Analysis of Glulisine Crystallisation Utilising Phase Diagrams and Nucleants
by
 , , , and
, , , and
Yanmin Li
1,2 ,
,
Lata Govada
1,
Hodaya V. Solomon
1,
Richard B. Gillis
3,
Gary G. Adams
3 and
Naomi E. Chayen
1,* 1
Biomolecular Medicine, Faculty of Medicine, Imperial College London, London SW7 2AZ, UK
2
Barts and The London School of Medicine and Dentistry, Queen Mary University of London, London E1 4NS, UK
3
Queen’s Medical Centre, Faculty of Medicine and Health Sciences, University of Nottingham, Nottingham NG7 2HA, UK
*
Author to whom correspondence should be addressed.
Crystals 2019, 9(9), 462; https://doi.org/10.3390/cryst9090462
Submission received: 1 August 2019
/
Revised: 29 August 2019
/
Accepted: 1 September 2019
/
Published: 3 September 2019
(This article belongs to the Special Issue Advanced Methods to Monitor and Control the Crystallisation Environment)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Glulisine is a US Food and Drug Administration (FDA) approved insulin analogue, used for controlling hyperglycaemia in patients with diabetes mellitus (DM). It is fast acting which better approximates physiological insulin secretion, improving patient outcome. Crystallisation of Glulisine was analysed by its crystallisation phase diagram and nucleation-inducing materials. Both the hanging drop vapour diffusion and microbatch-under-oil methods were used and compared. We have shown that the same protein can have different solubility behaviours depending on the nature of the salt in the precipitating agent. In the case of Glulisine with magnesium formate, lowering the precipitant concentration drove the system further into supersaturation resulting in the formation of crystals and precipitation. This was the opposite effect to the usual scenario where raising the precipitant concentration leads to supersaturation. Glulisine with sodium potassium tartrate tetrahydrate (NaKT) followed the expected trend of forming crystals or precipitate at higher concentrations and clear drops at lower concentrations of the precipitant. The outcomes of crystallisation using the different crystallisation methods is also described. Glulisine was successfully crystallised and the crystals diffracted up to a resolution limit of 1.4 Å.
Keywords:
proteins; macromolecules; crystals; crystallization; nucleation; phase diagram; insulin; Glulisine1. Introduction
High-resolution X-ray diffraction patterns require high-quality crystals and obtaining such crystals is often the bottleneck of the structural determination process [1]. Advances in automation and high throughput screening can increase the number of hits obtained but it does not address the core problem of reliance on trial and error [2]. For a more systematic approach to crystallisation, we must consider the principles underlining crystal formation. As the aqueous solution containing the protein reaches supersaturation, protein molecules will come out of the solution and under specific conditions form crystals. A variety of parameters such as protein concentration, crystallising agent or precipitant and additives can be adjusted to encourage the formation of crystals. A phase diagram can be experimentally generated and quantify the effect of such modifications. The phase diagram is a 2D representation of the distinct phases that occur and coexist at equilibrium [3].
1.1. The Application of the Phase Diagram and Nucleants
The zones of undersaturation and supersaturation are the main areas of the phase diagram, which is separated by the solubility curve. The supersaturation zone can be further divided into metastable, nucleation and precipitation zones depending on the levels of supersaturation. No crystals will form at conditions where the solution is undersaturated. By increasing the concentration of protein or another adjustable parameter, we can push the system towards crystallisation. The metastable zone is where crystals can grow but no nucleation will take place on the timescale of the experiment. The nucleation zone, as the name suggests is where nuclei form. Crystals can only grow from a nucleus so it is important to encourage nucleation but at the same time prevent excessive nucleation where a shower of microcrystals will form as a result [4]. The metastable and nucleation zone is separated by the supersolubility curve. For practical purposes, it is sufficient to experimentally determine the supersolubility curve as determining the solubility curve requires larger amounts of materials and increases the length of the experimental time by weeks or even months [5,6]. A diagram containing only the supersolubility curve is called a working phase diagram [6,7]. At even higher conditions, precipitates will form due to high supersaturation [6]. The trajectory to supersaturation of a crystallisation drop can have drastic effects on the outcome. It is advisable, therefore, to carry out crystallisation experiments with different methods.
