Abstract
Soluble fumarate reductase is essential for survival under anaerobic conditions. This enzyme can maintain the redox balance in the cell by catalyzing the reduction of fumarate to succinate. Although the overall reaction mechanism of soluble fumarate reductase in yeast, Osm1, has been proposed by a previous structural study, the details of the underlying mechanism are not completely elucidated. The present study provides the structural information regarding the active site mutant form of Osm1 (R326A), thus, revealing that R326A mutation does not affect the substrate binding. Structural alterations of the residues surrounding the active site, and the missing 2nd flavin adenine dinucleotide (FAD) in the previously defined 2nd FAD binding site, were observed as characteristic features of the Osm1 R326A crystal structure. Based on these findings, we provided a clue that can explain the loss of activity of Osm1 R326A.
1. Introduction
Fumarate reductase is an enzyme that can reduce fumarate to succinate using flavin adenine dinucleotide (FAD) as a cofactor [1,2]. This enzyme is particularly important for maintaining the redox balance in the cell during an oxygen deficient state, and is crucial in cell survival under anaerobic conditions [3]. Fumarate reductases can be divided into two different classes: The membrane-bound class, and the soluble class [2]. The clear differences between these two classes indicate that the membrane-bound form of fumarate reductase contains a covalently bound FAD cofactor, with a high redox potential, which catalyzes a reversible reaction. However, the soluble form contains noncovalently bound FAD, with a low redox potential, which catalyzes an irreversible reaction [4,5].
In yeast, two soluble forms of fumarate reductases, Osm1 and Frd1, were identified [6,7]. Genetic analysis revealed that both genes are upregulated under anaerobic conditions, and both fumarate reductases are crucial for the survival of yeast under anaerobic conditions [6,7,8,9]. Certain bacteria including Shewanella putrefaciens also contain soluble fumarate reductase (called the flavocytochrome c3 (Fcc3) family) [5,10,11]. This family of bacterial soluble fumarate reductase is different from the yeast soluble fumarate reductases (Osm1 and Frd1), wherein the Fcc3 family is organized into three distinct domains, the cytochrome domain, flavin domain, and clamp domain; whereas, Osm1 and Frd1 comprise two domains, the flavin domain and the clamp domain [11]. The reaction mechanism of Osm1 without the cytochrome domain, which is important for electron transfer, has been elucidated in the recent structural study of Osm1 [12]. This structural study indicated that Osm1 used a second FAD binding site, which is located at a similar position of the cytochrome site of bacterial soluble fumarate reductase, allowing for appropriate electron transfer. Yeast soluble fumarate reductase Osm1 is localized to both the mitochondria and the endoplasmic reticulum (ER) [6,13]. Two different forms, the ER-resident form and mitochondria-resident form, and the functional diversities of Osm1 at these two places, have been studied [12,13]. The structural and biochemical analysis suggested that Osm1 is an efficient redox regulator that can replenish the oxidized flavin molecules in both the ER and mitochondria [12].
Although the overall reaction mechanism of Osm1—which possesses two distinct FAD binding sites—has been reported in a previous a structural study, the details of the underlying mechanism remain unclear. Specifically, although the previous enzymatic studies showed that six residues around the active site, namely, H281, E301, R304, R326, H435, and R477, are crucial for the activity of Osm1, the functional role of each residue is not clearly elucidated. In this study, we report the crystal structure of the active site mutant form Osm1 (R326A). As dissimilar from the wild-type, two molecules are in the asymmetric unit of the crystal. Although Osm1 R326 is presumed to be efficient for substrate binding owing to the close location with its substrate, Osm1 R326A still contains fumarate in the substrate binding site, indicating that the loss of activity of the R326A mutant is not by substrate-binding defect. Based on the introduction of the R326A mutant on Osm1, the structural alteration of the surrounding residues was also detected. Interestingly, the Osm1 R326A structure does not contain the 2nd FAD at the 2nd FAD binding site, as defined in a previous study. Based on these findings, we suggested several possible models that can explain the loss of function of the R326A mutant of Osm1.
