Comparison of Candida Albicans Fatty Acid Amide Hydrolase Structure with Homologous Amidase Signature Family Enzymes




Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning of FAAH from Candida Albicans
2.2. Purification of Recombinant Proteins
2.3. Crystallization
2.4. Data Collection and Structure Determination
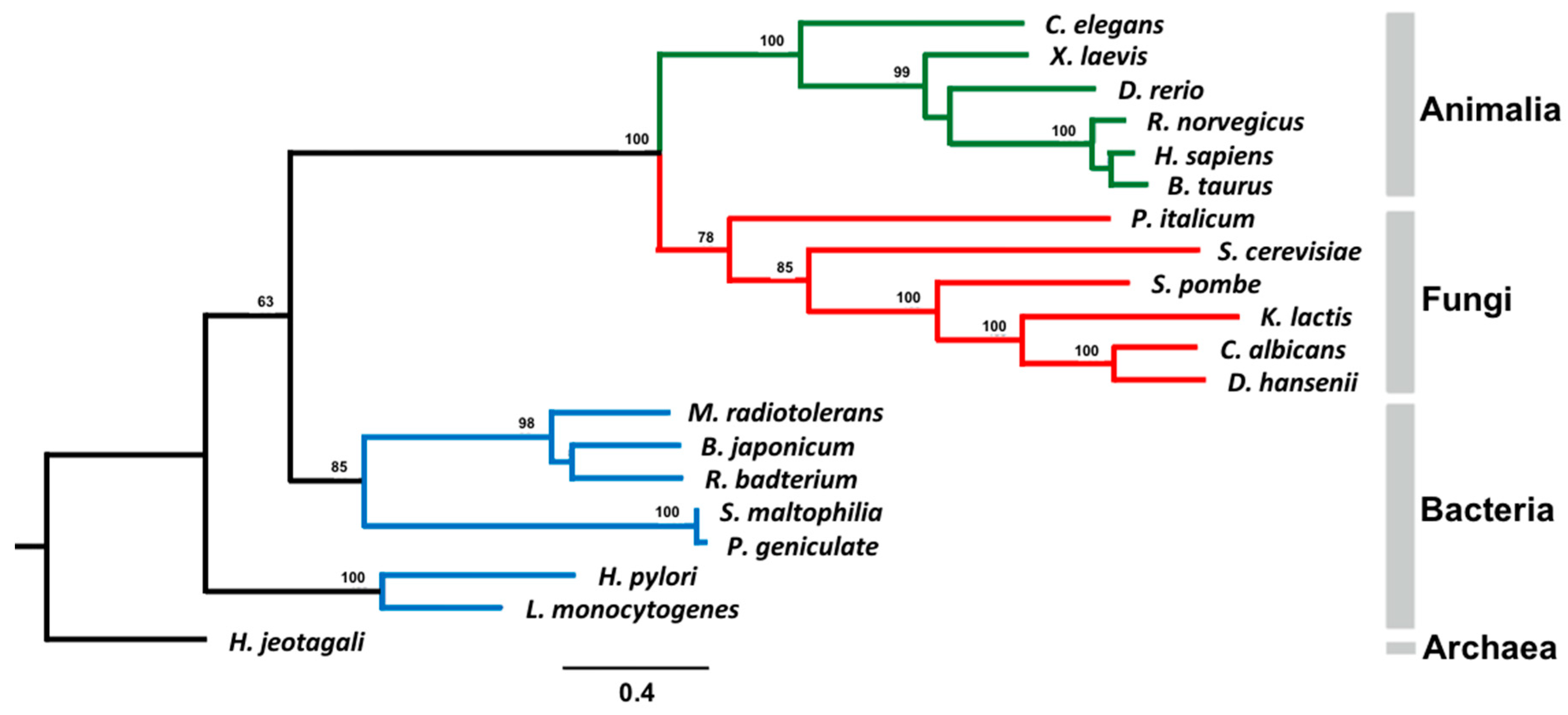
2.5. Phylogenetic Analysis
3. Results
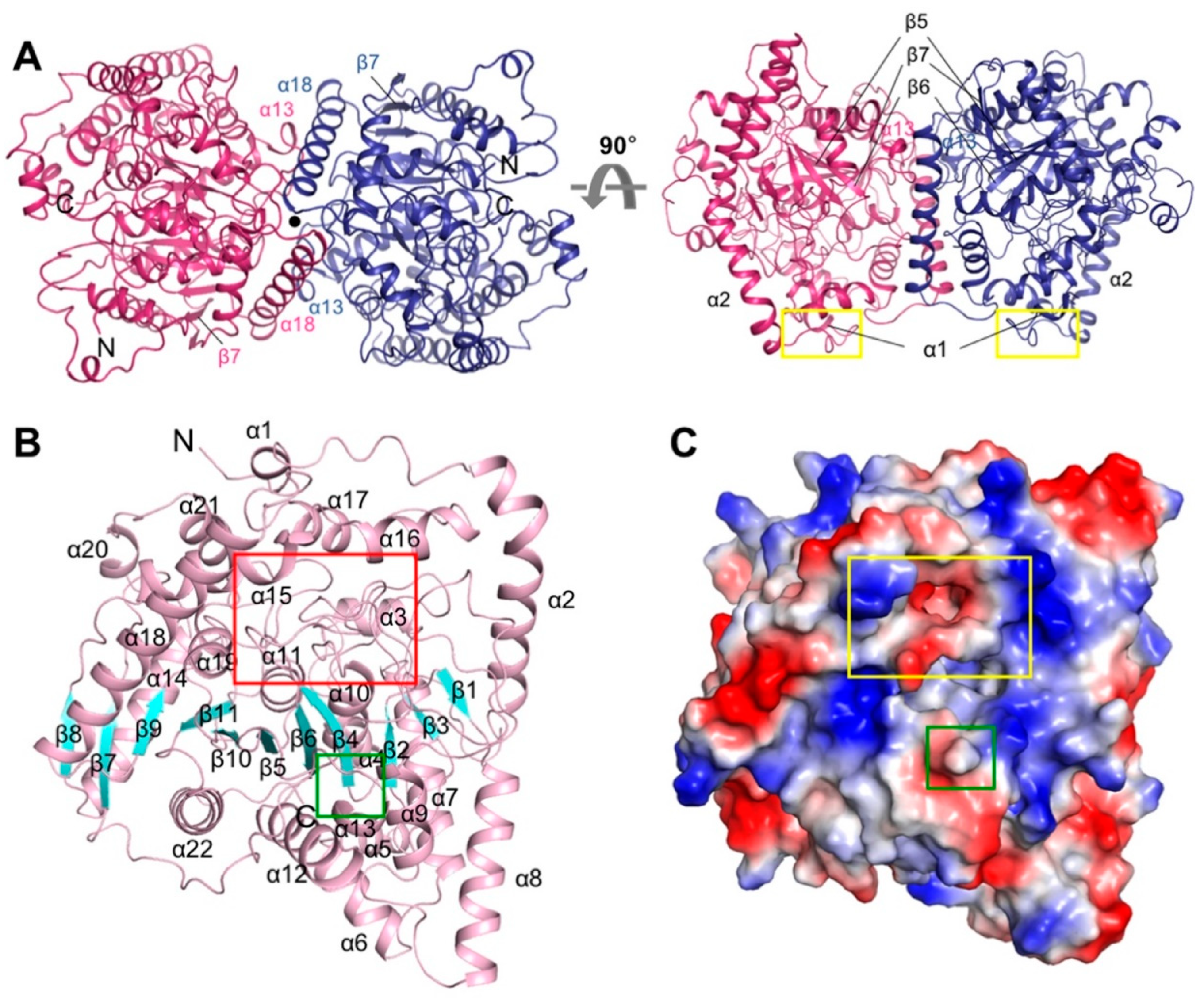
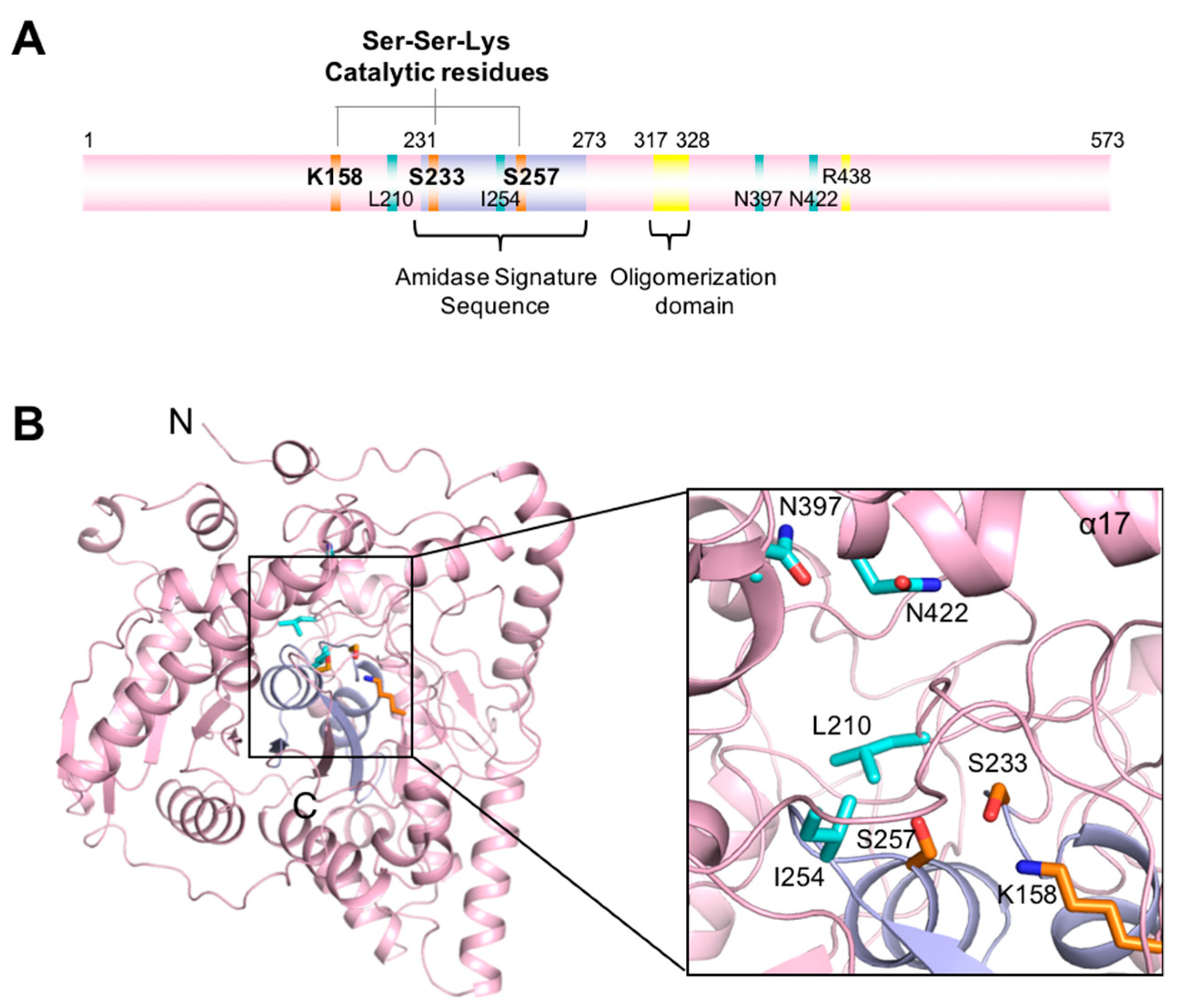
3.1. Overall Structure of CaFAAH
3.2. Active Site
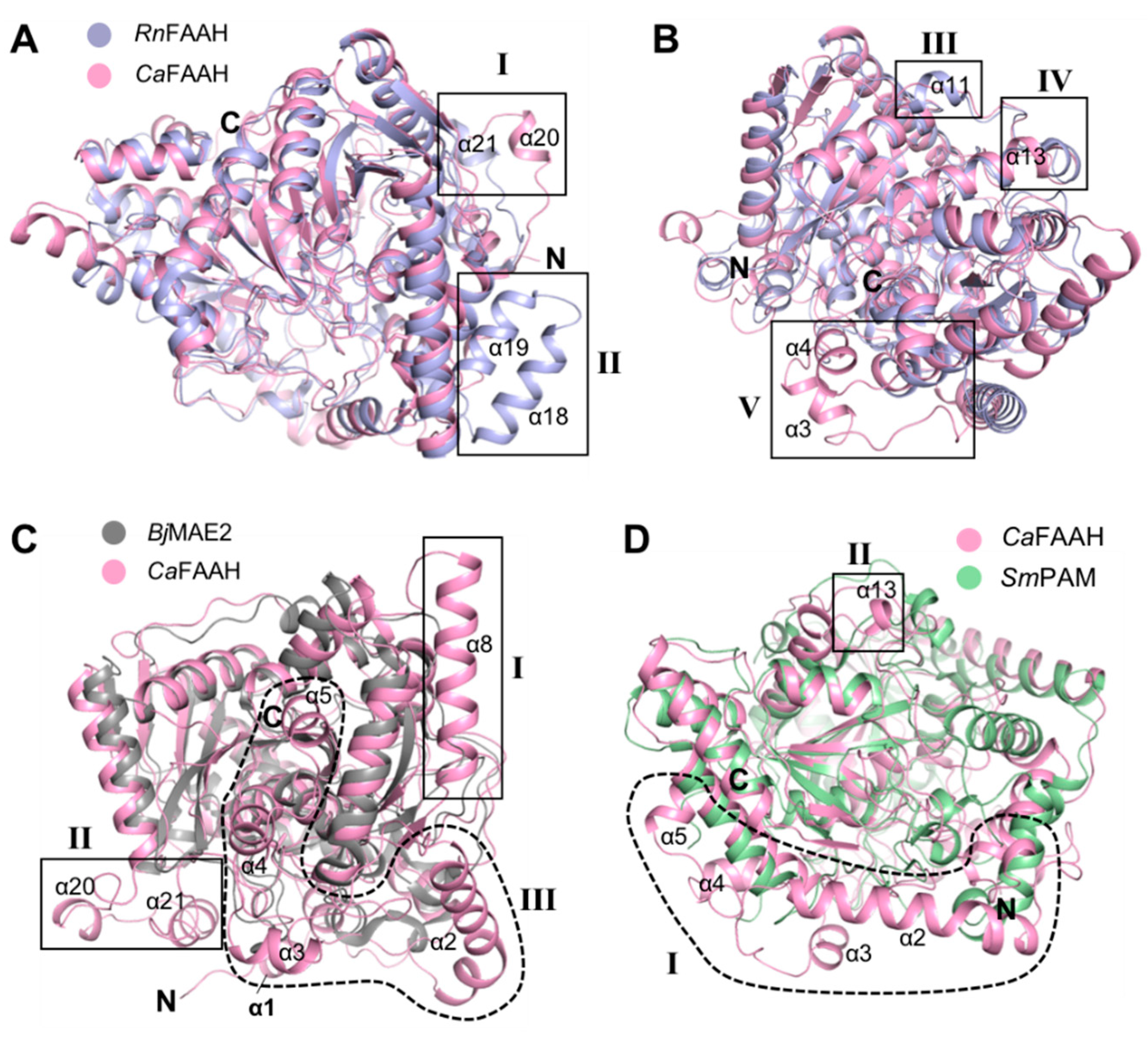
3.3. Comparison of CaFAAH Structure with the AS Family Protein RnFAAH
3.4. Comparison of CaFAAH Structure with AS Family Proteins BjMAE2 and SmFAM
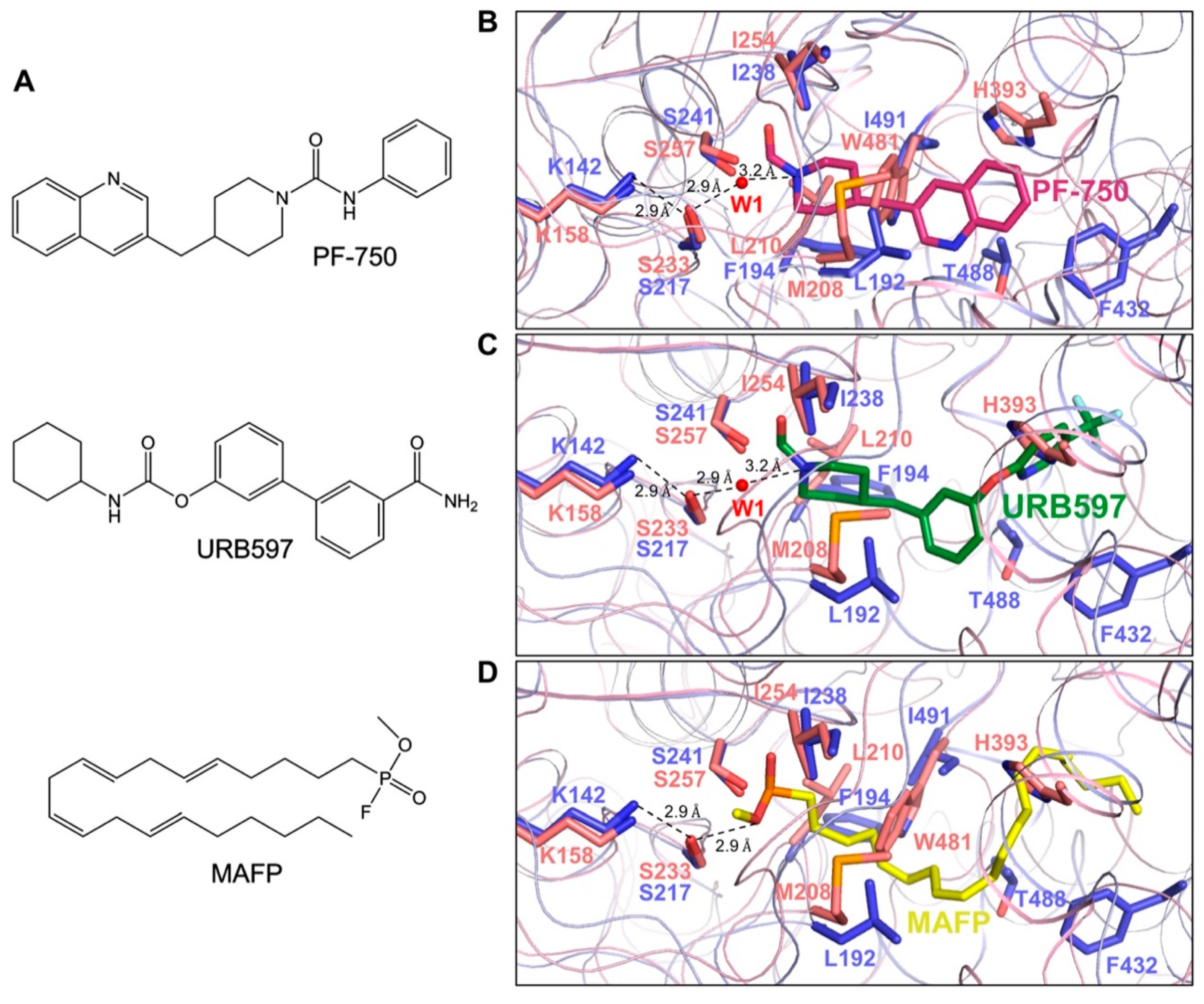
3.5. Comparison of CaFAAH Active Site with the RnFAAH-Inhibitor Complexes
3.6. Phylogenetic Analysis of AS Family Proteins
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patricelli, M.P.; Lovato, M.A.; Cravatt, B.F. Chemical and mutagenic investigations of fatty acid amide hydrolase: Evidence for a family of serine hydrolases with distinct catalytic properties. Biochemistry 1999, 38, 9804–9812. [Google Scholar] [CrossRef] [PubMed]
- Labahn, J.; Neumann, S.; Buldt, G.; Kula, M.R.; Granzin, J. An alternative mechanism for amidase signature enzymes. J. Mol. Biol. 2002, 322, 1053–1064. [Google Scholar] [CrossRef]
