.; Yang, X.; Wang, B.-H.; Yao, Y.-F.; Guo, B.; Yu, L.; Huang, Y.; Xu, J. Solvent-polymer guest exchange in a carbamazepine inclusion complex: Structure, kinetics and implication for guest selection. CrystEngComm 2019, 21, 2164–2173. [Google Scholar] [CrossRef]
- Kennedy, A.R.; Morrison, C.A.; Briggs, N.E.B.; Arbuckle, W. Density and Stability Differences Between Enantiopure and Racemic Salts: Construction and Structural Analysis of a Systematic Series of Crystalline Salt Forms of Methylephedrine. Cryst. Growth Des. 2011, 11, 1821–1834. [Google Scholar] [CrossRef]
- Tumanov, N.A.; Myz, S.A.; Shakhtshneider, T.P.; Boldyreva, E.V. Are meloxicam dimers really the structure-forming units in the ‘meloxicam–carboxylic acid’ co-crystals family? Relation between crystal structures and dissolution behaviour. CrystEngComm 2012, 14, 305–313. [Google Scholar] [CrossRef]
- Ueto, T.; Takata, N.; Muroyama, N.; Nedu, A.; Sasaki, A.; Tanida, S.; Terada, K. Polymorphs and a Hydrate of Furosemide–Nicotinamide 1:1 Cocrystal. Cryst. Growth Des. 2012, 12, 485–494. [Google Scholar] [CrossRef]
- Galcera, J.; Friščić, T.; Molins, E.; Jones, W. Isostructurality in three-component crystals achieved by the combination of persistent hydrogen bonding motifs and solvent inclusion. CrystEngComm 2013, 15, 1332–1338. [Google Scholar] [CrossRef]
- Briggs, N.E.; Kennedy, A.R.; Morrison, C.A. 42 salt forms of tyramine: Structural comparison and the occurrence of hydrate formation. Acta Cryst. Sect. B 2012, 68, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Salbego, P.R.S.; Bender, C.R.; Hörner, M.; Zanatta, N.; Frizzo, C.P.; Bonacorso, H.G.; Martins, M.A.P. Insights on the Similarity of Supramolecular Structures in Organic Crystals Using Quantitative Indexes. ACS Omega 2018, 3, 2569–2578. [Google Scholar] [CrossRef] [PubMed]
- Surov, A.O.; Simagina, A.A.; Manin, N.G.; Kuzmina, L.G.; Churakov, A.V.; Perlovich, G.L. Fenamate Cocrystals with 4,4′-Bipyridine: Structural and Thermodynamic Aspects. Cryst. Growth Des. 2015, 15, 228–238. [Google Scholar] [CrossRef]
- Sládková, V.; Skalická, T.; Skořepová, E.; Čejka, J.; Eigner, V.; Kratochvíl, B. Systematic solvate screening of trospium chloride: Discovering hydrates of a long-established pharmaceutical. CrystEngComm 2015, 17, 4712–4721. [Google Scholar] [CrossRef]
- Chennuru, R.; Muthudoss, P.; Voguri, R.S.; Ramakrishnan, S.; Vishweshwar, P.; Babu, R.R.C.; Mahapatra, S. Iso-Structurality Induced Solid Phase Transformations: A Case Study with Lenalidomide. Cryst. Growth Des. 2017, 17, 612–628. [Google Scholar] [CrossRef]
- Allen, F.H.; Groom, C.R.; Liebeschuetz, J.W.; Bardwell, D.A.; Olsson, T.S.G.; Wood, P.A. The Hydrogen Bond Environments of 1H-Tetrazole and Tetrazolate Rings: The Structural Basis for Tetrazole–Carboxylic Acid Bioisosterism. J. Chem. Inf. Model. 2012, 52, 857–866. [Google Scholar] [CrossRef]
- Chernyshov, I.Y.; Vener, M.V.; Prikhodchenko, P.V.; Medvedev, A.G.; Lev, O.; Churakov, A.V. Peroxosolvates: Formation Criteria, H2O2 Hydrogen Bonding, and Isomorphism with the Corresponding Hydrates. Cryst. Growth Des. 2017, 17, 214–220. [Google Scholar] [CrossRef]
- Cummings, M.D.; Sekharan, S. Structure-Based Macrocycle Design in Small-Molecule Drug Discovery and Simple Metrics to Identify Opportunities for Macrocyclization of Small-Molecule Ligands. J. Med. Chem. 2019, 62, 6843–6853. [Google Scholar] [CrossRef] [PubMed]
- Gavezzotti, A. Are Crystal Structures Predictable? Acc. Chem. Res. 1994, 27, 309–314. [Google Scholar] [CrossRef]
- Gavezzotti, A. The Crystal Packing of Organic Molecules: Challenge and Fascination Below 1000 Da. Crystallogr. Rev. 1998, 7, 5–121. [Google Scholar] [CrossRef]
- Chemla, D.S.; Zyss, J. Nonlinear Optical Properties of Organic Molecules and Crystals; Elsevier: Amsterdam, The Netherlands, 1987. [Google Scholar]
- Ostroverkhova, O. Organic Optoelectronic Materials: Mechanisms and Applications. Chem. Rev. 2016, 116, 13279–13412. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Sinha, N.; Yadav, H.; Kumar, B. Growth, morphology, structure and characterization of l-histidinium dihydrogen arsenate orthoarsenic acid single crystal. Acta Cryst. Sect. B 2016, 72, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Tian, X.; Zhang, J.; Wu, Q.; Lu, Q.; Tao, X. Large-Sized Crystal Growth and Electric-Elastic Properties of α-BaTeMo2O9 Single Crystal. Cryst. Growth Des. 2015, 15, 759–763. [Google Scholar] [CrossRef]
- Chen, F.; Jiang, C.; Tian, S.; Yu, F.; Cheng, X.; Duan, X.; Wang, Z.; Zhao, X. Electroelastic Features of Piezoelectric Bi2ZnB2O7 Crystal. Cryst. Growth Des. 2018, 18, 3988–3996. [Google Scholar] [CrossRef]
- Leclaire, N.A.; Li, M.; Véron, A.C.; Neels, A.; Heier, J.; Reimers, J.R.; Nüesch, F.A. Cyanine platelet single crystals: Growth, crystal structure and optical spectra. Phys. Chem. Chem. Phys. 2018, 20, 29166–29173. [Google Scholar] [CrossRef]
- Ozdemir, R.; Park, S.; Deneme, İ.; Park, Y.; Zorlu, Y.; Ardic Alidagi, H.; Harmandar, K.; Kim, C.; Usta, H. Triisopropylsilylethynyl-substituted indenofluorenes: Carbonyl versus dicyanovinylene functionalization in one-dimensional molecular crystals and solution-processed n-channel OFETs. Org. Chem. Front. 2018, 5, 2912–2924. [Google Scholar] [CrossRef]
- Glöcklhofer, F.; Morawietz, A.J.; Stöger, B.; Unterlass, M.M.; Fröhlich, J. Extending the Scope of a New Cyanation: Design and Synthesis of an Anthracene Derivative with an Exceptionally Low LUMO Level and Improved Solubility. ACS Omega 2017, 2, 1594–1600. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Dong, H.; Jiang, L.; Hu, W. Organic semiconductor crystals. Chem. Soc. Rev. 2018, 47, 422–500. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.S.; Walczak, M.; Jaye, C.; Fischer, D.A. In Situ Solid-State Reactions Monitored by X-ray Absorption Spectroscopy: Temperature-Induced Proton Transfer Leads to Chemical Shifts. Chem.—Eur. J. 2016, 22, 15600–15604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Liu, J.; Liu, Y.; Tao, X. Oriented Single-Crystal-to-Single-Crystal Phase Transition with Dramatic Changes in the Dimensions of Crystals. J. Am. Chem. Soc. 2014, 136, 590–593. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.-M.; Nobusue, S.; Saito, S.; Inoue, D.; Hashizume, D.; Yamaguchi, S. Highly bent crystals formed by restrained π-stacked columns connected via alkylene linkers with variable conformations. Chem. Sci. 2015, 6, 2354–2359. [Google Scholar] [CrossRef] [PubMed]
- Commins, P.; Desta, I.T.; Karothu, D.P.; Panda, M.K.; Naumov, P. Crystals on the move: Mechanical effects in dynamic solids. Chem. Commun. 2016, 52, 13941–13954. [Google Scholar] [CrossRef] [PubMed]
- Boldyreva, E.V. High-pressure diffraction studies of molecular organic solids. A personal view. Acta Cryst. Sect. A 2008, 64, 218–231. [Google Scholar] [CrossRef]