2. Materials and Methods
2.1. Site-Directed Mutagenesis
Site-directed mutagenesis was performed using the Quickchange kit (Stratagene, La Jolla, CA, USA), following the manufacturer’s protocols. The expression plasmid comprising the yeast OSM1 gene (corresponding to amino acids 32–501) was utilized here for the template. The mutagenesis of R326A was then confirmed by sequencing.
2.2. Protein Expression and Purification
The R326A mutant construct in the pET28a plasmid vector was transformed into BL21 (DE3) Escherichia coli competent cells, and their expression was then induced, by treating the bacteria with 0.25 mM isopropyl β-d-thiogalactopyranoside (IPTG) for 25 h at 18 °C. Cells were then harvested and sonicated in a resuspension buffer, containing 20 mM NaPO4, pH 7.4, 500 mM NaCl, 25 mM imidazole, and a protease inhibitor cocktail. Cell lysates were removed via centrifugation at 10,000× g for 1 h, and were filtered using a 0.22 µm filter. The target protein was purified by Ni-NTA affinity chromatography, followed by size-exclusion chromatography using a Superdex 200 gel filtration column 10/30 (GE Healthcare, Chicago, IL, USA) that had been pre-equilibrated with a solution of 20 mM Tris-HCl at pH 8.0 and 150 mM NaCl. The protein that eluted approximately 15–16 mL via size-exclusion chromatography was then collected, and concentrated to 7–8 mg/mL. The concentrated protein was treated with thrombin for 8 h. A digested sample was reapplied onto the Ni-NTA affinity column to remove the digested N-terminal part of the target protein and the undigested protein sample. The digested and unbound sample was collected, concentrated, and reapplied onto size-exclusion chromatography. The eluted peak fraction containing the target protein was pooled and concentrated to 7–8 mg/mL for crystallization.
2.3. Multi-Angle Light Scattering (MALS)
The absolute molar mass of the Osm1 R326A mutant was determined by MALS (Wyatt Technology, Santa Barbara, CA, USA). Briefly, the target protein was loaded onto a Superdex 200 HR 10/30 gel-filtration column (GE Healthcare, Chicago, IL, USA) that had been pre-equilibrated in the buffer containing 20 mM Tris-HCl, at pH 8.0, and 150 mM NaCl. The Acta chromatography system (GE Healthcare, Chicago, IL, USA) was coupled to a MALS detector (mini-DAWN Treos) and a refractive index detector (Optilab DSP; both Wyatt Technology).
2.4. Structure Determination and Analysis
Native diffraction data sets were collected on the beamline SBII-5C of the Pohang Accelerator Laboratory (PAL), Republic of Korea, at a wavelength of 0.9776 Å. The data set was indexed, and processed using HKL-2000. The structure was determined by the molecular replacement phasing method using Phaser [14]. The previously obtained wild-type of Osm1 structure (PDB code: 5ZYN) was used as a search model. Model building and refinement were conducted by COOT [15] and Refmac5 [16], respectively. The geometry was inspected using MolProbity [17], and was found to be acceptable. A total of 97.12% of the amino acids were located in the most favorable region, whereas 2.88% were in the allowed regions of the Ramachandran plot. All molecular figures were generated using the Pymol program [18].
2.5. Protein Data Bank Accession Code
Coordinates and structural factors were deposited in the Protein Data Bank under the PDB ID code 6KU6.