- Shin, S.; Lee, T.H.; Ha, N.C.; Koo, H.M.; Kim, S.Y.; Lee, H.S.; Kim, Y.S.; Oh, B.H. Structure of malonamidase E2 reveals a novel Ser-cisSer-Lys catalytic triad in a new serine hydrolase fold that is prevalent in nature. EMBO J. 2002, 21, 2509–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracey, M.H.; Hanson, M.A.; Masuda, K.R.; Stevens, R.C.; Cravatt, B.F. Structural adaptations in a membrane enzyme that terminates endocannabinoid signaling. Science 2002, 298, 1793–1796. [Google Scholar] [CrossRef] [PubMed]
- Yasuhira, K.; Shibata, N.; Mongami, G.; Uedo, Y.; Atsumi, Y.; Kawashima, Y.; Hibino, A.; Tanaka, Y.; Lee, Y.H.; Kato, D.; et al. X-ray crystallographic analysis of the 6-aminohexanoate cyclic dimer hydrolase: Catalytic mechanism and evolution of an enzyme responsible for nylon-6 byproduct degradation. J. Biol. Chem. 2010, 285, 1239–1248. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Yao, M.; Chimnaronk, S.; Sakai, N.; Tanaka, I. Ammonia channel couples glutaminase with transamidase reactions in GatCAB. Science 2006, 312, 1954–1958. [Google Scholar] [CrossRef]
- Ahn, K.; McKinney, M.K.; Cravatt, B.F. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem. Rev. 2008, 108, 1687–1707. [Google Scholar] [CrossRef]
- Bradshaw, H.B.; Rimmerman, N.; Hu, S.S.; Benton, V.M.; Stuart, J.M.; Masuda, K.; Cravatt, B.F.; O’Dell, D.K.; Walker, J.M. The endocannabinoid anandamide is a precursor for the signaling lipid N-arachidonoyl glycine by two distinct pathways. BMC Biochem. 2009, 10, 14. [Google Scholar] [CrossRef]
- Lu, H.C.; Mackie, K. An Introduction to the Endogenous Cannabinoid System. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef]
- Ahn, K.; Johnson, D.S.; Mileni, M.; Beidler, D.; Long, J.Z.; McKinney, M.K.; Weerapana, E.; Sadagopan, N.; Liimatta, M.; Smith, S.E.; et al. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem. Biol. 2009, 16, 411–420. [Google Scholar] [CrossRef]