Once metastable conditions have been identified with the aid of the phase diagram, the nucleation zone can be completely bypassed. If left undisturbed, no nucleus will form at metastable conditions, and the crystallisation drop will remain clear indefinitely. However, certain materials known as nucleants have the property to induce nucleation at metastable conditions as they provide more energetically favourable conditions for heterogeneous nucleation [8]. Porous materials have generated much interest as potential nucleants [9]. The cavities on porous materials have the ability to attract and entrap protein molecules [10]. A crystal can start growing in the pore after a critical nucleus is formed [11]. A silicate bioglass material (Naomi’s nucleant) has a highly porous surface with cavities of similar sizes to typical proteins (2–10 nm) [12]. The number of proteins crystallised using Naomi’s nucleant is continuously rising [1,13] and to date, it has assisted in the crystallisation of over 20 proteins. Another nucleating material currently being investigated is nanoporous gold foil containing 5–10 nm pores [14].
1.2. Glulisine
Glulisine/Apidra is a Food and Drug Administration (FDA) approved fast-acting insulin analogue, used for controlling hyperglycaemia in patients with diabetes mellitus. The aim of insulin therapy is to mimic physiological insulin secretion; this is better achieved by the use of modified insulin analogues [15]. The combination of fast and slow-acting insulin analogues can provide effective coverage to prevent the glucose spike immediately after meals and maintains the glucose concentration at a homeostatic level. In Glulisine, the B chain residues 3 and 29 were changed from asparagine to lysine and lysine to glutamic acid, respectively [16]. When administered, Glulisine is absorbed faster into the bloodstream than regular human insulin and better approximates microvascular flow and endothelial function [17,18]. To date, there is no published Glulisine structure other than the Protein Data Bank (PDB) deposition at 1.26 Å (PDB ID 6GV0) [19].
The aim of this work was to obtain diffracting crystals and to conduct a systematic study on the crystallisation of Glulisine by applying phase diagrams and controlling nucleation.
2. Materials and Methods
2.1. Protein and Precipitants
Pharmaceutical grade Glulisine/Apidra (Sanofi S.A., Paris, France) (5F337A) was obtained in solution at 3.49 mg/mL. Initial crystallisation screening trials were set up using the Index screen (Hampton Research, Aliso Viejo, CA, USA). Optimisation was performed with two promising screening conditions, one consisting of 0.1 M 2,2-Bis(hydroxymethyl)-2,2′,2″-nitrilotriethanol (BIS-TRIS) pH 5.5–6.2 and 0.1–0.9 M magnesium formate and the other of 0.1–0.8 M sodium potassium tartrate tetrahydrate, 0.1 M Tris pH 8.5 and 0.5% w/v polyethylene glycol monomethyl ether 5000 (Sigma Aldrich, St. Louis, MO, USA). The crystallisation solutions containing magnesium formate at pH 5.5 or sodium potassium tartrate tetrahydrate (NaKT) at pH 8.5 were used in all the experiments following screening. The concentration of protein was changed by adjusting the ratio of Glulisine and precipitant in the crystallisation drop.
All experiments were set up in duplicate drops. All incubations were carried out at 20 °C and observed daily for at least four weeks. Crystallisation drops were observed with a Leica M165 C microscope and images were captured with the Leica DFC295 camera and processed with Leica Application Suite software.
2.2. Crystallisation Methods
Hanging drop vapour diffusion experiments were set up using the EasyXtal plates with X-Seal crystallisation supports (Qiagen, Hilden, Germany). The reservoir contained 300 µL of the precipitant solution and 1 µL each of the protein and precipitant solution were mixed on the X-Seal supports. The X-Seal supports were then inverted and screwed above the precipitant reservoir creating an airtight seal. By changing the protein:precipitant ratio (1:2, 1:1.5, 1:1, 1.5:1 and 2:1), 1.16, 1.40, 1.75, 2.09 and 2.33 mg/mL working concentrations of Glulisine was achieved.
Microbatch experiments [20] were carried out in Nunc HLA Terasaki 72 well plates (Douglas Instruments, East Garston, UK). The plates were cleaned with compressed air and then filled with 100% paraffin oil. Protein and precipitant drops were dispensed under the paraffin oil at the ratios above.
2.3. Nucleants
The following nucleating agents were added to the crystallisation drop to encourage nucleation and the subsequent growth of crystals. Naomi’s nucleant was available in our laboratory. One grain was inserted into each crystallisation drop with fine-tip forceps. Nanoporous gold (provided by Prof. J. Erlerbacher, Johns Hopkins University, Baltimore, MD, USA) was also inserted one piece per drop.