3. Results
3.1. Overall Structure of the R326A Mutant Form of Osm1
Unlike bacterial soluble fumarate reductase that contains three functional domains, the eukaryotic soluble fumarate reductase in yeast (Osm1) is organized into two distinct domains, the flavin domain, which contains the noncovalently bound FAD as a cofactor, and the clamp domain, which is believed to be involved in controlling substrate access to the active site (Figure 1a). Previous enzymatic studies revealed that six residues around the active site, namely, H281, E301, R304, R326, H435, and R477, are crucial for the activity of Osm1 [12]. To understand the role of R326 residue on the function of Osm1, and the effect on the R326A mutation on Osm1, we characterized, solved the structure of Osm1 R326A, and compared it with those of the wild-type Osm1.
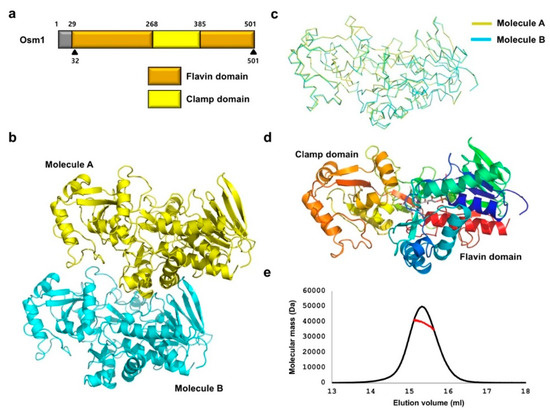
Figure 1.
Crystal structure of Osm1 R326A: (a) Domain boundary of Osm1; the range of amino acid residues that were used for the structural study is indicated by a black triangle; (b) crystal structure of Osm1 R326A; two molecules in an asymmetric unit are presented with the cartoon model; (c) structural superposition of the two molecules in the same asymmetric unit; (d) the structure of Osm1 R326A depicted with rainbow color; the structure from the N- to C-terminus is colored from blue to red; two distinct domains are indicated; (e) MALS profile; experimentally calculated molecular mass is indicated by a red line.
To obtain a soluble and crystallizable mutant protein, the FAD cofactor and fumarate substrate were added during the purification step, and limited proteolysis with trypsin was performed. As similar to the wild-type crystallization, only trypsin-resistant Osm1 was successfully crystallized. The crystal of Osm1 R326A was produced with a different space group (P2) from wild-type (P21). The structure was obtained by the molecular replacement phasing method, using the wild-type structure as a search model. The structure was refined to a Rwork = 20.0% and Rfree = 25.1%. The data collection and refinement statistics are summarized in Table 1. The asymmetric unit was comprised of two molecules, with the final model encompassing residues 32–501 for both molecules (Figure 1b). The structure of the two molecules in the asymmetric unit are nearly identical, having a 0.523 Å root mean square deviation (RMSD) and a 0.923 Template Modeling (TM)-Score (Figure 1c). The structure of Osm1 R326A was composed of two functional domains, a flavin domain (residues 32–268 and 386–501), and a clamp domain (residues 269–385) (Figure 1d). As two molecules are in the asymmetric unit and a previous study showed that soluble fumarate reductase in solution can exist as a dimer and highly oligomerized form [19], although the wild-type Osm1 was in monomeric form in the solution, we confirmed the stoichiometry of Osm1 R326A in solution using size-exclusion chromatography (SEC)-multiangle light scattering (MALS). The theoretically calculated molecular weight of monomeric Osm1 R326A without any tag is 51.4 kDa, and the experimental molecular weight from MALS was 41.9 kDa (3.3% fitting error), with a polydispersity of 1.0 (Figure 1e). Based on our analysis using SEC-MALS, we conclude that the R326A mutation did not affect the oligomeric state of Osm1.

Table 1.
Crystallographic statistics.
3.2. Comparison of the Structure of Osm1 R326A with the Structure of Wild-Type Osm1
The first analysis with the mutant structure was to identify the mutation site, R326, in the structure. The electron density of the arginine side chain was not detected, indicating that the R326 residue is mutated by alanine (Figure 2a). As the R326 residue is located in the active site, and seems to be crucial for substrate binding, we assessed whether the substrate and FAD cofactor are still located in the structure of the Osm1 R326A mutant. Based on the electron density, we noticed that fumarate and FAD are present in the structure of Osm1 R326A, indicating that the R326 residue is involved, however not critical, for the substrate binding (Figure 2b).