- Bull, C.L.; Flowitt-Hill, G.; de Gironcoli, S.; Küçükbenli, E.; Parsons, S.; Pham, C.H.; Playford, H.Y.; Tucker, M.G. ζ-Glycine: Insight into the mechanism of a polymorphic phase transition. IUCrJ 2017, 4, 569–574. [Google Scholar] [CrossRef]
- Giordano, N.; Beavers, C.M.; Kamenev, K.V.; Marshall, W.G.; Moggach, S.A.; Patterson, S.D.; Teat, S.J.; Warren, J.E.; Wood, P.A.; Parsons, S. High-pressure polymorphism in L-threonine between ambient pressure and 22 GPa. CrystEngComm 2019, 21, 4444–4456. [Google Scholar] [CrossRef]
- Sun, C.; Grant, D.J.W. Influence of Crystal Shape on the Tableting Performance of L-Lysine Monohydrochloride Dihydrate. J. Pharm. Sci. 2001, 90, 569–579. [Google Scholar] [CrossRef]
- Rosbottom, I.; Ma, C.Y.; Turner, T.D.; O’Connell, R.A.; Loughrey, J.; Sadiq, G.; Davey, R.J.; Roberts, K.J. Influence of Solvent Composition on the Crystal Morphology and Structure of p-Aminobenzoic Acid Crystallized from Mixed Ethanol and Nitromethane Solutions. Cryst. Growth Des. 2017, 17, 4151–4161. [Google Scholar] [CrossRef]
- Zhao, P.; Liu, X.; Wang, L.; Gao, Z.; Yang, Y.; Hao, H.; Xie, C.; Bao, Y. Predicting the crystal habit of photoinitiator XBPO and elucidating the solvent effect on crystal faces. CrystEngComm 2019, 21, 2422–2430. [Google Scholar] [CrossRef]
- O’Mahony, M.; Seaton, C.C.; Croker, D.M.; Veesler, S.; Rasmuson, Å.C.; Hodnett, B.K. Investigating the dissolution of the metastable triclinic polymorph of carbamazepine using in situ microscopy. CrystEngComm 2014, 16, 4133–4141. [Google Scholar] [CrossRef]
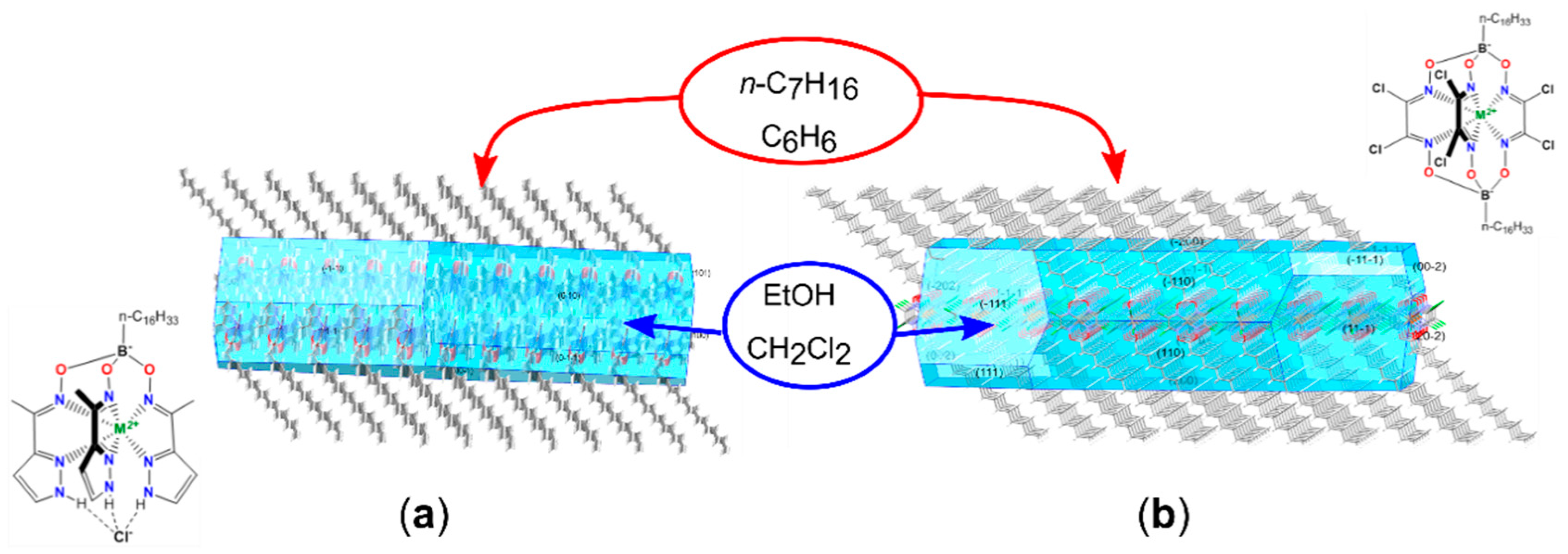
- Vologzhanina, A.V.; Belov, A.S.; Novikov, V.V.; Dolganov, A.V.; Romanenko, G.V.; Ovcharenko, V.I.; Korlyukov, A.A.; Buzin, M.I.; Voloshin, Y.Z. Synthesis and Temperature-Induced Structural Phase and Spin Transitions in Hexadecylboron-Capped Cobalt(II) Hexachloroclathrochelate and Its Diamagnetic Iron(II)-Encapsulating Analogue. Inorg. Chem. 2015, 54, 5827–5838. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, A.A.; Nelyubina, Y.V.; Kats, S.V.; Penkova, L.V.; Efimov, N.N.; Dmitrienko, A.O.; Vologzhanina, A.V.; Belov, A.S.; Voloshin, Y.Z.; Novikov, V.V. Polymorphism in a Cobalt-Based Single-Ion Magnet Tuning Its Barrier to Magnetization Relaxation. J. Phys. Chem. Lett. 2016, 7, 4111–4116. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.H.; Rosbottom, I.; Marziano, I.; Hammond, R.B.; Roberts, K.J. Crystal Morphology and Interfacial Stability of RS-Ibuprofen in Relation to Its Molecular and Synthonic Structure. Cryst. Growth Des. 2017, 17, 3088–3099. [Google Scholar] [CrossRef]
- Rosbottom, I.; Pickering, J.H.; Etbon, B.; Hammond, R.B.; Roberts, K.J. Examination of inequivalent wetting on the crystal habit surfaces of RS-ibuprofen using grid-based molecular modelling. Phys. Chem. Chem. Phys. 2018, 20, 11622–11633. [Google Scholar] [CrossRef] [Green Version]
- Turner, T.D.; Hatcher, L.E.; Wilson, C.C.; Roberts, K.J. Habit Modification of the Active Pharmaceutical Ingredient Lovastatin Through a Predictive Solvent Selection Approach. J. Pharm. Sci. 2019, 108, 1779–1787. [Google Scholar] [CrossRef] [Green Version]
- Gajda, R.; Domański, M.A.; Malinska, M.; Makal, A. Crystal morphology fixed by interplay of π-stacking and hydrogen bonds – the case of 1-hydroxypyrene. CrystEngComm 2019, 21, 1701–1717. [Google Scholar] [CrossRef]
- Han, D.; Karmakar, T.; Bjelobrk, Z.; Gong, J.; Parrinello, M. Solvent-mediated morphology selection of the active pharmaceutical ingredient isoniazid: Experimental and simulation studies. Chem. Eng. Sci. 2019, 204, 320–328. [Google Scholar] [CrossRef]
- Destri, G.L.; Marrazzo, A.; Rescifina, A.; Punzo, F. Crystal Morphologies and Polymorphs in Tolbutamide Microcrystalline Powder. J. Pharm. Sci. 2013, 102, 73–83. [Google Scholar] [CrossRef]
- Thirupugalmani, K.; Venkatesh, M.; Karthick, S.; Maurya, K.K.; Vijayan, N.; Chaudhary, A.K.; Brahadeeswaran, S. Influence of polar solvents on growth of potentially NLO active organic single crystals of N-benzyl-2-methyl-4-nitroaniline and their efficiency in terahertz generation. CrystEngComm 2017, 19, 2623–2631. [Google Scholar] [CrossRef]
- Croker, D.M.; Kelly, D.M.; Horgan, D.E.; Hodnett, B.K.; Lawrence, S.E.; Moynihan, H.A.; Rasmuson, Å.C. Demonstrating the Influence of Solvent Choice and Crystallization Conditions on Phenacetin Crystal Habit and Particle Size Distribution. Org. Process Res. Dev. 2015, 19, 1826–1836. [Google Scholar] [CrossRef]
- Poornachary, S.K.; Lau, G.; Chow, P.S.; Tan, R.B.H.; George, N. The Effect and Counter-Effect of Impurities on Crystallization of an Agrochemical Active Ingredient: Stereochemical Rationalization and Nanoscale Crystal Growth Visualization. Cryst. Growth Des. 2011, 11, 492–500. [Google Scholar] [CrossRef]
- Anuar, N.; Wan Daud, W.R.; Roberts, K.J.; Kamarudin, S.K.; Tasirin, S.M. Morphology and Associated Surface Chemistry of l-Isoleucine Crystals Modeled under the Influence of l-Leucine Additive Molecules. Cryst. Growth Des. 2012, 12, 2195–2203. [Google Scholar] [CrossRef]
- Tang, Y.; Gao, J. Investigation of the Effects of Sodium Dicarboxylates on the Crystal Habit of Calcium Sulfate α-Hemihydrate. Langmuir 2017, 33, 9637–9644. [Google Scholar] [CrossRef]
- Schmidt, C.; Jones, M.J.; Ulrich, J. The Influence of Additives and Impurities on Crystallization. In Crystallization; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2013; pp. 105–127. [Google Scholar]
- Liu, D.; Liu, Y.; Dai, F.; Zhao, J.; Yang, K.; Liu, C. Size- and morphology-controllable synthesis of MIL-96 (Al) by hydrolysis and coordination modulation of dual aluminium source and ligand systems. Dalton Trans. 2015, 44, 16421–16429. [Google Scholar] [CrossRef]
- Liu, D.; Yan, L.; Li, L.; Gu, X.; Dai, P.; Yang, L.; Liu, Y.; Liu, C.; Zhao, G.; Zhao, X. Impact of moderative ligand hydrolysis on morphology evolution and the morphology-dependent breathing effect performance of MIL-53(Al). CrystEngComm 2018, 20, 2102–2111. [Google Scholar] [CrossRef]
- Zelinskii, G.E.; Belov, A.S.; Lebed, E.G.; Vologzhanina, A.V.; Novikov, V.V.; Voloshin, Y.Z. Synthesis, structure and reactivity of iron(II) clathrochelates with terminal formyl (acetal) groups. Inorg. Chim. Acta 2016, 440, 154–164. [Google Scholar] [CrossRef]
- Koradia, V.; de Diego, H.L.; Elema, M.R.; Rantanen, J. Integrated Approach to Study the Dehydration Kinetics of Nitrofurantoin Monohydrate. J. Pharm. Sci. 2010, 99, 3966–3976. [Google Scholar] [CrossRef]
- Thakuria, R.; Eddleston, M.D.; Chow, E.H.H.; Lloyd, G.O.; Aldous, B.J.; Krzyzaniak, J.F.; Bond, A.D.; Jones, W. Use of in Situ Atomic Force Microscopy to Follow Phase Changes at Crystal Surfaces in Real Time. Angew. Chem. Int. Ed. 2013, 52, 10541–10544. [Google Scholar] [CrossRef]