2.4. Crystal Harvesting and Data Collection
Appropriately sized nylon loops (Hampton Research, Aliso Viejo, CA, USA) were used to harvest crystals from the drop. A cryoprotectant composed of 20% glycerol and 80% precipitant was used. Crystals were then flash-frozen and stored in liquid nitrogen. X-ray diffraction was performed remotely at Imperial College using the I04-1 beamline at the Diamond Light Source, Oxford.
3. Results
3.1. Glulisine and Magnesium Formate
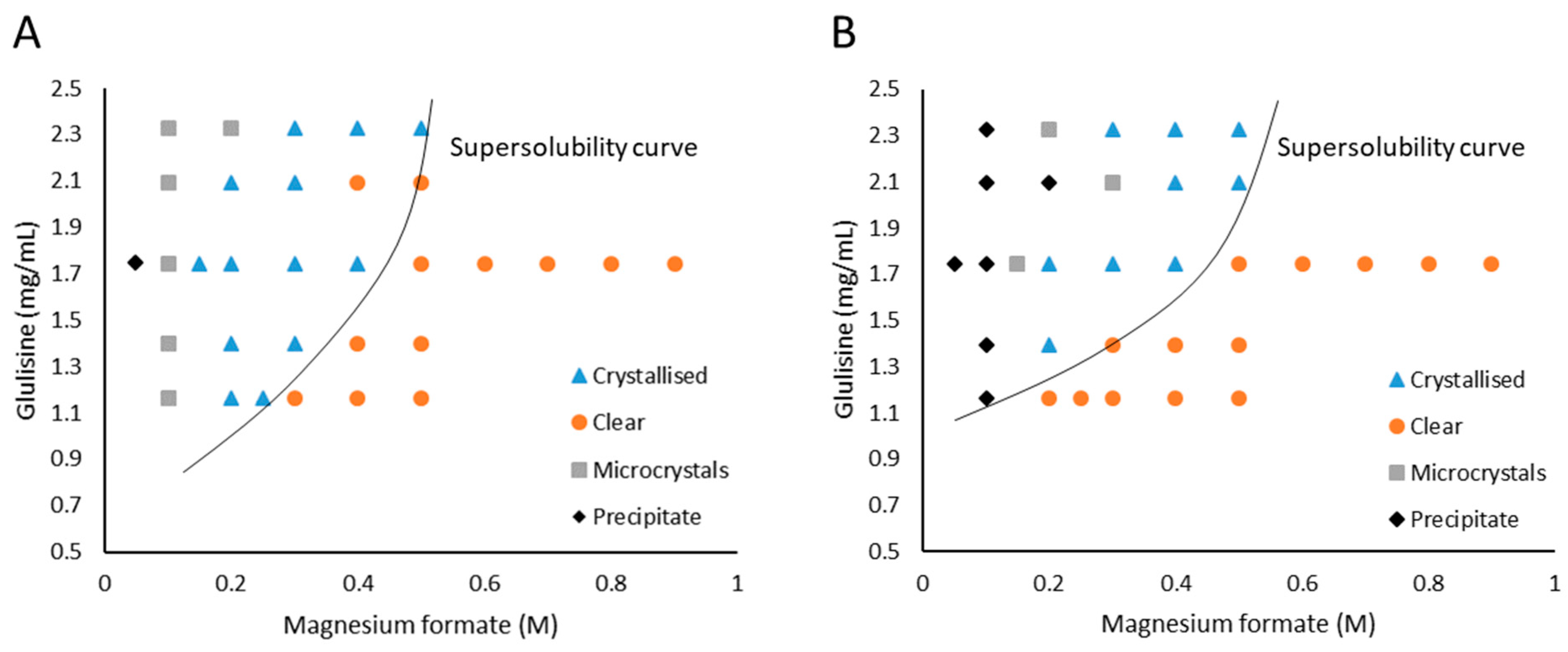
Working phase diagrams for Glulisine with magnesium formate as the precipitant were constructed for both the vapour diffusion and microbatch methods (Figure 1) by varying the concentration of Glulisine (1.16–2.33 mg/mL) and magnesium formate in the precipitant solution (0.05–0.9 M). The solubility of magnesium formate is 1 M in water, hence no trials were set up beyond the concentration of 0.9 M.
