Crystal Structure of NADPH-Dependent Methylglyoxal Reductase Gre2 from Candida Albicans


Abstract
1. Instruction
2. Materials and Methods
2.1. Overexpression and Purification of CaGre2
2.2. Crystallization
2.3. Data Collection, Structure Solution, and Refinement
3. Results
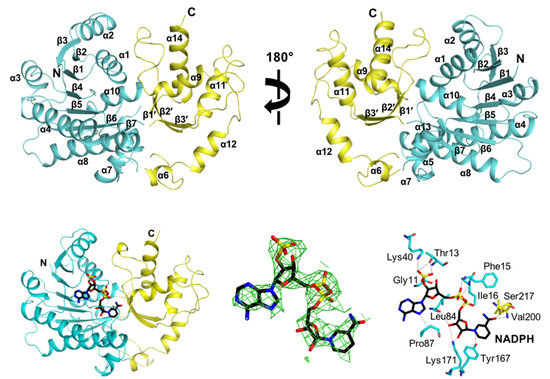
3.1. Overall Structure of CaGre2
3.2. Structural Comparisons between CaGre2 and Homologous Proteins
3.3. NADPH Binding Site
3.4. Comparisons of NAD(P)H Binding Sites
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Murata, K.; Fukuda, Y.; Simosaka, M.; Watanabe, K.; Saikusa, T.; Kimura, A. Metabolism of 2-oxoaldehyde in yeasts. Purification and characterization of NADPH-dependent methylglyoxal-reducing enzyme from Saccharomyces cerevisiae. J. Boil. Inorg. Chem. 1985, 151, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.C.; Bao, Z.Z.; Ma, X.X.; Xia, Q.; Li, W.F. Structural insights into the cofactor-assisted substrate recognition of yeast methylglyoxal/isovaleraldehyde reductase Gre2. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2014, 1844, 1486–1492. [Google Scholar] [CrossRef] [PubMed]
- Dafré, A.; Goldberg, J.; Wang, T.; Spiegel, D.; Maher, P. Methylglyoxal, the foe and friend of glyoxalase and Trx/TrxR systems in HT22 nerve cells. Free. Radic. Boil. Med. 2015, 89, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Maeta, K.; Izawa, S.; Inoue, Y.J. Methylglyoxal, a metabolite derived from glycolysis, functions as a signal initiator of the high osmolarity glycerol-mitogen-activated protein kinase cascade and calcineurin/Crz1-mediated pathway in Saccharomyces cerevisiae. J. Biol. Chem. 2005, 280, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Gomes, R.A.; Miranda, H.V.; Silva, M.S.; Graã§a, G.; Coelho, A.V.; Ferreira, A.E.D.N.; Cordeiro, C.; Freire, A.P.; Graça, G.; Ferreira, A.E.D.N. Protein glycation and methylglyoxal metabolism in yeast: Finding peptide needles in protein haystacks. FEMS Yeast Res. 2008, 8, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Lee, S.; Ku, M.; Kwak, M.K.; Kang, S.O. Cytochrome c peroxidase regulates intracellular reactive oxygen species and methylglyoxal via enzyme activities of erythroascorbate peroxidase and glutathione-related enzymes in Candida albicans. Int. J. Biochem. Cell Boil. 2017, 92, 183–201. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Ku, M.; Kang, S.O. Inducible NAD(H)-linked methylglyoxal oxidoreductase regulates cellular methylglyoxal and pyruvate through enhanced activities of alcohol dehydrogenase and methylglyoxal-oxidizing enzymes in glutathione-depleted Candida albicans. Biochim. Biophys. Acta (BBA) Gen. Subj. 2018, 1862, 18–39. [Google Scholar] [CrossRef] [PubMed]
- Islam, T.; Ghosh, A. Genome-wide dissection and expression profiling of unique glyoxalase III genes in soybean reveal the differential pattern of transcriptional regulation. Sci. Rep. 2018, 8, 4848. [Google Scholar] [CrossRef] [PubMed]
- Gomes, R.A.; Miranda, H.V.; Silva, M.S.; Graça, G.; Coelho, A.V.; Ferreira, A.E.; Cordeiro, C.; Freire, A.P.J.T.F.j. Yeast protein glycation in vivo by methylglyoxal: Molecular modification of glycolytic enzymes and heat shock proteins. FEBS J. 2006, 273, 5273–5287. [Google Scholar] [CrossRef]
- Chen, C.N.; Porubleva, L.; Shearer, G.; Svrakic, M.; Holden, L.G.; Dover, J.L.; Johnston, M.; Chitnis, P.R.; Kohl, D.H. Associating protein activities with their genes: Rapid identification of a gene encoding a methylglyoxal reductase in the yeastSaccharomyces cerevisiae. Yeast 2003, 20, 545–554. [Google Scholar] [CrossRef]
- Oppermann, U.; Filling, C.; Hult, M.; Shafqat, N.; Wu, X.; Lindh, M.; Shafqat, J.; Nordling, E.; Kallberg, Y.; Persson, B.; et al. Short-chain dehydrogenases/reductases (SDR): The 2002 update. Chem. Interact. 2003, 143, 247–253. [Google Scholar] [CrossRef]