- Park, Y.; Boerrigter, S.X.M.; Yeon, J.; Lee, S.H.; Kang, S.K.; Lee, E.H. New Metastable Packing Polymorph of Donepezil Grown on Stable Polymorph Substrates. Cryst. Growth Des. 2016, 16, 2552–2560. [Google Scholar] [CrossRef]
- Bond, A.D.; Boese, R.; Desiraju, G.R. On the Polymorphism of Aspirin: Crystalline Aspirin as Intergrowths of Two “Polymorphic” Domains. Angew. Chem. Int. Ed. 2007, 46, 618–622. [Google Scholar] [CrossRef]
- Mishra, M.K.; Desiraju, G.R.; Ramamurty, U.; Bond, A.D. Studying Microstructure in Molecular Crystals with Nanoindentation: Intergrowth Polymorphism in Felodipine. Angew. Chem. Int. Ed. 2014, 53, 13102–13105. [Google Scholar] [CrossRef]
- Croker, D.M.; Davey, R.J.; Rasmuson, Å.C.; Seaton, C.C. Solution mediated phase transformations between co-crystals. CrystEngComm 2013, 15, 2044–2047. [Google Scholar] [CrossRef] [Green Version]
- Bhandary, S.; Chopra, D. Silicone Oil Induced Spontaneous Single-Crystal-to-Single-Crystal Phase Transitions in Ethynyl Substituted ortho- and meta-Fluorinated Benzamides. Cryst. Growth Des. 2017, 17, 4533–4540. [Google Scholar] [CrossRef]
- Ravi, A.; Sureshan, K.M. Tunable Mechanical Response from a Crystal Undergoing Topochemical Dimerization: Instant Explosion at a Faster Rate and Chemical Storage of a Harvestable Explosion at a Slower Rate. Angew. Chem. Int. Ed. 2018, 57, 9362–9366. [Google Scholar] [CrossRef]
- Lee, A.Y.; Ulman, A.; Myerson, A.S. Crystallization of Amino Acids on Self-Assembled Monolayers of Rigid Thiols on Gold. Langmuir 2002, 18, 5886–5898. [Google Scholar] [CrossRef]
- Cox, J.R.; Dabros, M.; Shaffer, J.A.; Thalladi, V.R. Selective Crystal Growth of the Anhydrous and Monohydrate Forms of Theophylline on Self-Assembled Monolayers. Angew. Chem. Int. Ed. 2007, 46, 1988–1991. [Google Scholar] [CrossRef]
- Urbelis, J.H.; Swift, J.A. Phase-Selective Crystallization of Perylene on Monolayer Templates. Cryst. Growth Des. 2014, 14, 5244–5251. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, A.; Han, Y.; Ren, Y.; Gong, J.; Li, W.; Wang, J. Effects of Self-Assembled Monolayers on Selective Crystallization of Tolbutamide. Cryst. Growth Des. 2011, 11, 5498–5506. [Google Scholar] [CrossRef]
- Solomos, M.A.; Capacci-Daniel, C.; Rubinson, J.F.; Swift, J.A. Polymorph Selection via Sublimation onto Siloxane Templates. Cryst. Growth Des. 2018, 18, 6965–6972. [Google Scholar] [CrossRef]
- Serrano, D.R.; Mugheirbi, N.A.; O’Connell, P.; Leddy, N.; Healy, A.M.; Tajber, L. Impact of Substrate Properties on the Formation of Spherulitic Films: A Case Study of Salbutamol Sulfate. Cryst. Growth Des. 2016, 16, 3853–3858. [Google Scholar] [CrossRef]
- Patel, M.A.; Nguyen, B.; Chadwick, K. Predicting the Nucleation Induction Time Based on Preferred Intermolecular Interactions. Cryst. Growth Des. 2017, 17, 4613–4621. [Google Scholar] [CrossRef]
- Telford, R.; Seaton, C.C.; Clout, A.; Buanz, A.; Gaisford, S.; Williams, G.R.; Prior, T.J.; Okoye, C.H.; Munshi, T.; Scowen, I.J. Stabilisation of metastable polymorphs: The case of paracetamol form III. Chem. Commun. 2016, 52, 12028–12031. [Google Scholar] [CrossRef]
- Zhang, K.; Xu, S.; Liu, S.; Tang, W.; Fu, X.; Gong, J. Novel Strategy to Control Polymorph Nucleation of Gamma Pyrazinamide by Preferred Intermolecular Interactions during Heterogeneous Nucleation. Cryst. Growth Des. 2018, 18, 4874–4879. [Google Scholar] [CrossRef]
- Rao, K.P.; Higuchi, M.; Sumida, K.; Furukawa, S.; Duan, J.; Kitagawa, S. Design of Superhydrophobic Porous Coordination Polymers through the Introduction of External Surface Corrugation by the Use of an Aromatic Hydrocarbon Building Unit. Angew. Chem. Int. Ed. 2014, 53, 8225–8230. [Google Scholar] [CrossRef]
- Zhang, J.; Mitchell, L.A.; Parrish, D.A.; Shreeve, J.M. Enforced Layer-by-Layer Stacking of Energetic Salts towards High-Performance Insensitive Energetic Materials. J. Am. Chem. Soc. 2015, 137, 10532–10535. [Google Scholar] [CrossRef]
- Wang, K.; Mishra, M.K.; Sun, C.C. Exceptionally Elastic Single-Component Pharmaceutical Crystals. Chem. Mater. 2019, 31, 1794–1799. [Google Scholar] [CrossRef]
- Devarapalli, R.; Kadambi, S.B.; Chen, C.-T.; Krishna, G.R.; Kammari, B.R.; Buehler, M.J.; Ramamurty, U.; Reddy, C.M. Remarkably Distinct Mechanical Flexibility in Three Structurally Similar Semiconducting Organic Crystals Studied by Nanoindentation and Molecular Dynamics. Chem. Mater. 2019, 31, 1391–1402. [Google Scholar] [CrossRef]
- SeethaLekshmi, S.; Kiran, M.S.R.N.; Ramamurty, U.; Varughese, S. Molecular Basis for the Mechanical Response of Sulfa Drug Crystals. Chem.—Eur. J. 2019, 25, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Krishna, G.R.; Shi, L.; Bag, P.P.; Sun, C.C.; Reddy, C.M. Correlation Among Crystal Structure, Mechanical Behavior, and Tabletability in the Co-Crystals of Vanillin Isomers. Cryst. Growth Des. 2015, 15, 1827–1832. [Google Scholar] [CrossRef]
- Zolotarev, P.N.; Moret, M.; Rizzato, S.; Proserpio, D.M. Searching New Crystalline Substrates for OMBE: Topological and Energetic Aspects of Cleavable Organic Crystals. Cryst. Growth Des. 2016, 16, 1572–1582. [Google Scholar] [CrossRef] [Green Version]
- Reddy, C.M.; Gundakaram, R.C.; Basavoju, S.; Kirchner, M.T.; Padmanabhan, K.A.; Desiraju, G.R. Structural basis for bending of organic crystals. Chem. Commun. 2005, 3945–3947. [Google Scholar] [CrossRef] [PubMed]
- Chattoraj, S.; Shi, L.; Sun, C.C. Understanding the relationship between crystal structure, plasticity and compaction behaviour of theophylline, methyl gallate, and their 1:1 co-crystal. CrystEngComm 2010, 12, 2466–2472. [Google Scholar] [CrossRef]
- Veits, G.K.; Carter, K.K.; Cox, S.J.; McNeil, A.J. Developing a Gel-Based Sensor Using Crystal Morphology Prediction. J. Am. Chem. Soc. 2016, 138, 12228–12233. [Google Scholar] [CrossRef] [PubMed]
- Bučar, D.-K.; Lancaster, R.W.; Bernstein, J. Disappearing Polymorphs Revisited. Angew. Chem. Int. Ed. 2015, 54, 6972–6993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
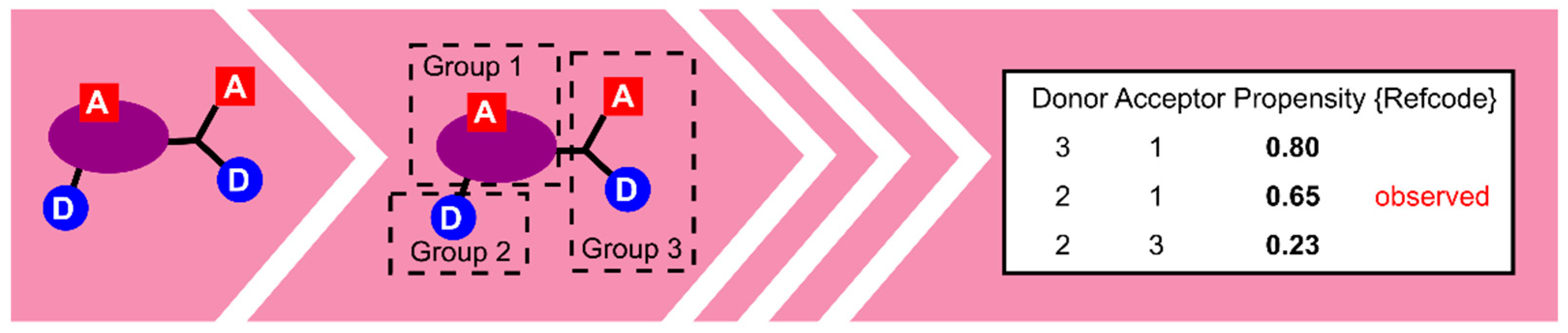
- Galek, P.T.A.; Allen, F.H.; Fábián, L.; Feeder, N. Knowledge-based H-bond prediction to aid experimental polymorph screening. CrystEngComm 2009, 11, 2634–2639. [Google Scholar] [CrossRef]
- Galek, P.T.A.; Fábián, L.; Motherwell, W.D.S.; Allen, F.H.; Feeder, N. Knowledge-based model of hydrogen-bonding propensity in organic crystals. Acta Cryst. Sect. B 2007, 63, 768–782. [Google Scholar] [CrossRef]
- Galek, P.T.A.; Chisholm, J.A.; Pidcock, E.; Wood, P.A. Hydrogen-bond coordination in organic crystal structures: Statistics, predictions and applications. Acta Cryst. Sect. B 2014, 70, 91–105. [Google Scholar] [CrossRef]
- Galek, P.T.A.; Pidcock, E.; Wood, P.A.; Bruno, I.J.; Groom, C.R. One in half a million: A solid form informatics study of a pharmaceutical crystal structure. CrystEngComm 2012, 14, 2391–2403. [Google Scholar] [CrossRef]