At 1.75 mg/mL Glulisine crystal formation was observed within 16 hours at 0.2 M magnesium formate concentrations in both vapour diffusion and microbatch. At 0.3 M and 0.4 M magnesium formate crystals formed within seven days in microbatch and over two weeks in vapour diffusion. At concentrations below 0.2 M magnesium formate, Glulisine precipitated out of solution while at concentrations above 0.6 M magnesium formate all drops remained clear for four weeks. Trials with 0.8 and 0.9 M magnesium formate remained clear for over 10 weeks.
At 2.33 mg/mL Glulisine, microcrystals and precipitate formed within 24 hours at 0.1 and 0.2 M magnesium formate concentrations in both vapour diffusion and microbatch. At 2.33 mg/mL Glulisine and magnesium formate concentrations above 0.3 M, crystals formed within two weeks in vapour diffusion and within seven days in microbatch.
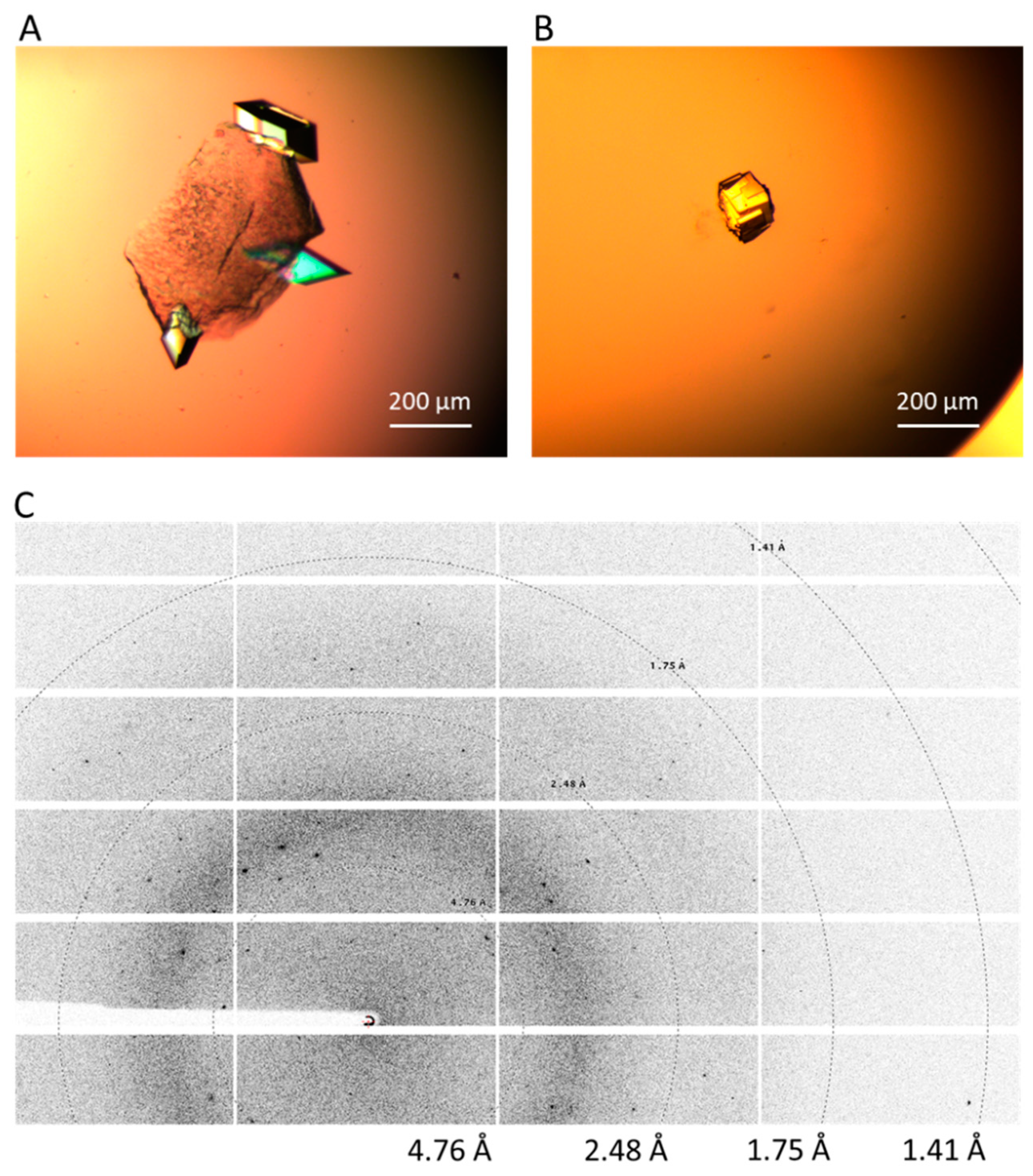
Having identified metastable regions, Naomi’s nucleant and nanoporous gold were inserted into the crystallisation drops at conditions just below the supersolubility curve. At 1.16 mg/mL Glulisine and 0.3 M magnesium formate, Naomi’s nucleant produced crystals (200 × 100 µm) on day seven in vapour diffusion (Figure 2A). The drops at 1.16 mg/mL Glulisine and 0.2 M magnesium formate (these were supersaturated conditions where crystals form spontaneously) produced crystals (100 × 100 µm) without the addition of nucleants on day one in vapour diffusion, however, they were stacked and not single (Figure 2C) whilst the crystals obtained with Naomi’s nucleant were single and diffracted to 1.4 Å (Figure 2B). Naomi’s nucleant did not produce crystals in microbatch under the same conditions and nanoporous gold was ineffective in both methods. Control drops at 1.16 mg/mL Glulisine and 0.3 M magnesium formate without the addition of nucleants remained clear over the four-week incubation period.