- Ogawa, S.; Kunugi, H. Inhibitors of Fatty Acid Amide Hydrolase and Monoacylglycerol Lipase: New Targets for Future Antidepressants. Curr. Neuropharm. 2015, 13, 760–775. [Google Scholar] [CrossRef] [Green Version]
- Deutsch, D.G.; Ueda, N.; Yamamoto, S. The fatty acid amide hydrolase (FAAH). Prostaglandins Leukot. Essent. Fat. Acids 2002, 66, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Mileni, M.; Johnson, D.S.; Wang, Z.; Everdeen, D.S.; Liimatta, M.; Pabst, B.; Bhattacharya, K.; Nugent, R.A.; Kamtekar, S.; Cravatt, B.F.; et al. Structure-guided inhibitor design for human FAAH by interspecies active site conversion. Proc. Natl. Acad. Sci. USA 2008, 105, 12820–12824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mileni, M.; Kamtekar, S.; Wood, D.C.; Benson, T.E.; Cravatt, B.F.; Stevens, R.C. Crystal structure of fatty acid amide hydrolase bound to the carbamate inhibitor URB597: Discovery of a deacylating water molecule and insight into enzyme inactivation. J. Mol. Biol. 2010, 400, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Mileni, M.; Garfunkle, J.; Ezzili, C.; Cravatt, B.F.; Stevens, R.C.; Boger, D.L. Fluoride-mediated capture of a noncovalent bound state of a reversible covalent enzyme inhibitor: X-ray crystallographic analysis of an exceptionally potent alpha-ketoheterocycle inhibitor of fatty acid amide hydrolase. J. Am. Chem. Soc. 2011, 133, 4092–4100. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Matsumoto, T.; Kawamura, T.; Nishimura, A.; Kiyota, Y.; Oki, H.; Miyazaki, J.; Igaki, S.; Behnke, C.A.; Shimojo, M.; et al. Synthesis, SAR study, and biological evaluation of a series of piperazine ureas as fatty acid amide hydrolase (FAAH) inhibitors. Bioorg. Med. Chem. 2013, 21, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Bertolacci, L.; Romeo, E.; Veronesi, M.; Magotti, P.; Albani, C.; Dionisi, M.; Lambruschini, C.; Scarpelli, R.; Cavalli, A.; De Vivo, M.; et al. A binding site for nonsteroidal anti-inflammatory drugs in fatty acid amide hydrolase. J. Am. Chem. Soc. 2013, 135, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Ogasawara, D.; Seneviratne, U.I.; Cognetta, A.B., 3rd; Am Ende, C.W.; Nason, D.M.; Lapham, K.; Litchfield, J.; Johnson, D.S.; Cravatt, B.F. Global Portrait of Protein Targets of Metabolites of the Neurotoxic Compound BIA 10-2474. ACS Chem. Biol. 2019, 14, 192–197. [Google Scholar] [CrossRef]
- Alasmari, M.; Bhlke, M.; Kelley, C.; Maher, T.; Pino-Figueroa, A. Inhibition of Fatty Acid Amide Hydrolase (FAAH) by Macamides. Mol. Neurobiol. 2019, 56, 1770–1781. [Google Scholar] [CrossRef]
- Pacher, P.; Batkai, S.; Kunos, G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef]
- Cravatt, B.F.; Demarest, K.; Patricelli, M.P.; Bracey, M.H.; Giang, D.K.; Martin, B.R.; Lichtman, A.H. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc. Natl. Acad. Sci. USA 2001, 98, 9371–9376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, C.A.; Yun, J.S.; Yoon, J.H.; Chang, J.H. Purification, crystallization, and X-ray crystallographic analysis of fatty acid amide hydrolase from Candida albicans. Biodesign 2019, 7, 38–41. [Google Scholar] [CrossRef]