- Abramov, Y.A. Current Computational Approaches to Support Pharmaceutical Solid Form Selection. Org. Process Res. Dev. 2013, 17, 472–485. [Google Scholar] [CrossRef]
- Feeder, N.; Pidcock, E.; Reilly, A.M.; Sadiq, G.; Doherty, C.L.; Back, K.R.; Meenan, P.; Docherty, R. The integration of solid-form informatics into solid-form selection. J. Pharm. Pharm. 2015, 67, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.H.; Wood, P.A.; Galek, P.T.A. The versatile role of the ethynyl group in crystal packing: An interaction propensity study. Acta Cryst. Sect. B 2013, 69, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.H.; Wood, P.A.; Galek, P.T.A. Role of chloroform and dichloromethane solvent molecules in crystal packing: An interaction propensity study. Acta Cryst. Sect. B 2013, 69, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Srirambhatla, V.K.; Kraft, A.; Watt, S.; Powell, A.V. Crystal Design Approaches for the Synthesis of Paracetamol Co-Crystals. Cryst. Growth Des. 2012, 12, 4870–4879. [Google Scholar] [CrossRef]
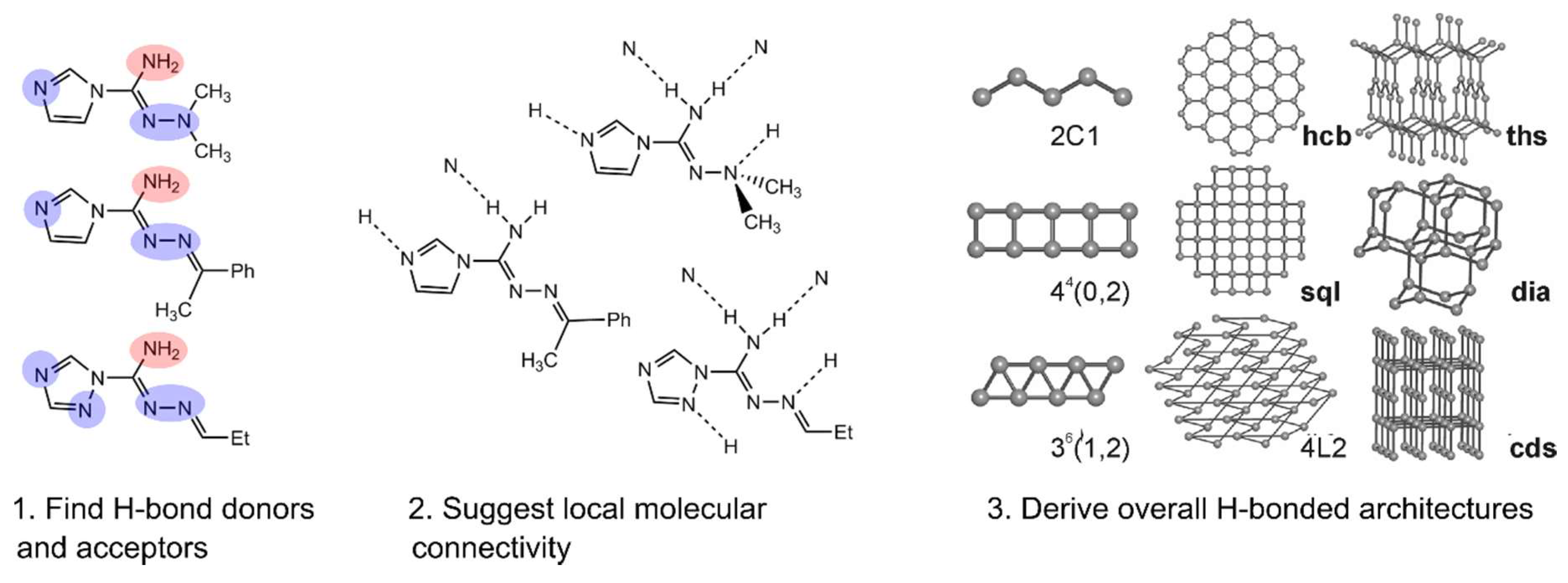
- Vologzhanina, A.V.; Sokolov, A.V.; Purygin, P.P.; Zolotarev, P.N.; Blatov, V.A. Knowledge-Based Approaches to H-Bonding Patterns in Heterocycle-1-Carbohydrazoneamides. Cryst. Growth Des. 2016, 16, 6354–6362. [Google Scholar] [CrossRef]
- Hulme, A.T.; Johnston, A.; Florence, A.J.; Fernandes, P.; Shankland, K.; Bedford, C.T.; Welch, G.W.A.; Sadiq, G.; Haynes, D.A.; Motherwell, W.D.S.; et al. Search for a Predicted Hydrogen Bonding Motif—A Multidisciplinary Investigation into the Polymorphism of 3-Azabicyclo[3.3.1]nonane-2,4-dione. J. Am. Chem. Soc. 2007, 129, 3649–3657. [Google Scholar] [CrossRef] [PubMed]
- Nauha, E.; Bernstein, J. “Predicting” Polymorphs of Pharmaceuticals Using Hydrogen Bond Propensities: Probenecid and Its Two Single-Crystal-to-Single-Crystal Phase Transitions. J. Pharm. Sci. 2015, 104, 2056–2061. [Google Scholar] [CrossRef]
- Nauha, E.; Bernstein, J. “Predicting” Crystal Forms of Pharmaceuticals Using Hydrogen Bond Propensities: Two Test Cases. Cryst. Growth Des. 2014, 14, 4364–4370. [Google Scholar] [CrossRef]
- Jones, W.; Motherwell, W.D.S.; Trask, A.V. Pharmaceutical Cocrystals: An Emerging Approach to Physical Property Enhancement. MRS Bull. 2006, 31, 875–879. [Google Scholar] [CrossRef] [Green Version]
- Sathisaran, I.; Dalvi, S.V. Engineering Cocrystals of Poorly Water-Soluble Drugs to Enhance Dissolution in Aqueous Medium. Pharmaceutics 2018, 10, 108. [Google Scholar] [CrossRef] [PubMed]
- Schultheiss, N.; Newman, A. Pharmaceutical Cocrystals and Their Physicochemical Properties. Cryst. Growth Des. 2009, 9, 2950–2967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karki, S.; Friščić, T.; Fábián, L.; Laity, P.R.; Day, G.M.; Jones, W. Improving Mechanical Properties of Crystalline Solids by Cocrystal Formation: New Compressible Forms of Paracetamol. Adv. Mater. 2009, 21, 3905–3909. [Google Scholar] [CrossRef]
- Saha, S.; Desiraju, G.R. Acid···Amide Supramolecular Synthon in Cocrystals: From Spectroscopic Detection to Property Engineering. J. Am. Chem. Soc. 2018, 140, 6361–6373. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shreeve, J.M. Time for pairing: Cocrystals as advanced energetic materials. CrystEngComm 2016, 18, 6124–6133. [Google Scholar] [CrossRef]
- Bolton, O.; Simke, L.R.; Pagoria, P.F.; Matzger, A.J. High Power Explosive with Good Sensitivity: A 2:1 Cocrystal of CL-20: HMX. Cryst. Growth Des. 2012, 12, 4311–4314. [Google Scholar] [CrossRef]
- Gryl, M.; Seidler, T.; Stadnicka, K.; Matulková, I.; Němec, I.; Tesařová, N.; Němec, P. The crystal structure and optical properties of a pharmaceutical co-crystal—The case of the melamine–barbital addition compound. CrystEngComm 2014, 16, 5765–5768. [Google Scholar] [CrossRef]
- Christopherson, J.-C.; Topić, F.; Barrett, C.J.; Friščić, T. Halogen-Bonded Cocrystals as Optical Materials: Next-Generation Control over Light–Matter Interactions. Cryst. Growth Des. 2018, 18, 1245–1259. [Google Scholar] [CrossRef]
- Noa, F.M.A.; Mehlana, G. Co-crystals and salts of vanillic acid and vanillin with amines. CrystEngComm 2018, 20, 896–905. [Google Scholar]
- Walsh, R.D.B.; Bradner, M.W.; Fleischman, S.; Morales, L.A.; Moulton, B.; Rodríguez-Hornedo, N.; Zaworotko, M.J. Crystal engineering of the composition of pharmaceutical phases. Chem. Commun. 2003, 186–187. [Google Scholar] [CrossRef] [PubMed]
- Price(Sally), S.L. Computed Crystal Energy Landscapes for Understanding and Predicting Organic Crystal Structures and Polymorphism. Acc. Chem. Res. 2009, 42, 117–126. [Google Scholar]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
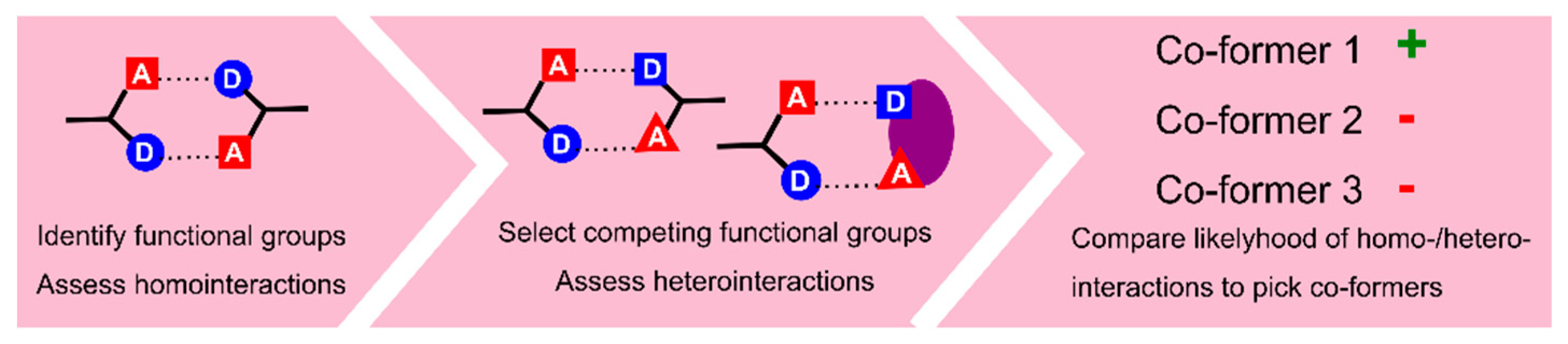
- Delori, A.; Galek, P.T.A.; Pidcock, E.; Jones, W. Quantifying Homo- and Heteromolecular Hydrogen Bonds as a Guide for Adduct Formation. Chem. Eur. J. 2012, 18, 6835–6846. [Google Scholar] [CrossRef] [PubMed]
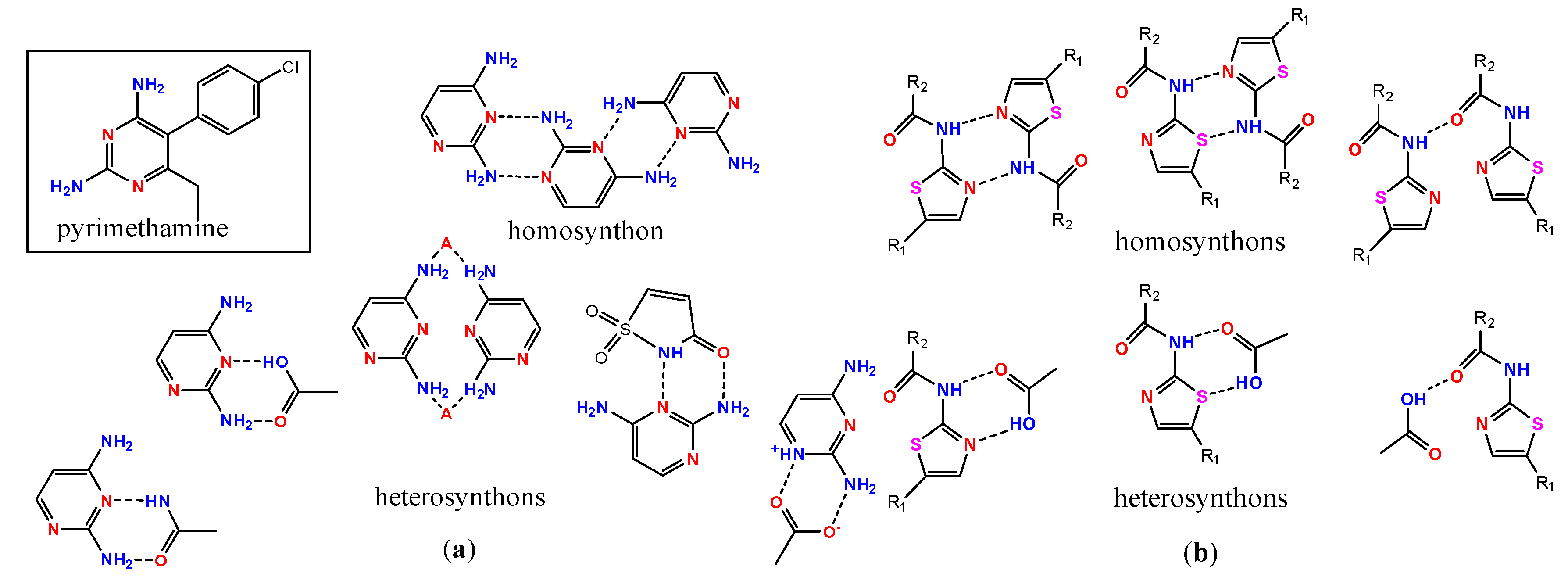
- Delori, A.; Galek, P.T.A.; Pidcock, E.; Patni, M.; Jones, W. Knowledge-based hydrogen bond prediction and the synthesis of salts and cocrystals of the anti-malarial drug pyrimethamine with various drug and GRAS molecules. CrystEngComm 2013, 15, 2916–2928. [Google Scholar] [CrossRef]
- Bhogala, B.R.; Basavoju, S.; Nangia, A. Tape and layer structures in cocrystals of some di- and tricarboxylic acids with 4,4′-bipyridines and isonicotinamide. From binary to ternary cocrystals. CrystEngComm 2005, 7, 551–562. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J. Acid–base crystalline complexes and the pKa rule. CrystEngComm 2012, 14, 6362–6365. [Google Scholar] [CrossRef]
- Chamorro Orué, A.I.; Boese, R.; Schauerte, C.; Merz, K. An Experimental and Theoretical Approach to Control Salt vs Cocrystal vs Hybrid Formation—Crystal Engineering of an E/Z-Butenedioic Acid/Phthalazine System. Cryst. Growth Des. 2019, 19, 1616–1620. [Google Scholar] [CrossRef]
- Sandhu, B.; McLean, A.; Sinha, A.S.; Desper, J.; Sarjeant, A.A.; Vyas, S.; Reutzel-Edens, S.M.; Aakeröy, C.B. Evaluating Competing Intermolecular Interactions through Molecular Electrostatic Potentials and Hydrogen-Bond Propensities. Cryst. Growth Des. 2018, 18, 466–478. [Google Scholar] [CrossRef]
- Wang, J.-R.; Ye, C.; Mei, X. Structural and physicochemical aspects of hydrochlorothiazide co-crystals. CrystEngComm 2014, 16, 6996–7003. [Google Scholar] [CrossRef]
- Eddleston, M.D.; Arhangelskis, M.; Fábián, L.; Tizzard, G.J.; Coles, S.J.; Jones, W. Investigation of an Amide-Pseudo Amide Hydrogen Bonding Motif within a Series of Theophylline: Amide Cocrystals. Cryst. Growth Des. 2016, 16, 51–58. [Google Scholar] [CrossRef]
- Corpinot, M.K.; Stratford, S.A.; Arhangelskis, M.; Anka-Lufford, J.; Halasz, I.; Judaš, N.; Jones, W.; Bučar, D.-K. On the predictability of supramolecular interactions in molecular cocrystals—the view from the bench. CrystEngComm 2016, 18, 5434–5439. [Google Scholar] [CrossRef]