3.2. Glulisine and Sodium Potassium Tartrate Tetrahydrate (NaKT)
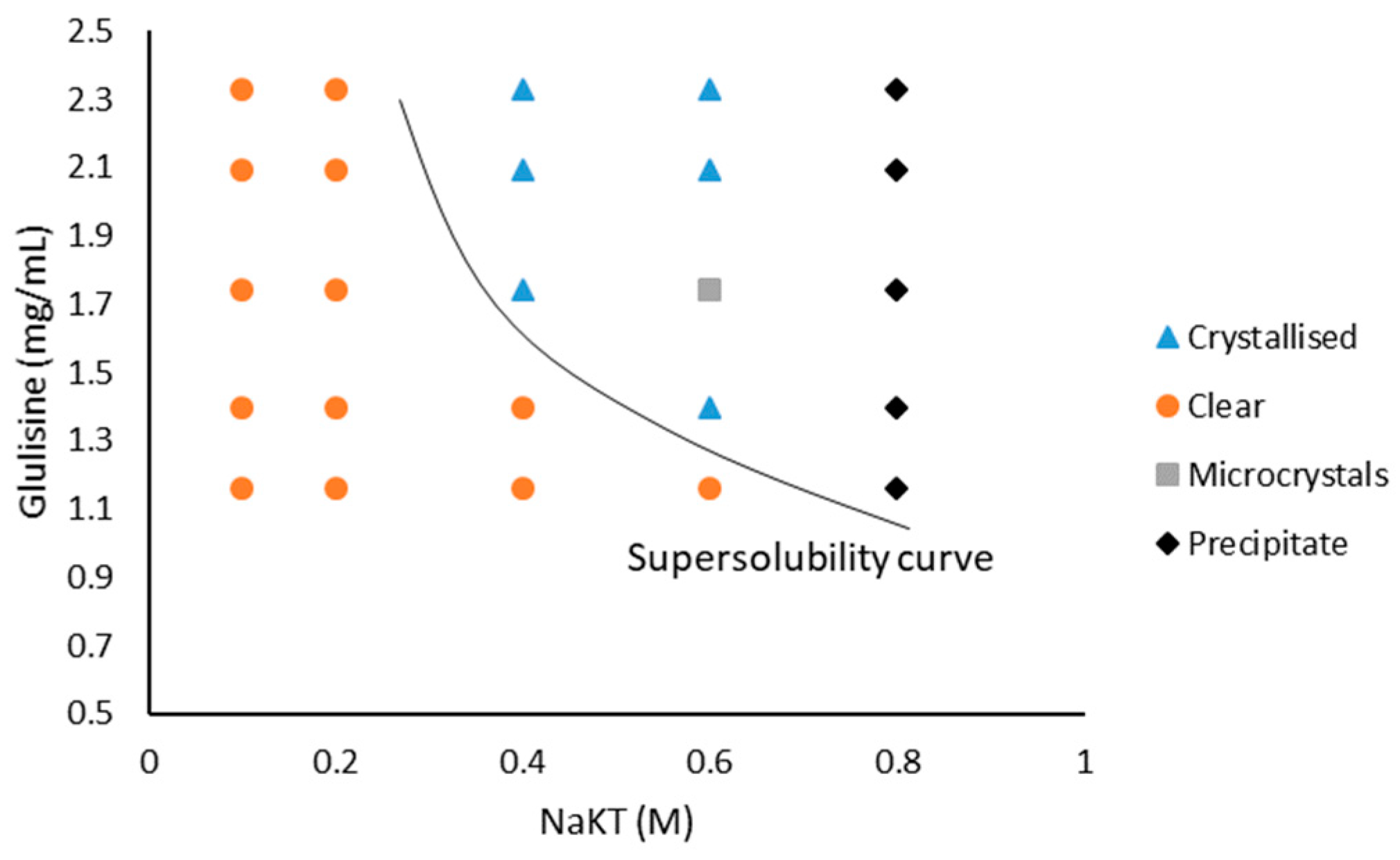
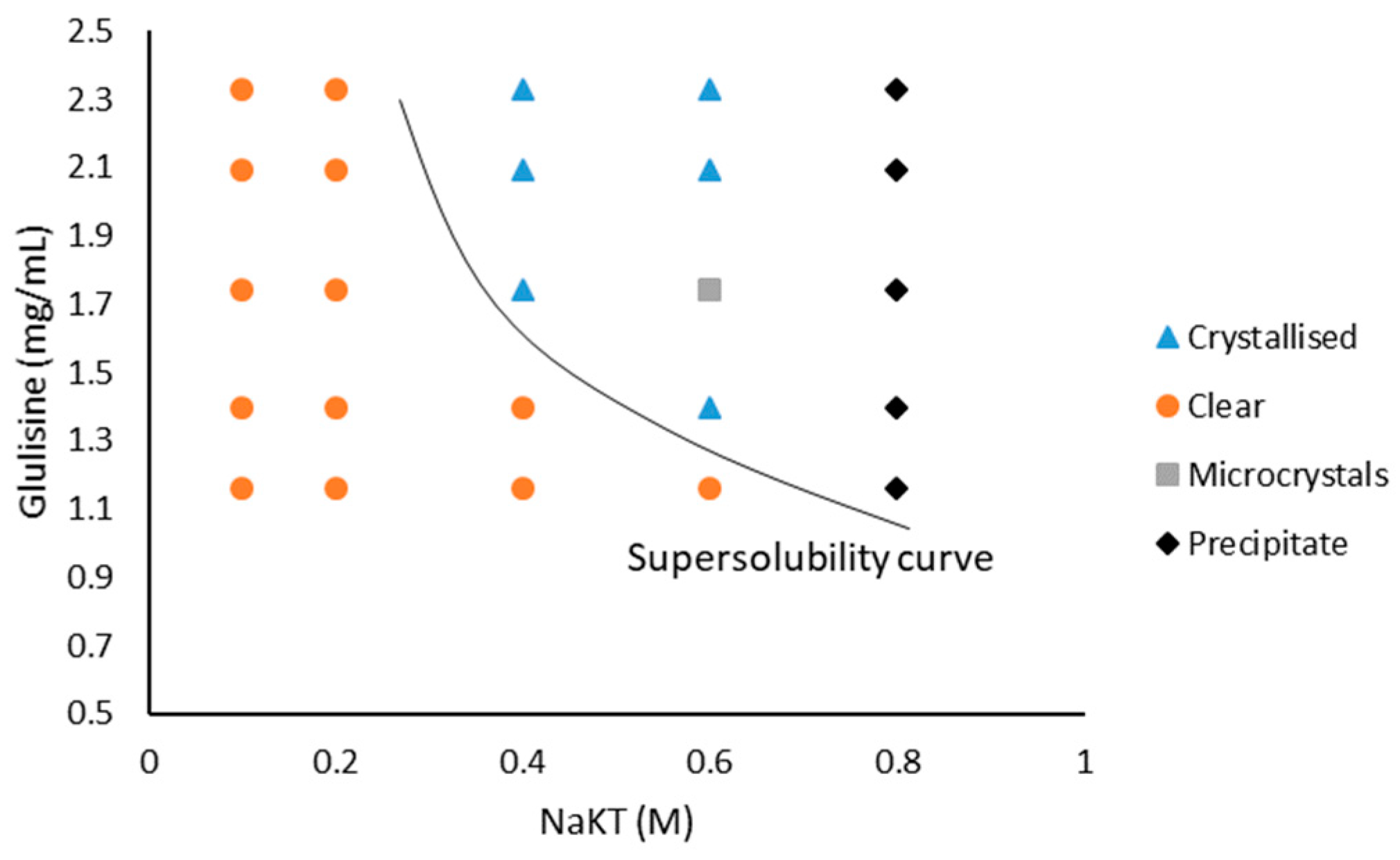
A working phase diagram was generated by varying the concentration of Glulisine (1.16–2.33 mg/mL) and NaKT (0.1–0.8 M) in the precipitant solution (Figure 3). Microbatch was ineffective at crystallising Glulisine with NaKT as precipitant. At concentrations below 0.2 M NaKT, drops remained clear for four weeks for all Glulisine concentrations. At NaKT concentrations above 0.4 M, precipitates and microcrystals formed overnight, and larger crystals (100 × 100 µm) were produced three weeks after set-up. Some crystals took over 40 days to form. Crystals grown with 0.4 M NaKT and 1.75 mg/mL Glulisine in vapour diffusion were large and single (100 × 100 µm) compared to the crystals grown in the microbatch. They were X-rayed and the diffraction at 2.3 Å was inferior to Glulisine crystals grown in magnesium formate. The crystals obtained at 0.4 M NaKT in microbatch were not large enough for diffraction.
Having identified metastable regions, Naomi’s nucleant and nanoporous gold were inserted into the crystallisation drops at 0.2 M NaKT but no crystals were obtained under these conditions.
4. Discussion
Macromolecular crystallisation still presents a major challenge for structural determination projects. The unpredictability and almost randomness of crystal formation have prevented the success of many projects [1]. Over the years, a substantial amount of research has been directed to better understand the crystallisation process and to encourage the formation of crystals.