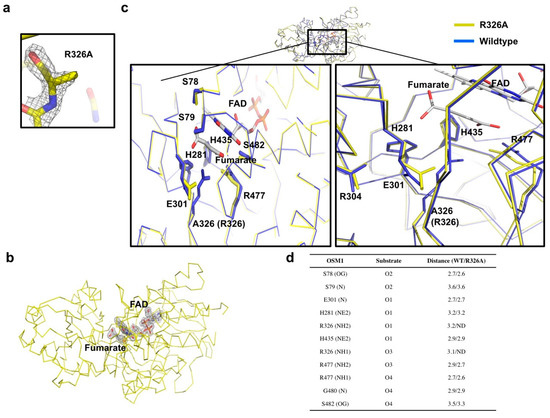
Figure 2.
Active site of Osm1 R326A: (a) Omit density map contoured at the 1-σ level around the mutated residue, R326A; (b) omit density map contoured at the 1-σ level around the active site of Osm1 R326A; (c) details of the active site structure; the active site is presented by a black rectangle; the magnified structures of the R326A region, the FAD, and the substrate binding site are depicted at the left and right panels, respectively; (d) details of the interaction between the substrate and surrounding residues.
Structural comparison between the wild-type and mutant with superimposition analysis indicated that the structures are nearly identical, thus, exhibiting an RMSD of 0.523 Å, and a TM-score of 0.923 (Figure 2c). Furthermore, structural analysis of the substrate binding site and the active site revealed that the side chains of the E301 and R477 residues are altered by R326A mutagenesis (Figure 2c). Without the long side chain of R326 by mutagenesis, both side chains of E301 and R477 moved toward the substrate, indicating that the exact position of residues that are crucial for the electron transfer is disrupted by R326A mutagenesis (Figure 2c,d).
3.3. Undetectable 2nd FAD on Osm1 R326A at the Previously Defined 2nd FAD Binding Site
Interestingly, the structure of Osm1 R326A does not contain the 2nd FAD at the 2nd FAD binding site, although the FAD was added during the purification steps. The mutation of R326A may not contribute to the loss of the 2nd FAD, owing to the large distance between R326 or A326 and the 2nd FAD (Figure 3a). An indirect mutation effect, which disrupts the 2nd FAD binding pocket by mutation, can inhibit the binding of the 2nd FAD. To analyze the putative disrupted 2nd FAD binding pocket by R326A mutation, we superposed both structures, and analyzed the residues that are involved in the formation of the 2nd FAD binding pocket. A previous structural study showed that hydrophobic pocket formed by six amino acid residues, namely, W295, F297, L298, N359, F362, and Y363, from the clamp domain, and three amino acid residues, namely, K76, S78, and P162 from the flavin domain, is mainly involved in the interaction with the isoalloxazine ring of the 2nd FAD [12]. Based on the observation of the 2nd FAD binding pocket with structural superposition, we noticed that the structure of the majority of the residues in the 2nd FAD binding pocket remained unchanged by R326A mutation (Figure 3b), indicating that the loss of the 2nd FAD might not be due to R326A mutation.
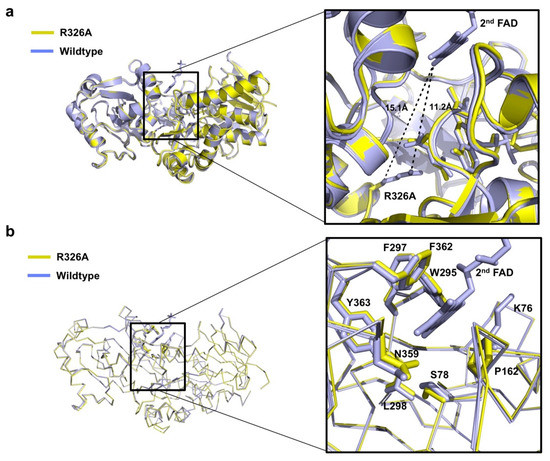
Figure 3.