- Ramakrishnan, V.; Finch, J.T.; Graziano, V.; Lee, P.L.; Sweet, R.M. Crystal structure of globular domain of histone H5 and its implications for nucleosome binding. Nature 1993, 362, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997, 276, 307–326. [Google Scholar]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evolut. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef]
- Cravatt, B.F.; Giang, D.K.; Mayfield, S.P.; Boger, D.L.; Lerner, R.A.; Gilula, N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 1996, 384, 83–87. [Google Scholar] [CrossRef]
- Holm, L.; Rosenstrom, P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010, 38, W545–W549. [Google Scholar] [CrossRef] [PubMed]
- Valina, A.L.; Mazumder-Shivakumar, D.; Bruice, T.C. Probing the Ser-Ser-Lys catalytic triad mechanism of peptide amidase: Computational studies of the ground state, transition state, and intermediate. Biochemistry 2004, 43, 15657–15672. [Google Scholar] [CrossRef] [PubMed]
- Chebrou, H.; Bigey, F.; Arnaud, A.; Galzy, P. Study of the amidase signature group. Biochim. Biophys. Acta 1996, 1298, 285–293. [Google Scholar] [CrossRef]
- Cilia, E.; Fabbri, A.; Uriani, M.; Scialdone, G.G.; Ammendola, S. The signature amidase from Sulfolobus solfataricus belongs to the CX3C subgroup of enzymes cleaving both amides and nitriles. Ser195 and Cys145 are predicted to be the active site nucleophiles. FEBS J. 2005, 272, 4716–4724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.; Yun, Y.S.; Koo, H.M.; Kim, Y.S.; Choi, K.Y.; Oh, B.H. Characterization of a novel Ser-cisSer-Lys catalytic triad in comparison with the classical Ser-His-Asp triad. J. Biol. Chem. 2003, 278, 24937–24943. [Google Scholar] [CrossRef]