- Breicha, K.; Müller, M.; Hummel, W.; Niefind, K.J.A.C.S.F. Crystallization and preliminary crystallographic analysis of Gre2p, an NADP+-dependent alcohol dehydrogenase from Saccharomyces cerevisiae. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2010, 66, 838–841. [Google Scholar] [CrossRef] [PubMed]
- Persson, B.; Kallberg, Y. Classification and nomenclature of the superfamily of short-chain dehydrogenases/reductases (SDRs). Chem. Interact. 2013, 202, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.; Liu, Z.L. Engineered NADH-dependent GRE2 from Saccharomyces cerevisiae by directed enzyme evolution enhances HMF reduction using additional cofactor NADPH. Enzym. Microb. Technol. 2012, 50, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Warringer, J.; Blomberg, A. Involvement of yeastYOL151W/GRE2 in ergosterol metabolism. Yeast 2006, 23, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Garay-Arroyo, A.; Covarrubias, A.A. Three genes whose expression is induced by stress in Saccharomyces cerevisiae. Yeast 1999, 15, 879–892. [Google Scholar] [CrossRef]
- Akita, H.; Watanabe, M.; Suzuki, T.; Nakashima, N.; Hoshino, T. Characterization of the Kluyveromyces marxianus strain DMB1 YGL157w gene product as a broad specificity NADPH-dependent aldehyde reductase. AMB Express 2015, 5, 991. [Google Scholar] [CrossRef]
- Nguyen, G.T.; Kim, S.A.; Chun, H.S.; Kim, W.K.; Chang, J.H. Purification, crystallization and X-ray crystallographic analysis of methylglyoxal reductase Gre2 from Candida albicans. Korean Soc. Struct. Boil. 2019, 7, 42–46. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J Appl. Cryst. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Karplus, P.A.; Diederichs, K. Linking crystallographic model and data quality. Science 2012, 336, 1030–1033. [Google Scholar] [CrossRef]
- Madden, T. The BLAST Sequence Analysis Tool. In The NCBI Handbook [Internet], 2nd ed.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2013. Available online: https://www.ncbi.nlm.nih.gov/books/NBK153387/ (accessed on 9 September 2019).
- Matthews, B. Solvent content of protein crystals. J. Mol. Boil. 1968, 33, 491–497. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Boil. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Holm, L. Benchmarking fold detection by DaliLite v.5. Bioinformatics 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinform. 2003, 00, 2.3.1–2.3.22. [Google Scholar] [CrossRef] [PubMed]
- Courcelle, E.; Gouet, P.; Stuart, D.I.; Métoz, F. ESPript: Analysis of multiple sequence alignments in PostScript. Bioinformatics 1999, 15, 305–308. [Google Scholar]
- Zhou, J.; Wang, Y.; Xu, G.; Wu, L.; Han, R.; Schwaneberg, U.; Rao, Y.; Zhao, Y.L.; Zhou, J.; Ni, Y. Structural Insight into Enantioselective Inversion of an Alcohol Dehydrogenase Reveals a “Polar Gate” in Stereorecognition of Diaryl Ketones. J. Am. Chem. Soc. 2018, 140, 12645–12654. [Google Scholar] [CrossRef]
- Shang, Y.P.; Chen, Q.; Kong, X.D.; Zhang, Y.J.; Xu, J.H.; Yu, H.L. Efficient Synthesis of (R)-2-Chloro-1-(2,4-dichlorophenyl)ethanol with a Ketoreductase fromScheffersomyces stipitisCBS 6045. Adv. Synth. Catal. 2017, 359, 426–431. [Google Scholar] [CrossRef]
- Filling, C.; Berndt, K.D.; Benach, J.; Knapp, S.; Prozorovski, T.; Nordling, E.; Ladenstein, R.; Jörnvall, H.; Oppermann, U. Critical Residues for Structure and Catalysis in Short-chain Dehydrogenases/Reductases. J. Boil. Chem. 2002, 277, 25677–25684. [Google Scholar] [CrossRef]
- Kamitori, S.; Iguchi, A.; Ohtaki, A.; Yamada, M.; Kita, K. X-ray Structures of NADPH-dependent Carbonyl Reductase from Sporobolomyces salmonicolor Provide Insights into Stereoselective Reductions of Carbonyl Compounds. J. Mol. Boil. 2005, 352, 551–558. [Google Scholar] [CrossRef]