- Wang, T.; Stevens, J.S.; Vetter, T.; Whitehead, G.F.S.; Vitorica-Yrezabal, I.J.; Hao, H.; Cruz-Cabeza, A.J. Salts, Cocrystals, and Ionic Cocrystals of a “Simple” Tautomeric Compound. Cryst. Growth Des. 2018, 18, 6973–6983. [Google Scholar] [CrossRef]
- Mapp, L.K.; Coles, S.J.; Aitipamula, S. Design of Cocrystals for Molecules with Limited Hydrogen Bonding Functionalities: Propyphenazone as a Model System. Cryst. Growth Des. 2017, 17, 163–174. [Google Scholar] [CrossRef]
- Liu, F.; Song, Y.; Liu, Y.-N.; Li, Y.-T.; Wu, Z.-Y.; Yan, C.-W. Drug-Bridge-Drug Ternary Cocrystallization Strategy for Antituberculosis Drugs Combination. Cryst. Growth Des. 2018, 18, 1283–1286. [Google Scholar] [CrossRef]
- Almansa, C.; Mercè, R.; Tesson, N.; Farran, J.; Tomàs, J.; Plata-Salamán, C.R. Co-crystal of Tramadol Hydrochloride–Celecoxib (ctc): A Novel API–API Co-crystal for the Treatment of Pain. Cryst. Growth Des. 2017, 17, 1884–1892. [Google Scholar] [CrossRef]
- Skořepová, E.; Hušák, M.; Čejka, J.; Zámostný, P.; Kratochvíl, B. Increasing dissolution of trospium chloride by co-crystallization with urea. J. Cryst. Growth 2014, 399, 19–26. [Google Scholar] [CrossRef]
- Chandra, A.; Ghate, M.V.; Aithal, K.S.; Lewis, S.A. In silico prediction coupled with in vitro experiments and absorption modeling to study the inclusion complex of telmisartan with modified beta-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2018, 91, 47–60. [Google Scholar] [CrossRef]
- Tothadi, S.; Desiraju, G.R. Designing ternary cocrystals with hydrogen bonds and halogen bonds. Chem. Commun. 2013, 49, 7791–7793. [Google Scholar] [CrossRef]
- Arman, H.D.; Gieseking, R.L.; Hanks, T.W.; Pennington, W.T. Complementary halogen and hydrogen bonding: Sulfur⋯iodine interactions and thioamide ribbons. Chem. Commun. 2010, 46, 1854–1856. [Google Scholar] [CrossRef]
- Topić, F.; Rissanen, K. Systematic Construction of Ternary Cocrystals by Orthogonal and Robust Hydrogen and Halogen Bonds. J. Am. Chem. Soc. 2016, 138, 6610–6616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seaton, C.C.; Blagden, N.; Munshi, T.; Scowen, I.J. Creation of Ternary Multicomponent Crystals by Exploitation of Charge-Transfer Interactions. Chem.—Eur. J. 2013, 19, 10663–10671. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.H.; Blagden, N.; Gutmann, M.J.; Kallay, A.A.; Parkin, A.; Seaton, C.C.; Wilson, C.C. Tuning Proton Behavior in a Ternary Molecular Complex. Cryst. Growth Des. 2010, 10, 2770–2774. [Google Scholar] [CrossRef]
- Mishra, M.K.; Mukherjee, A.; Ramamurty, U.; Desiraju, G.R. Crystal chemistry and photomechanical behavior of 3,4-dimethoxycinnamic acid: Correlation between maximum yield in the solid-state topochemical reaction and cooperative molecular motion. IUCrJ 2015, 2, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Mir, N.A.; Dubey, R.; Desiraju, G.R. Four- and five-component molecular solids: Crystal engineering strategies based on structural inequivalence. IUCrJ 2016, 3, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.; Chakraborty, S.; Desiraju, G.R. Six-Component Molecular Solids: ABC[D1–(x+y)ExFy]2. J. Am. Chem. Soc. 2018, 140, 2309–2315. [Google Scholar] [CrossRef]
- Sander, J.R.G.; Bučar, D.-K.; Henry, R.F.; Giangiorgi, B.N.; Zhang, G.G.Z.; MacGillivray, L.R. ‘Masked synthons’ in crystal engineering: Insulated components in acetaminophen cocrystal hydrates. CrystEngComm 2013, 15, 4816–4822. [Google Scholar] [CrossRef]
- Oswald, I.D.H.; Allan, D.R.; McGregor, P.A.; Motherwell, W.D.S.; Parsons, S.; Pulham, C.R. The formation of paracetamol (acetaminophen) adducts with hydrogen-bond acceptors. Acta Cryst. Sect. B 2002, 58, 1057–1066. [Google Scholar] [CrossRef] [Green Version]
- Oswald, I.D.H.; Motherwell, W.D.S.; Parsons, S.; Pidcock, E.; Pulham, C.R. Rationalisation of Co-Crystal Formation Through Knowledge-Mining. Crystallogr. Rev. 2004, 10, 57–66. [Google Scholar] [CrossRef]
- Wicker, J.G.P.; Crowley, L.M.; Robshaw, O.; Little, E.J.; Stokes, S.P.; Cooper, R.I.; Lawrence, S.E. Will they co-crystallize? CrystEngComm 2017, 19, 5336–5340. [Google Scholar] [CrossRef] [Green Version]
- Fábián, L. Cambridge Structural Database Analysis of Molecular Complementarity in Cocrystals. Cryst. Growth Des. 2009, 9, 1436–1443. [Google Scholar] [CrossRef]
- Surov, A.O.; Churakov, A.V.; Proshin, A.N.; Dai, X.-L.; Lu, T.; Perlovich, G.L. Cocrystals of a 1,2,4-thiadiazole-based potent neuroprotector with gallic acid: Solubility, thermodynamic stability relationships and formation pathways. Phys. Chem. Chem. Phys. 2018, 20, 14469–14481. [Google Scholar] [CrossRef] [PubMed]
- Karki, S.; Friščić, T.; Fábián, L.; Jones, W. New solid forms of artemisinin obtained through cocrystallisation. CrystEngComm 2010, 12, 4038–4041. [Google Scholar] [CrossRef]
- Alsubaie, M.; Aljohani, M.; Erxleben, A.; McArdle, P. Cocrystal Forms of the BCS Class IV Drug Sulfamethoxazole. Cryst. Growth Des. 2018, 18, 3902–3912. [Google Scholar] [CrossRef] [Green Version]
- Cadden, J.; Klooster, W.T.; Coles, S.J.; Aitipamula, S. Cocrystals of Leflunomide: Design, Structural, and Physicochemical Evaluation. Cryst. Growth Des. 2019, 19, 3923–3933. [Google Scholar] [CrossRef]
- Gryl, M.; Rydz, A.; Wojnarska, J.; Krawczuk, A.; Kozieł, M.; Seidler, T.; Ostrowska, K.; Marzec, M.; Stadnicka, K.M. Origin of chromic effects and crystal-to-crystal phase transition in the polymorphs of tyraminium violurate. IUCrJ 2019, 6, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.C.; Wright, J.S.; Carrington, E.J.; Perutz, R.N.; Hunter, C.A.; Brammer, L. Hydrogen bonding vs. halogen bonding: The solvent decides. Chem. Sci. 2017, 8, 5392–5398. [Google Scholar] [CrossRef]
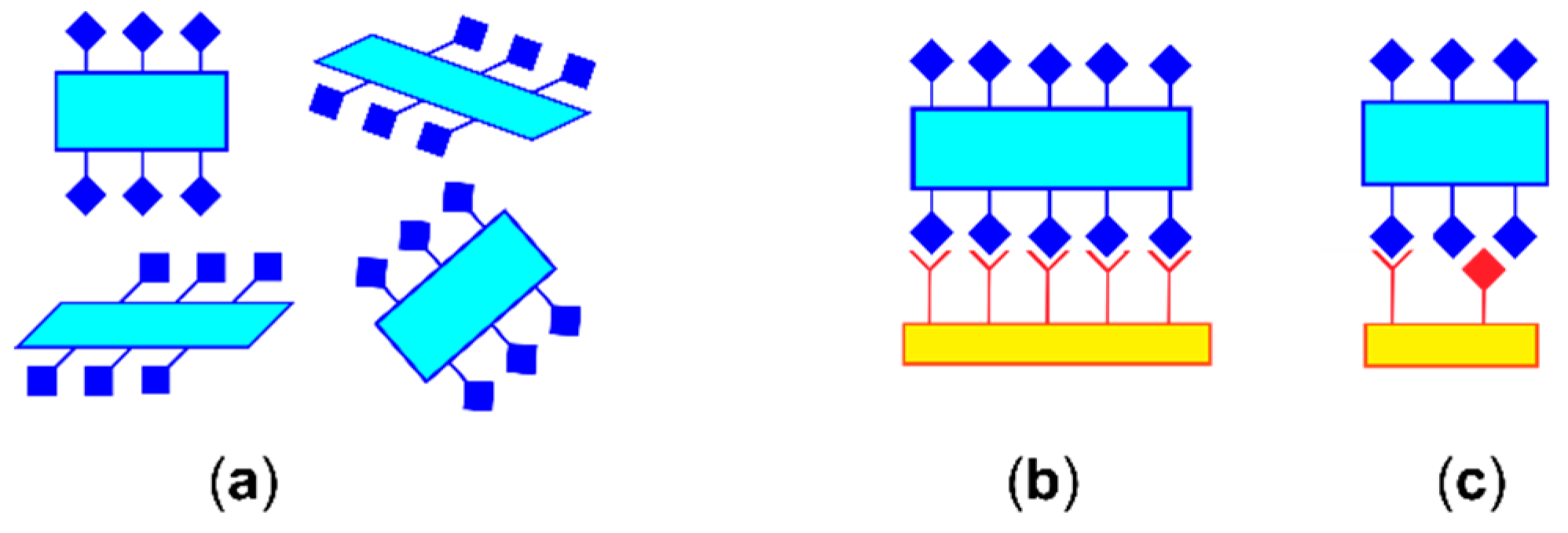
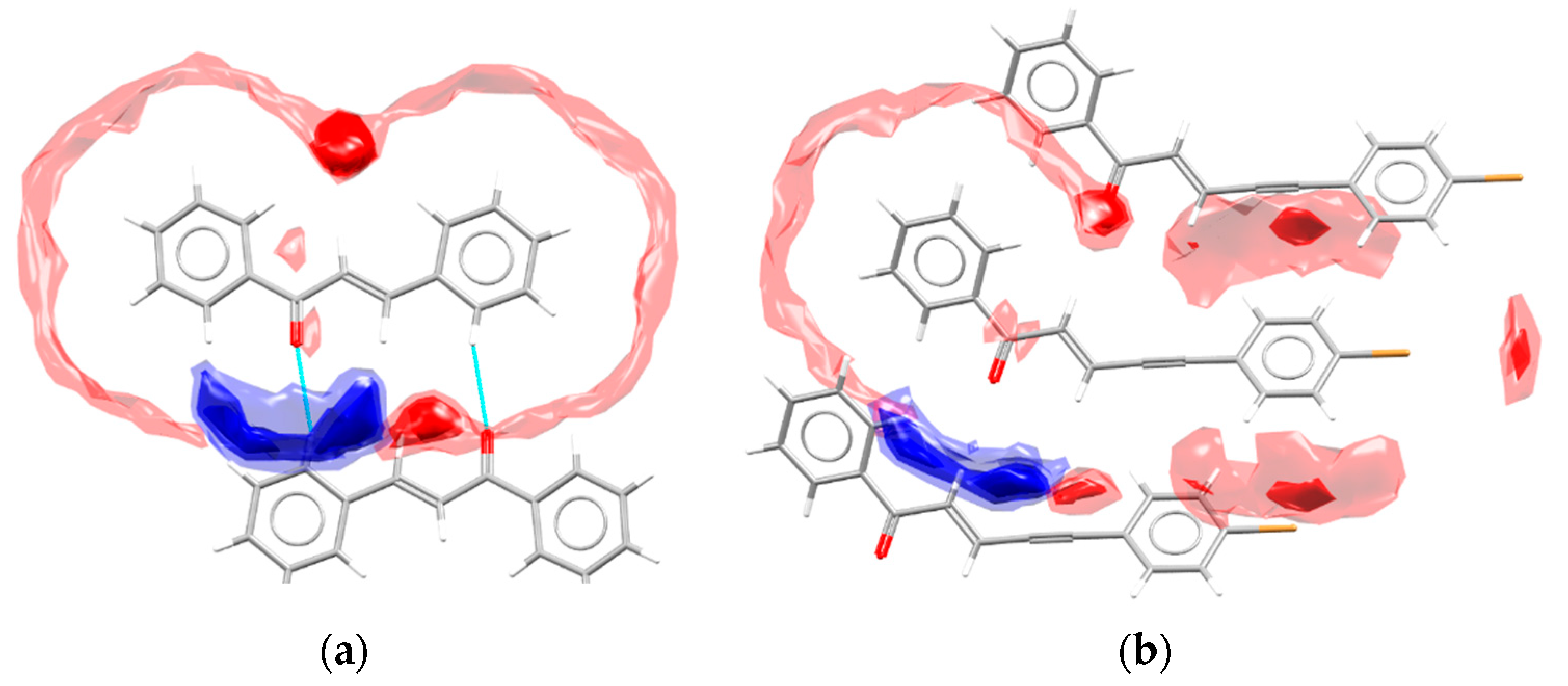
- Wood, P.A.; Olsson, T.S.G.; Cole, J.C.; Cottrell, S.J.; Feeder, N.; Galek, P.T.A.; Groom, C.R.; Pidcock, E. Evaluation of molecular crystal structures using Full Interaction Maps. CrystEngComm 2013, 15, 65–72. [Google Scholar] [CrossRef]
- Golovanov, A.A.; Latypova, D.R.; Bekin, V.V.; Pisareva, V.S.; Vologzhanina, A.V.; Dokichev, V.A. Synthesis of 1,5-disubstituted (E)-pent-2-en-4-yn-1-ones. Russ. J. Org. Chem. 2013, 49, 1264–1269. [Google Scholar] [CrossRef]
- Vologzhanina, A.V.; Golovanov, A.A.; Gusev, D.M.; Odin, I.S.; Apreyan, R.A.; Suponitsky, K.Yu. Intermolecular Interactions and Second-Harmonic Generation Properties of (E)-1,5-Diarylpentenyn-1-ones. Cryst. Growth Des. 2014, 14, 4402–4410. [Google Scholar] [CrossRef]
- Mugheirbi, N.A.; Tajber, L. Crystal Habits of Itraconazole Microcrystals: Unusual Isomorphic Intergrowths Induced via Tuning Recrystallization Conditions. Mol. Pharm. 2015, 12, 3468–3478. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, B.; Sinha, A.S.; Desper, J.; Aakeröy, C.B. Modulating the physical properties of solid forms of urea using co-crystallization technology. Chem. Commun. 2018, 54, 4657–4660. [Google Scholar] [CrossRef] [PubMed]