- Patricelli, M.P.; Cravatt, B.F. Clarifying the catalytic roles of conserved residues in the amidase signature family. J. Biol. Chem. 2000, 275, 19177–19184. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishna, K.N.; Stewart, B.H.; Kneen, M.M.; Andricopulo, A.D.; Kenyon, G.L.; McLeish, M.J. Mandelamide hydrolase from Pseudomonas putida: Characterization of a new member of the amidase signature family. Biochemistry 2004, 43, 7725–7735. [Google Scholar] [CrossRef]
- Kobayashi, M.; Fujiwara, Y.; Goda, M.; Komeda, H.; Shimizu, S. Identification of active sites in amidase: Evolutionary relationship between amide bond- and peptide bond-cleaving enzymes. Proc. Natl. Acad. Sci. USA 1997, 94, 11986–11991. [Google Scholar] [CrossRef] [Green Version]
- Mileni, M.; Garfunkle, J.; Ezzili, C.; Kimball, F.S.; Cravatt, B.F.; Stevens, R.C.; Boger, D.L. X-ray crystallographic analysis of alpha-ketoheterocycle inhibitors bound to a humanized variant of fatty acid amide hydrolase. J. Med. Chem. 2010, 53, 230–240. [Google Scholar] [CrossRef]
- Tarzia, G.; Duranti, A.; Gatti, G.; Piersanti, G.; Tontini, A.; Rivara, S.; Lodola, A.; Plazzi, P.V.; Mor, M.; Kathuria, S.; et al. Synthesis and structure-activity relationships of FAAH inhibitors: Cyclohexylcarbamic acid biphenyl esters with chemical modulation at the proximal phenyl ring. ChemMedChem 2006, 1, 130–139. [Google Scholar] [CrossRef]