Details of the 2nd FAD binding site of Osm1 R326A: (a) Position of R326A and 2nd FAD with distances indicated; (b) structural comparison of the 2nd FAD binding site of Osm1 R326A with that of the wild-type.
Because the 2nd FAD is not detected in the previously identified 2nd FAD binding site of the mutant, we investigated the 2nd FAD binding cavity to find the clue that can explain the loss of the 2nd FAD by mutation. This investigation revealed that there are five water molecules in the 2nd FAD binding site in the mutant (Figure 4a). Based on the B-factor comparison, we realized that the B-factor of the 2nd FAD binding site of Osm1R326A is much higher than that of the wild-type (Figure 4b). These findings indicate that the 2nd FAD is lost in the structure of Osm1R326A, and is replaced by five waters. Without the 2nd FAD, although the B-factor is increased, the overall structure of 2nd FAD binding site remains intact.
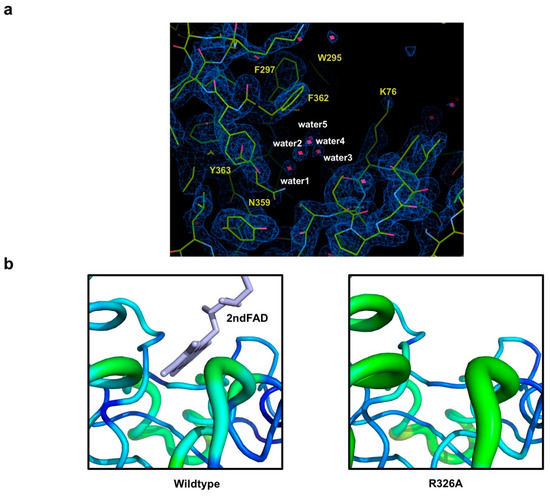
Figure 4.
Analysis of the feature of the 2nd FAD binding site in Osm1 R326A: (a) An omit density map contoured at the 1-σ level around the 2nd FAD binding site in Osm1 R326A; red star marks indicate water molecules; five waters in the 2nd FAD binding cavity are numbered; (b) B-factor distribution of the 2nd FAD binding site; the structure is presented in a putty representation and rainbow-colored from red to violet in B-factor value order.
4. Discussion
It has been reported that soluble fumarate reductase is particularly important for survival under anaerobic conditions. In the case of Caenorhabditis elegans, putative soluble fumarate reductase was involved in the aging process [20,21,22]. Despite the crucial roles of soluble fumarate reductases for survival of the organism, the molecular reaction mechanism of eukaryotic soluble fumarate reductase has not been fully elucidated. Recent structural and functional studies of Osm1, a soluble fumarate reductase form of yeast, showed that Osm1 comprises two FAD cofactors for appropriate electron transfer [12,23]. With two FAD binding sites, Osm1 was able to oxidize environments under anaerobic conditions, and suggested Osm1 as a master redox regulator in the cell [12]. Although these previous studies indicated that six residues in the active site, namely, H281, E301, R304, R326, H435, and R477, are crucial for the reductase activity of Osm1, the contribution of each residue in the activity of Osm1 remains unclear.