- Wojnarska, J.; Gryl, M.; Seidler, T.; Stadnicka, K.M. Crystal engineering, optical properties and electron density distribution of polar multicomponent materials containing sulfanilamide. CrystEngComm 2018, 20, 3638–3646. [Google Scholar] [CrossRef]
- Xing, G.; Bassanetti, I.; Ben, T.; Bracco, S.; Sozzani, P.; Marchiò, L.; Comotti, A. Multifunctional Organosulfonate Anions Self-Assembled with Organic Cations by Charge-Assisted Hydrogen Bonds and the Cooperation of Water. Cryst. Growth Des. 2018, 18, 2082–2092. [Google Scholar] [CrossRef]
- Honorato, J.; Colina-Vegas, L.; Correa, R.S.; Guedes, A.P.M.; Miyata, M.; Pavan, F.R.; Ellena, J.; Batista, A.A. Esterification of the free carboxylic group from the lutidinic acid ligand as a tool to improve the cytotoxicity of Ru(II) complexes. Inorg. Chem. Front. 2019, 6, 376–390. [Google Scholar] [CrossRef]
- Kodrin, I.; Soldin, Ž.; Aakeröy, C.B.; Đaković, M. Role of the “Weakest Link” in a Pressure-Driven Phase Transition of Two Polytypic Polymorphs. Cryst. Growth Des. 2016, 16, 2040–2051. [Google Scholar] [CrossRef]
- Moggach, S.A.; Marshall, W.G.; Rogers, D.M.; Parsons, S. How focussing on hydrogen bonding interactions in amino acids can miss the bigger picture: A high-pressure neutron powder diffraction study of ε-glycine. CrystEngComm 2015, 17, 5315–5328. [Google Scholar] [CrossRef]
- Voronova, E.D.; Golovanov, A.A.; Suponitsky, K.Yu.; Fedyanin, I.V.; Vologzhanina, A.V. Theoretical Charge Density Analysis and Nonlinear Optical Properties of Quasi-Planar 1-Aryl(hetaryl)-5-phenylpent-1-en-4-yn-3-ones. Cryst. Growth Des. 2016, 16, 3859–3868. [Google Scholar] [CrossRef]
- Vráblová, A.; Černák, J.; Falvello, L.R.; Tomás, M. Polymorphism of the dinuclear CoIII–Schiff base complex [Co2(o-van-en)3]·4CH3CN (o-van-en is a salen-type ligand). Acta Cryst. Sect. C 2019, 75, 433–442. [Google Scholar] [CrossRef]
- Voronova, E.D.; Golovanov, A.A.; Odin, I.S.; Anisimov, M.A.; Dorovatovskii, P.V.; Zubavichus, Y.V.; Vologzhanina, A.V. Peculiarities of supramolecular organization of cyclic ketones with vinylacetylene fragments. Acta Cryst. Sect. C 2018, 74, 1674–1683. [Google Scholar] [CrossRef]
- Dotsenko, V.V.; Frolov, K.А.; Krivokolysko, S.G.; Chigorina, E.A.; Pekhtereva, T.M.; Suykov, S.Yu.; Papayanina, E.S.; Dmitrienko, A.О.; Bushmarinov, I.S. Aminomethylation of morpholinium and N-methylmorpholinium 3,5-dicyano-4,4-dimethyl-6-oxo-1,4,5,6-tetrahydropyridine-2-thiolates. Chem. Heterocycl. Comp. 2016, 52, 116–127. [Google Scholar] [CrossRef]
- Carletta, A.; Zbačnik, M.; Van Gysel, M.; Vitković, M.; Tumanov, N.; Stilinović, V.; Wouters, J.; Cinčić, D. Playing with Isomerism: Cocrystallization of Isomeric N-Salicylideneaminopyridines with Perfluorinated Compounds as Halogen Bond Donors and Its Impact on Photochromism. Cryst. Growth Des. 2018, 18, 6833–6842. [Google Scholar] [CrossRef]
- Mugheirbi, N.A.; Tajber, L. Mesophase and size manipulation of itraconazole liquid crystalline nanoparticles produced via quasi nanoemulsion precipitation. Eur. J. Pharm. Biopharm. 2015, 96, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.C.; Kabova, E.A.; Shankland, K. Utilizing organic and organometallic structural data in powder diffraction. Powder Diffr. 2014, 29, S19–S30. [Google Scholar] [CrossRef]
- Anisimov, A.A.; Zhemchugov, P.V.; Milenin, S.A.; Goloveshkin, A.S.; Tsareva, U.S.; Bushmarinov, I.S.; Korlyukov, A.A.; Takazova, R.U.; Molodtsova, Y.A.; Muzafarov, A.M.; et al. Sodium cis-tetratolylcyclotetrasiloxanolate and cis-tritolylcyclotrisiloxanolate: Synthesis, structure and their mutual transformations. J. Organomet. Chem. 2016, 823, 103–111. [Google Scholar] [CrossRef]
- Taylor, R.; Cole, J.; Korb, O.; McCabe, P. Knowledge-Based Libraries for Predicting the Geometric Preferences of Druglike Molecules. J. Chem. Inf. Model. 2014, 54, 2500–2514. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.C.; Korb, O.; McCabe, P.; Read, M.G.; Taylor, R. Knowledge-Based Conformer Generation Using the Cambridge Structural Database. J. Chem. Inf. Model. 2018, 58, 615–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giangreco, I.; Olsson, T.S.G.; Cole, J.C.; Packer, M.J. Assessment of a Cambridge Structural Database-Driven Overlay Program. J. Chem. Inf. Model. 2014, 54, 3091–3098. [Google Scholar] [CrossRef] [PubMed]
- Capucci, D.; Balestri, D.; Mazzeo, P.P.; Pelagatti, P.; Rubini, K.; Bacchi, A. Liquid Nicotine Tamed in Solid Forms by Cocrystallization. Cryst. Growth Des. 2017, 17, 4958–4964. [Google Scholar] [CrossRef]
- Habgood, M. Bioactive focus in conformational ensembles: A pluralistic approach. J. Comput. Aided Mol. Des. 2017, 31, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- Iuzzolino, L.; Reilly, A.M.; McCabe, P.; Price, S.L. Use of Crystal Structure Informatics for Defining the Conformational Space Needed for Predicting Crystal Structures of Pharmaceutical Molecules. J. Chem. Theory Comput. 2017, 13, 5163–5171. [Google Scholar] [CrossRef]
- Iuzzolino, L.; McCabe, P.; Price, S.L.; Brandenburg, J.G. Crystal structure prediction of flexible pharmaceutical-like molecules: Density functional tight-binding as an intermediate optimisation method and for free energy estimation. Faraday Discuss. 2018, 211, 275–296. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.K.; Zewail, A.H. Dynamics of Water in Biological Recognition. Chem. Rev. 2004, 104, 2099–2124. [Google Scholar] [CrossRef]
- Terao, H.; Sugawara, T.; Kita, Y.; Sato, N.; Kaho, E.; Takeda, S. Proton Relay in a One-Dimensional Hydrogen-Bonded Chain Composed of Water Molecules and a Squaric Acid Derivative. J. Am. Chem. Soc. 2001, 123, 10468–10474. [Google Scholar] [CrossRef] [PubMed]
- Ananyev, I.V.; Bushmarinov, I.S.; Ushakov, I.E.; Aitkulova, A.I.; Lyssenko, K.A. Tuning of the double-well potential of short strong hydrogen bonds by ionic interactions in alkali metal hydrodicarboxylates. RSC Adv. 2015, 5, 97495–97502. [Google Scholar] [CrossRef]
- Hickey, M.B.; Peterson, M.L.; Manas, E.S.; Alvarez, J.; Haeffner, F.; Almarsson, Ö. Hydrates and Solid-State Reactivity: A Survey of β-Lactam Antibiotics. J. Pharm. Sci. 2007, 96, 1090–1099. [Google Scholar] [CrossRef]
- Xu, H.-R.; Zhang, Q.-C.; Ren, Y.-P.; Zhao, H.-X.; Long, L.-S.; Huang, R.-B.; Zheng, L.-S. The influence of water on dielectric property in cocrystal compound of [orotic acid] [melamine]·H2O. CrystEngComm 2011, 13, 6361–6364. [Google Scholar] [CrossRef]
- Ananyev, I.V.; Barzilovich, P.Yu.; Lyssenko, K.A. Evidence for the Zundel-like Character of Oxoethylidenediphosphonic Acid Hydrate. Mendeleev Commun. 2012, 22, 242–244. [Google Scholar] [CrossRef]