- Otrubova, K.; Brown, M.; McCormick, M.S.; Han, G.W.; O’Neal, S.T.; Cravatt, B.F.; Stevens, R.C.; Lichtman, A.H.; Boger, D.L. Rational design of fatty acid amide hydrolase inhibitors that act by covalently bonding to two active site residues. J. Am. Chem. Soc. 2013, 135, 6289–6299. [Google Scholar] [CrossRef] [PubMed]
- Gustin, D.J.; Ma, Z.; Min, X.; Li, Y.; Hedberg, C.; Guimaraes, C.; Porter, A.C.; Lindstrom, M.; Lester-Zeiner, D.; Xu, G.; et al. Identification of potent, noncovalent fatty acid amide hydrolase (FAAH) inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2492–2496. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.L.; McMahon, L.R. The fatty acid amide hydrolase inhibitor URB 597: Interactions with anandamide in rhesus monkeys. Br. J. Pharmacol. 2011, 164, 655–666. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystallographic Data | CaFAAH |
|---|---|
| Data collection | |
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 84.8, 68.7, 100.9 |
| α, β, γ (˚) | 90, 99.6, 90 |
| Resolution range (Å) | 50.0–2.28 (2.28–2.20) a |
| Rmerge (%) b | 10.4 (64.6) |
| I / sigmaI | 13.03 (4.66) |
| Completeness (%) | 99.8 (98.5) |
| Redundancy | 7.0 (7.4) |
| Refinement | |
| Resolution | 50.0–2.28 |
| No. of reflections | 58833 |
| Rworkc / Rfree (%) d | 17.27 / 21.97 |
| No. of atoms | 9560 |
| Protein | 8962 |
| Water | 598 |
| B-factors | |
| Protein | 33.40 |
| R.M.S. deviation | |
| Bond lengths (Å) | 0.009 |
| Bond angles (˚) | 1.21 |
| PDB code | 6KVR |
| Protein | Species | NCBI Reference Sequence |
|---|---|---|
| FAAH (Fatty acid amide hydrolase) | Danio rerio | NP_001103295.1 |
| Homo sapiens | NP_001432.2 | |
| Xenopus laevis | OCT82705.1 | |
| Bos Taurus | DAA31035.1 | |