- Thoden, J.B.; Wohlers, T.M.; Fridovich-Keil, J.L.; Holden, H.M. Crystallographic Evidence for Tyr 157 Functioning as the Active Site Base in Human UDP−Galactose 4-Epimerase. Biochemistry 2000, 39, 5691–5701. [Google Scholar] [CrossRef]
- Thoden, J.B.; Frey, P.A.; Holden, H.M. Crystal Structures of the Oxidized and Reduced Forms of UDP-galactose 4-Epimerase Isolated from Escherichia coli. Biochemistry 1996, 35, 2557–2566. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Zhou, R.; Louie, G.V.; Muhlemann, J.K.; Bomati, E.K.; Bowman, M.E.; Dudareva, N.; Dixon, R.A.; Noel, J.P.; Wang, X. Structural Studies of Cinnamoyl-CoA Reductase and Cinnamyl-Alcohol Dehydrogenase, Key Enzymes of Monolignol Biosynthesis. Plant Cell 2014, 26, 3709–3727. [Google Scholar] [CrossRef] [PubMed]
- Sattler, S.A.; Walker, A.M.; Vermerris, W.; Sattler, S.E.; Kang, C. Structural and biochemical characterization of cinnamoyl-CoA reductases. Plant Physiol. 2017, 173, 1031–1044. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystallographic Data | CaGre2-apo | CaGre2-NADPH Complex |
|---|---|---|
| Data Collection | ||
| Space group | I23 | I23 |
| a, b, c (Å) | 149.59, 149.59, 149.59 | 152.45, 152.45, 152.45 |
| α, β, γ (˚) | 90, 90, 90 | 90, 90, 90 |
| Resolution range (Å) | 50.0–2.8 (2.9−2.8)a | 50.0–3.02 (3.07−3.02) |
| No. of total reflections | 144,851 | 482,172 |
| No. of unique reflections | 13,918 | 11,745 |
| Completeness (%) | 100 (100) | 100 (100) |
| Multiplicity | 10.4 | 41.1 |
| I/σI | 43.8 (8.8) | 32 (1.9) |
| Rmerge (%)b | 11.4 (53.1) | 14.2 (186.8) |
| CC1/2f | 0.998 (0.949) | 0.999 (0.815) |
| Refinement | ||
| Resolution range (Å) | 35.0–2.8 | 50.0–3.02 |
| No. of reflections | 13,912 | 11,745 |
| Rworkc/Rfreed (%) | 17.2/21.1 | 19.8/26.2 |
| R.M.S. deviation | ||
| Bond lengths (Å) | 0.010 | 0.009 |
| Bond angles (o) | 1.034 | 1.034 |
| Average B-factor | ||
| Protein | 47.9 | 91.2 |
| Solvent | 39.2 | |
| Ligand | 136.3 | |
| Ramachandran plotg | ||
| Most favored (%) | 97.89 | 92.26 |
| Additional allowed (%) | 1.51 | 5.95 |
| PDB code | 6KWS | 6KWT |
| Protein Name | Species | Z-Score | r.m.s.d (Å) | Structural Features a,b | |||
|---|---|---|---|---|---|---|---|
| β7 | β3′ | α11 | α12 | ||||
| ScGre2 | S. cerevisiae | 47.2 | 1.3 | s | s A268–T271 | h | l |
| SsCR | S. stipitis | 45.3 | 1.4 | s | s R265–N267 | h | l |
| MtCAD2 | M. truncatula | 35.4 | 2.2 | s | s V257–H259 | h | l |
| SbCCR1 | S. bicolor | 33.1 | 2.5 | s | s R275–H278 | h | l |
| HsUGE | H. sapiens | 26.1 | 3.1 | s | s Y275–S276 | h | s Y294–V297 |
| EcUGE | E. coli | 26 | 3.0 | s | l | h | s Y286–A289 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, G.T.; Kim, S.; Jin, H.; Cho, D.-H.; Chun, H.-S.; Kim, W.-K.; Chang, J.H. Crystal Structure of NADPH-Dependent Methylglyoxal Reductase Gre2 from Candida Albicans. Crystals 2019, 9, 471. https://doi.org/10.3390/cryst9090471
Nguyen GT, Kim S, Jin H, Cho D-H, Chun H-S, Kim W-K, Chang JH. Crystal Structure of NADPH-Dependent Methylglyoxal Reductase Gre2 from Candida Albicans. Crystals. 2019; 9(9):471. https://doi.org/10.3390/cryst9090471
Chicago/Turabian StyleNguyen, Giang Thu, Shinae Kim, Hyeonseok Jin, Dong-Hyung Cho, Hang-Suk Chun, Woo-Keun Kim, and Jeong Ho Chang. 2019. "Crystal Structure of NADPH-Dependent Methylglyoxal Reductase Gre2 from Candida Albicans" Crystals 9, no. 9: 471. https://doi.org/10.3390/cryst9090471
APA StyleNguyen, G. T., Kim, S., Jin, H., Cho, D.-H., Chun, H.-S., Kim, W.-K., & Chang, J. H. (2019). Crystal Structure of NADPH-Dependent Methylglyoxal Reductase Gre2 from Candida Albicans. Crystals, 9(9), 471. https://doi.org/10.3390/cryst9090471