In this study, we proposed a tentative role of the function of the R326 residue of Osm1, based on the structure of the Osm1 R326A mutant form. Although the crystal of the Osm1 R326A mutant form was different from that of the wild-type, possessing a distinct unit cell parameter with two molecules in the asymmetric unit, SEC-MALS analysis revealed that Osm1 R326A was a monomer in the solution, indicating that R326A mutagenesis did not alter the stoichiometry of Osm1. Structural analysis of Osm1 R326A revealed that the mutant form still has a substrate in the active site, indicating that R326 residue is not crucial for substrate accommodation, although it is involved in substrate interaction, as it is located approximately 3.1 Å away from substrate binding site. Structural comparison of Osm1 R326A with the wild-type structure revealed that the side chains of the E301 and R477 residues, which are located at the active site and known to be crucial for the activity of Osm1, are slightly dislocated from the typical location detected from the wild-type structure by mutagenesis. This indicates that the loss of activity of the R326A mutant is due to both the direct role of R326 on the electron transfer, and the indirect role on the disrupted structure of the side chains of E301 and R477, which might be crucial for the Osm1 activity.
Interestingly, the structure of Osm1 R326A does not contain the 2nd FAD at the 2nd FAD binding site, which is formed between the flavin and clamp domains. As the binding of the 2nd FAD is necessary for the Osm1 activity, the loss of the 2nd FAD by R326A mutation might be another reason for the Osm1 R326A mutant to lose its activity. It is difficult to infer that the loss of the 2nd FAD is due to the R326A mutation, because of the large distance between the R326 residue and the 2nd FAD binding site, and also because the structure of the 2nd FAD binding site remained unaltered by mutagenesis, as judged by the structure of Osm1 R326A. The relationship between the R326A mutation effect and the loss of the 2nd FAD needs to be further studied, in order to fully understand the role of the R326 residue in the Osm1 function.
Author Contributions
H.H.P. designed and supervised the project; C.M.K., H.L.C., and K.H.J. performed cloning, expression, and protein purification. C.M.K. and S.K. crystallized and collected the X-ray data; C.M.K. and S.K. solved the protein structure; K.H.J. and H.J.H. performed MALS; H.H.P., C.M.K., and S.K. wrote the manuscript. All the authors discussed the results, commented on the manuscript, and approved the manuscript.
Funding
This study was supported by the Chung-Ang University Research Scholarship Grants in 2018, and a grant from the Korea Healthcare Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI17C0155).
Acknowledgments
The authors thank the park’s laboratory member for the helpful discussion.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Iverson, T.M.; Luna-Chavez, C.; Cecchini, G.; Rees, D.C. Structure of the escherichia coli fumarate reductase respiratory complex. Science 1999, 284, 1961–1966. [Google Scholar] [CrossRef] [PubMed]
- Reid, G.A.; Miles, C.S.; Moysey, R.K.; Pankhurst, K.L.; Chapman, S.K. Catalysis in fumarate reductase. Biochim. Biophys. Acta 2000, 1459, 310–315. [Google Scholar] [CrossRef]
- Tielens, A.G.; Van Hellemond, J.J. The electron transport chain in anaerobically functioning eukaryotes. Biochim. Biophys. Acta 1998, 1365, 71–78. [Google Scholar] [CrossRef]
- Lancaster, C.R.; Kroger, A.; Auer, M.; Michel, H. Structure of fumarate reductase from Wolinella succinogenes at 2.2 Å resolution. Nature 1999, 402, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Leys, D.; Tsapin, A.S.; Nealson, K.H.; Meyer, T.E.; Cusanovich, M.A.; Van Beeumen, J.J. Structure and mechanism of the flavocytochrome c fumarate reductase of Shewanella putrefaciens MR-1. Nat. Struct. Biol. 1999, 6, 1113–1117. [Google Scholar]