- Liu, F.; Hooks, D.E.; Li, N.; Mara, N.A.; Swift, J.A. Mechanical Properties of Anhydrous and Hydrated Uric Acid Crystals. Chem. Mater. 2018, 30, 3798–3805. [Google Scholar] [CrossRef]
- Silverstein, K.A.T.; Haymet, A.D.J.; Dill, K.A. A Simple Model of Water and the Hydrophobic Effect. J. Am. Chem. Soc. 1998, 120, 3166–3175. [Google Scholar] [CrossRef]
- Ludwig, R. Water: From Clusters to the Bulk. Angew. Chem. Int. Ed. 2001, 40, 1808–1827. [Google Scholar] [CrossRef]
- Takeuchi, F.; Hiratsuka, M.; Ohmura, R.; Alavi, S.; Sum, A.K.; Yasuoka, K. Water proton configurations in structures I, II, and H clathrate hydrate unit cells. J. Chem. Phys. 2013, 138, 124504. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.; Alves, J.S.R.; Bernardes, C.E.S.; Piedade, M.F.M.; Piedade, M.E.M. da Tautomer selection through solvate formation: The case of 5-hydroxynicotinic acid. CrystEngComm 2019, 21, 2220–2233. [Google Scholar] [CrossRef]
- Medvedev, A.G.; Mikhailov, A.A.; Prikhodchenko, P.V.; Tripol’skaya, T.A.; Lev, O.; Churakov, A.V. Crystal structures of pyridinemonocarboxylic acid peroxosolvates. Russ. Chem. Bull. 2013, 62, 1871–1876. [Google Scholar] [CrossRef]
- Gillon, A.L.; Feeder, N.; Davey, R.J.; Storey, R. Hydration in Molecular Crystals. A Cambridge Structural Database Analysis. Cryst. Growth Des. 2003, 3, 663–673. [Google Scholar] [CrossRef]
- Puntus, L.N.; Lyssenko, K.A.; Pekareva, I.S.; Bünzli, J.-C.G. Intermolecular Interactions as Actors in Energy-Transfer Processes in Lanthanide Complexes with 2,2′-Bipyridine. J. Phys. Chem. B 2009, 113, 9265–9277. [Google Scholar] [CrossRef]
- Nelyubina, Y.V.; Puntus, L.N.; Lyssenko, K.A. The Dark Side of Hydrogen Bonds in the Design of Optical Materials: A Charge-Density Perspective. Chem. Eur. J. 2014, 20, 2860–2865. [Google Scholar] [CrossRef]
- Banaru, A.M.; Slovokhotov, Y.L. Crystal hydrates of organic compounds. J. Struct. Chem. 2015, 56, 967–982. [Google Scholar] [CrossRef]
- Dobrzycki, Ł.; Socha, P.; Ciesielski, A.; Boese, R.; Cyrański, M.K. Formation of Crystalline Hydrates by Nonionic Chaotropes and Kosmotropes: Case of Piperidine. Cryst. Growth Des. 2019, 19, 1005–1020. [Google Scholar] [CrossRef]
- Siddiqui, K.A. Structural Diversity of Metal–organic Hydrates: A Crystallographic Structural Database Study. J Struct. Chem. 2018, 59, 106–113. [Google Scholar] [CrossRef]
- Infantes, L.; Fábián, L.; Motherwell, W.D.S. Organic crystal hydrates: What are the important factors for formation. CrystEngComm 2007, 9, 65–71. [Google Scholar] [CrossRef]
- Desiraju, G.R. Hydration in organic crystals: Prediction from molecular structure. J. Chem. Soc. Chem. Commun. 1991, 426–428. [Google Scholar] [CrossRef]
- Van de Streek, J.; Motherwell, S. New software for searching the Cambridge Structural Database for solvated and unsolvated crystal structures applied to hydrates. CrystEngComm 2007, 9, 55–64. [Google Scholar] [CrossRef]
- Bajpai, A.; Scott, H.S.; Pham, T.; Chen, K.-J.; Space, B.; Lusi, M.; Perry, M.L.; Zaworotko, M.J. Towards an understanding of the propensity for crystalline hydrate formation by molecular compounds. IUCrJ 2016, 3, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Brychczynska, M.; Davey, R.J.; Pidcock, E. A study of methanol solvates using the Cambridge structural database. New J. Chem. 2008, 32, 1754–1760. [Google Scholar] [CrossRef]
- Brychczynska, M.; Davey, R.J.; Pidcock, E. A study of dimethylsulfoxide solvates using the Cambridge Structural Database (CSD). CrystEngComm 2012, 14, 1479–1484. [Google Scholar] [CrossRef]
- Spiteri, L.; Baisch, U.; Vella-Zarb, L. Correlations and statistical analysis of solvent molecule hydrogen bonding—a case study of dimethyl sulfoxide (DMSO). CrystEngComm 2018, 20, 1291–1303. [Google Scholar] [CrossRef]
- Navasardyan, M.A.; Grishanov, D.A.; Tripol’skaya, T.A.; Kuz’mina, L.G.; Prikhodchenko, P.V.; Churakov, A.V. Crystal structures of non-proteinogenic amino acid peroxosolvates: Rare example of H-bonded hydrogen peroxide chains. CrystEngComm 2018, 20, 7413–7416. [Google Scholar] [CrossRef]
- Inokuma, Y.; Matsumura, K.; Yoshioka, S.; Fujita, M. Finding a New Crystalline Sponge from a Crystallographic Database. Chem.—Asian J. 2017, 12, 208–211. [Google Scholar] [CrossRef]
- Byrn, S.R.; Lin, C.-T. The effect of crystal packing and defects on desolvation of hydrate crystals of caffeine and L-(-)-1,4-cyclohexadiene-1-alanine. J. Am. Chem. Soc. 1976, 98, 4004–4005. [Google Scholar] [CrossRef]
- Leung, S.S.; Padden, B.E.; Munson, E.J.; Grant, D.J.W. Hydration and Dehydration Behavior of Aspartame Hemihydrate. J. Pharm. Sci. 1998, 87, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.; Biswas, S.; Steele, I.M.; Dey, K.; Jana, A.D.; Kumar, S. Stabilization of 2D water sheets in a supramolecular metal–organic Schiff base complex: Reversible structural transformation upon dehydration–rehydration. Inorg. Chim. Acta 2013, 399, 200–207. [Google Scholar] [CrossRef]
- Zhang, H.; Yin, Z. Discrete cage form of water hexamer in the hydrophilic channels assembled by heterocyclic azopyrrole. J. Mol. Struct. 2015, 1092, 9–13. [Google Scholar] [CrossRef]
- Vologzhanina, A.V.; Zorina-Tikhonova, E.N.; Matyukhina, A.K.; Sidorov, A.A.; Dorovatovskii, P.V.; Eremenko, I.L. 36-Nuclear anionic cobalt(II) and nickel(II) complexes in solid-phase insertion reactions. Russ. J. Coord. Chem. 2017, 43, 801–806. [Google Scholar] [CrossRef]
- Willart, J.F.; Danede, F.; De Gusseme, A.; Descamps, M.; Neves, C. Origin of the Dual Structural Transformation of Trehalose Dihydrate upon Dehydration. J. Phys. Chem. B 2003, 107, 11158–11162. [Google Scholar] [CrossRef]
- Gillon, A.L.; Davey, R.J.; Storey, R.; Feeder, N.; Nichols, G.; Dent, G.; Apperley, D.C. Solid State Dehydration Processes: Mechanism of Water Loss from Crystalline Inosine Dihydrate. J. Phys. Chem. B 2005, 109, 5341–5347. [Google Scholar] [CrossRef] [PubMed]
- Fucke, K.; Steed, J.W. X-ray and Neutron Diffraction in the Study of Organic Crystalline Hydrates. Water 2010, 2, 333–350. [Google Scholar] [CrossRef] [Green Version]
- Braun, D.E.; Griesser, U.J. Supramolecular Organization of Nonstoichiometric Drug Hydrates: Dapsone. Front. Chem. 2018, 6, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Białońska, A.; Ciunik, Z.; Ilczyszyn, M.M.; Siczek, M. Discrete Cuboidal 15- and 16-Membered Water Clusters in Brucine 3.86-Hydrate, Water Release and Its Consequences. Cryst. Growth Des. 2014, 14, 6537–6541. [Google Scholar] [CrossRef]
- Febles, M.; Pérez-Hernández, N.; Pérez, C.; Rodríguez, M.L.; Foces-Foces, C.; Roux, M.V.; Morales, E.Q.; Buntkowsky, G.; Limbach, H.-H.; Martín, J.D. Distinct Dynamic Behaviors of Water Molecules in Hydrated Pores. J. Am. Chem. Soc. 2006, 128, 10008–10009. [Google Scholar] [CrossRef]
- CSD Python API Forum. Available online: https://www.ccdc.cam.ac.uk/forum/csd_python_api (accessed on 14 August 2019).