| Rattus norvegicus | NP_077046.1 | |
| Glutamyl-tRNA (Gln) Amidotransferase | Helicobacter pylori | WP_000631451.1 |
| Listeria monocytogenes | WP_061104924.1 | |
| Malonamidase | Bradyrhizobium Japonicum | WP_011087863.1 |
| Peptide Amidase | Stenotrophomonas maltophilia | CAC93616.1 |
| Amidase | Rhizobiales bacterium | WP_037017835.1 |
| Pseudomonas geniculate | WP_057502485.1 | |
| Haloterrigena jeotgali | WP_049965501.1 | |
| Methylobacterium radiotolerans | WP_012319231.1 | |
| Saccharomyces cerevisiae * | CAA39514.1 | |
| Penicillium italicum | KGO69811 | |
| Debaryomyces hansenii * | XP_457833.2 | |
| Kluyveromyces lactis * | XP_451920.1 | |
| Candida albicans * | XP_716991.1 | |
| Schizosaccharomyces pombe * | NP_588099.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Min, C.-A.; Yun, J.-S.; Choi, E.H.; Hwang, U.W.; Cho, D.-H.; Yoon, J.-H.; Chang, J.H. Comparison of Candida Albicans Fatty Acid Amide Hydrolase Structure with Homologous Amidase Signature Family Enzymes. Crystals 2019, 9, 472. https://doi.org/10.3390/cryst9090472
Min C-A, Yun J-S, Choi EH, Hwang UW, Cho D-H, Yoon J-H, Chang JH. Comparison of Candida Albicans Fatty Acid Amide Hydrolase Structure with Homologous Amidase Signature Family Enzymes. Crystals. 2019; 9(9):472. https://doi.org/10.3390/cryst9090472
Chicago/Turabian StyleMin, Cho-Ah, Ji-Sook Yun, Eun Hwa Choi, Ui Wook Hwang, Dong-Hyung Cho, Je-Hyun Yoon, and Jeong Ho Chang. 2019. "Comparison of Candida Albicans Fatty Acid Amide Hydrolase Structure with Homologous Amidase Signature Family Enzymes" Crystals 9, no. 9: 472. https://doi.org/10.3390/cryst9090472