- Arikawa, Y.; Enomoto, K.; Muratsubaki, H.; Okazaki, M. Soluble fumarate reductase isoenzymes from Saccharomyces cerevisiae are required for anaerobic growth. FEMS Microbiol. Lett. 1998, 165, 111–116. [Google Scholar] [CrossRef][Green Version]
- Enomoto, K.; Ohki, R.; Muratsubaki, H. Cloning and sequencing of the gene encoding the soluble fumarate reductase from Saccharomyces cerevisiae. DNA Res. 1996, 3, 263–267. [Google Scholar] [CrossRef]
- Muratsubaki, H.; Enomoto, K. One of the fumarate reductase isoenzymes from Saccharomyces cerevisiae is encoded by the OSM1 gene. Arch. Biochem. Biophys. 1998, 352, 175–181. [Google Scholar] [CrossRef]
- Camarasa, C.; Faucet, V.; Dequin, S. Role in anaerobiosis of the isoenzymes for saccharomyces cerevisiae fumarate reductase encoded by OSM1 and FRDS1. Yeast 2007, 24, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Doherty, M.K.; Pealing, S.L.; Miles, C.S.; Moysey, R.; Taylor, P.; Walkinshaw, M.D.; Reid, G.A.; Chapman, S.K. Identification of the active site acid/base catalyst in a bacterial fumarate reductase: A kinetic and crystallographic study. Biochemistry 2000, 39, 10695–10701. [Google Scholar] [CrossRef]
- Taylor, P.; Pealing, S.L.; Reid, G.A.; Chapman, S.K.; Walkinshaw, M.D. Structural and mechanistic mapping of a unique fumarate reductase. Nat. Struct. Biol. 1999, 6, 1108–1112. [Google Scholar] [PubMed]
- Kim, S.; Kim, C.M.; Son, Y.J.; Choi, J.Y.; Siegenthaler, R.K.; Lee, Y.; Jang, T.H.; Song, J.; Kang, H.; Kaiser, C.A.; et al. Molecular basis of maintaining an oxidizing environment under anaerobiosis by soluble fumarate reductase. Nat. Commun. 2018, 9, 4867. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.C.; Jan, C.H.; Weissman, J.S. Targeting and plasticity of mitochondrial proteins revealed by proximity-specific ribosome profiling. Science 2014, 346, 748–751. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J. Solving structures of protein complexes by molecular replacement with phaser. Acta Cryst. D Biol. Cryst. 2007, 63, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-Building tools for molecular graphics. Acta Cryst. D Biol. Cryst. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Vagin, A.A.; Steiner, R.A.; Lebedev, A.A.; Potterton, L.; McNicholas, S.; Long, F.; Murshudov, G.N. REFMAC5 dictionary: Organization of prior chemical knowledge and guidelines for its use. Acta Cryst. D Biol. Cryst. 2004, 60, 2184–2195. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-Atom structure validation for macromolecular crystallography. Acta Cryst. D Biol. Cryst. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Delano, W.L. The PyMol Molecular Graphics System. 2002. Available online: http://pymol.sourceforge.net/overview/index.htm (accessed on 27 September 2019).
- Bamford, V.; Dobbin, P.S.; Richardson, D.J.; Hemmings, A.M. Open conformation of a flavocytochrome c3 fumarate reductase. Nat. Struct. Biol. 1999, 6, 1104–1107. [Google Scholar]
- Edwards, C.B.; Copes, N.; Brito, A.G.; Canfield, J.; Bradshaw, P.C. Malate and fumarate extend lifespan in caenorhabditis elegans. PLoS ONE 2013, 8, e58345. [Google Scholar] [CrossRef]
- Butler, J.A.; Ventura, N.; Johnson, T.E.; Rea, S.L. Long-Lived mitochondrial (Mit) mutants of caenorhabditis elegans utilize a novel metabolism. FASEB J. 2010, 24, 4977–4988. [Google Scholar] [CrossRef]
- Rea, S.; Johnson, T.E. A metabolic model for life span determination in Caenorhabditis elegans. Dev. Cell 2003, 5, 197–203. [Google Scholar] [CrossRef]
- Enomoto, K.; Arikawa, Y.; Muratsubaki, H. Physiological role of soluble fumarate reductase in redox balancing during anaerobiosis in Saccharomyces cerevisiae. FEMS Microbiol. Lett. 2002, 215, 103–108. [Google Scholar] [CrossRef] [PubMed][Green Version]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).