- Moghadam, P.Z.; Li, A.; Wiggin, S.B.; Tao, A.; Maloney, A.G.P.; Wood, P.A.; Ward, S.C.; Fairen-Jimenez, D. Development of a Cambridge Structural Database Subset: A Collection of Metal–Organic Frameworks for Past, Present, and Future. Chem. Mater. 2017, 29, 2618–2625. [Google Scholar] [CrossRef]
- Dolinar, B.S.; Samedov, K.; Maloney, A.G.P.; West, R.; Khrustalev, V.N.; Guzei, I.A. A chiral diamine: Practical implications of a three-stereoisomer cocrystallization. Acta Cryst. Sect. C 2018, 74, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Bryant, M.J.; Maloney, A.G.P.; Sykes, R.A. Predicting mechanical properties of crystalline materials through topological analysis. CrystEngComm 2018, 20, 2698–2704. [Google Scholar] [CrossRef]
- Miklitz, M.; Jelfs, K.E. pywindow: Automated Structural Analysis of Molecular Pores. J. Chem. Inf. Model. 2018, 58, 2387–2391. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kim, B.; Choi, S.; Boyd, P.G.; Smit, B.; Kim, J. Text Mining Metal–Organic Framework Papers. J. Chem. Inf. Model. 2018, 58, 244–251. [Google Scholar] [CrossRef] [PubMed]
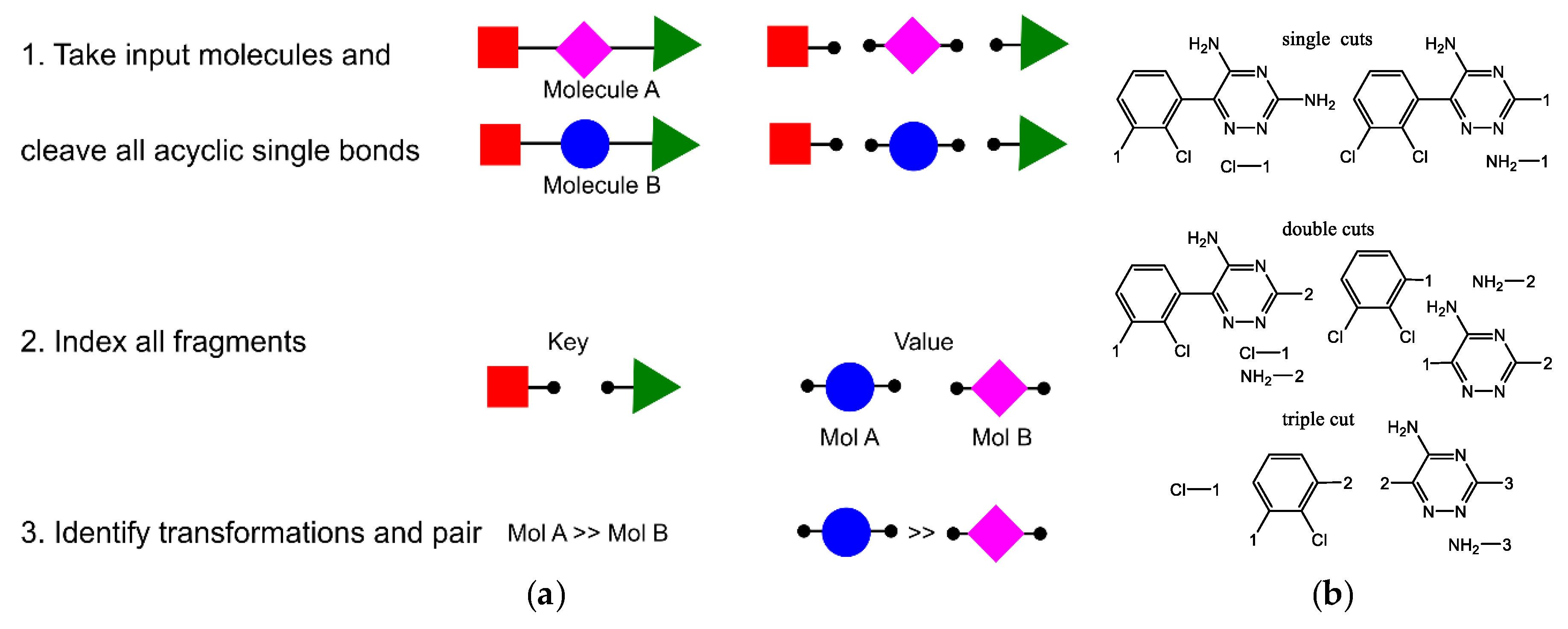
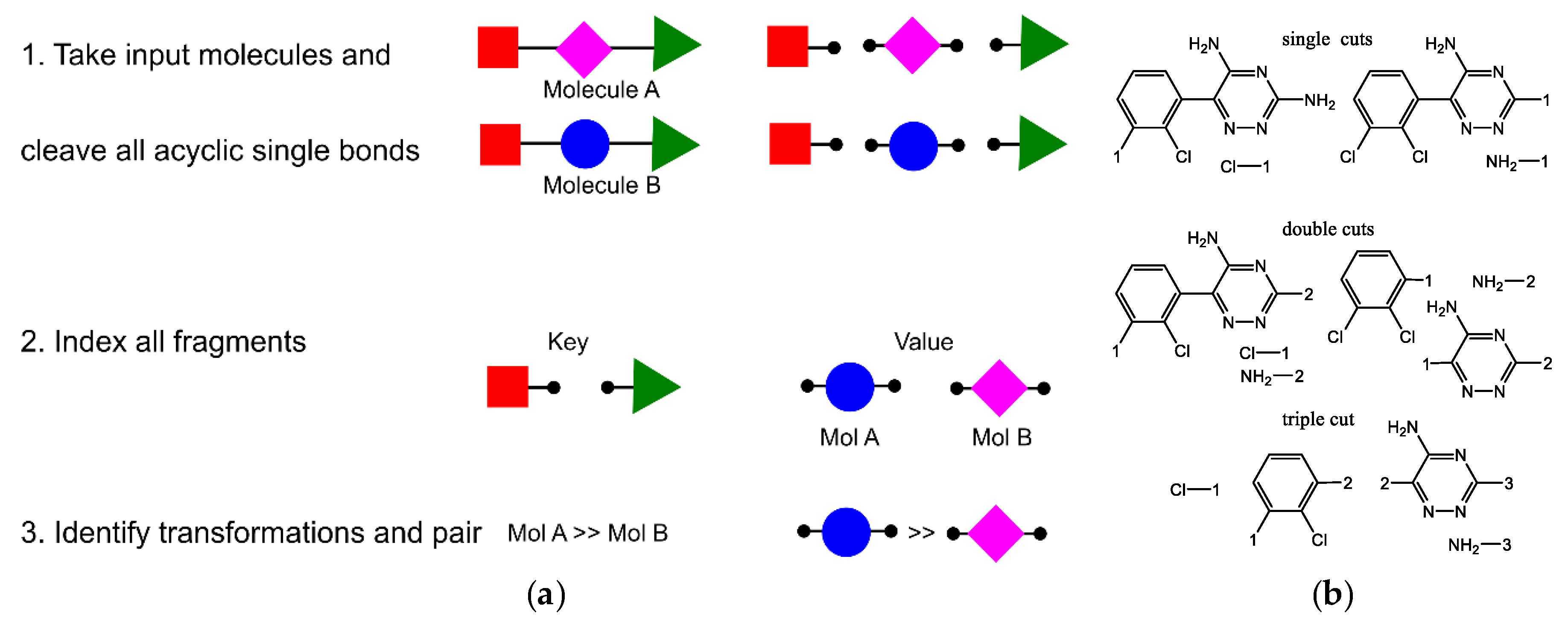
- Hussain, J.; Rea, C. Computationally Efficient Algorithm to Identify Matched Molecular Pairs (MMPs) in Large Data Sets. J. Chem. Inf. Model. 2010, 50, 339–348. [Google Scholar] [CrossRef] [PubMed]
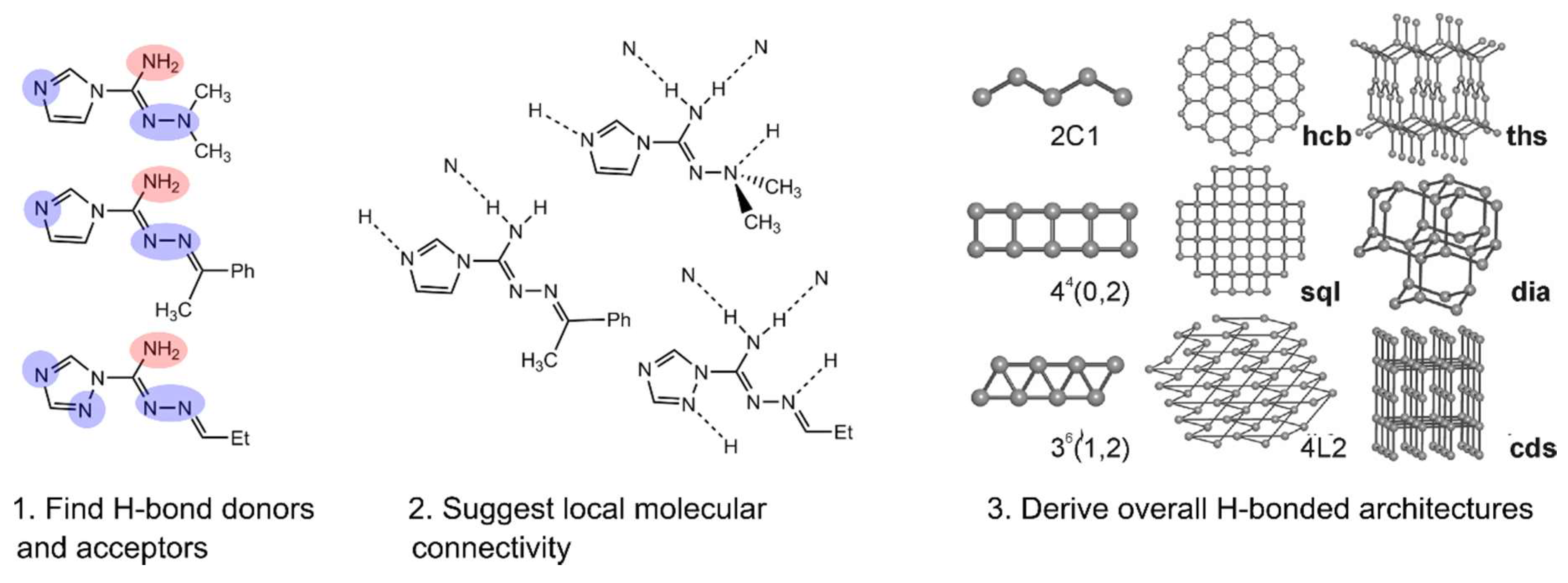
- Zolotarev, P.N.; Arshad, M.N.; Asiri, A.M.; Al-amshany, Z.M.; Blatov, V.A. A Possible Route toward Expert Systems in Supramolecular Chemistry: 2-Periodic H-Bond Patterns in Molecular Crystals. Cryst. Growth Des. 2014, 14, 1938–1949. [Google Scholar] [CrossRef]
- Blatova, O.A.; Asiri, A.M.; Al-amshany, Z.M.; Arshad, M.N.; Blatov, V.A. Molecular packings and specific-bonding patterns in sulfonamides. New J. Chem. 2014, 38, 4099–4106. [Google Scholar] [CrossRef]
- Baburin, I.A.; Blatov, V.A.; Carlucci, L.; Ciani, G.; Proserpio, D.M. Interpenetrated Three-Dimensional Networks of Hydrogen-Bonded Organic Species: A Systematic Analysis of the Cambridge Structural Database. Cryst. Growth Des. 2008, 8, 519–539. [Google Scholar] [CrossRef]
- Klamt, A. Conductor-like Screening Model for Real Solvents: A New Approach to the Quantitative Calculation of Solvation Phenomena. J. Phys. Chem. 1995, 99, 2224–2235. [Google Scholar] [CrossRef]
- Klamt, A.; Jonas, V.; Bürger, T.; Lohrenz, J.C.W. Refinement and Parametrization of COSMO-RS. J. Phys. Chem. A 1998, 102, 5074–5085. [Google Scholar] [CrossRef]
- Tilbury, C.J.; Chen, J.; Mattei, A.; Chen, S.; Sheikh, A.Y. Combining Theoretical and Data-Driven Approaches To Predict Drug Substance Hydrate Formation. Cryst. Growth Des. 2018, 18, 57–67. [Google Scholar] [CrossRef]
- Chem3D, version 18.2 Suite; Perkin Elmer Corporation: Waltham, MA, USA, 2016.
- CCDC Documentation and Resources. Available online: https://ccdc.cam.ac.uk/support-and-resources/ccdcresources/ (accessed on 14 August 2019).
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vologzhanina, A.V. Intermolecular Interactions in Functional Crystalline Materials: From Data to Knowledge. Crystals 2019, 9, 478. https://doi.org/10.3390/cryst9090478
Vologzhanina AV. Intermolecular Interactions in Functional Crystalline Materials: From Data to Knowledge. Crystals. 2019; 9(9):478. https://doi.org/10.3390/cryst9090478
Chicago/Turabian StyleVologzhanina, Anna V. 2019. "Intermolecular Interactions in Functional Crystalline Materials: From Data to Knowledge" Crystals 9, no. 9: 478. https://doi.org/10.3390/cryst9090478
APA StyleVologzhanina, A. V. (2019). Intermolecular Interactions in Functional Crystalline Materials: From Data to Knowledge. Crystals, 9(9), 478. https://doi.org/10.3390/cryst9090478