4.1. The Solubility of Glulisine
The phase diagram provides a systematic approach to optimise promising results from the screening process. In the case of Glulisine with magnesium formate, lowering the precipitant concentration drove the system further into supersaturation resulting in the formation of crystals and precipitation. This was the opposite effect to the usual scenario where raising the precipitant concentration leads to supersaturation. The solubility of a protein in solution is governed by two principles: “salting in” and “salting out” [21]. The ability of cations and anions to salt in or salt out proteins is organised as the Hofmeister series where ions are ranked by their ability to solubilise and precipitate proteins [22]. As salt ions are added into the solution at low concentrations, the electrostatic energy between protein molecules is reduced, stabilising and increasing the solubility of the protein—this is salting in. As the concentration of the salt in solution rises, the excess ions compete with the protein for interactions with water molecules, effectively strengthening the hydrophobic interaction between proteins molecules. Without a layer of water molecules surrounding each protein molecule, proteins will start to aggregate and eventually precipitate out of the solution—this is salting out [21].
It is likely that magnesium formate is necessary for Glulisine to solubilise. The magnesium ion in solution has a high kosmotropic effect as the charge density on the small ion is high. This allows it to form interactions with the surrounding water molecules [23]. The bonding between a kosmotrope and water is greater than the bonding between water molecules themselves, therefore, the water–water hydrogen bonding is disrupted. The density of water is reduced as hydrogen bonds are broken allowing for more protein molecules to be dissolved. Without magnesium, the water starts to reorganise and Glulisine starts to aggregate and precipitate out of solution. As the solubility of magnesium formate in water is relatively low at 1 M [24], it might not be possible for the magnesium formate precipitant to cause supersaturation thus the crystallisation drop remains clear. Crystals are formed by reducing the solubility of the protein rather than the conventional process of increasing saturation of the drop.
The crystallisation of Glulisine with NaKT followed the usual trend where increased salt concentration resulted in the formation of crystals and amorphous precipitate. The charge density of sodium and potassium ions are lower compared to that of magnesium ions and do not affect the structuring of the water molecules as much Reference [25]. Thus, the solubility of Glulisine was less affected by the concentration of NaKT. It was interesting to see contrasting trends in the same protein. Moreover, crystals formed much more slowly with NaKT as precipitant, often taking weeks rather than days to form (four weeks after setting up the trials with NaKT at 3.49 mg/mL of the protein), whereas magnesium formate produced crystals in as little as 16 hours. It was interesting to note that despite the slow formation and growth of crystals in NaKT, their diffraction quality was much worse than crystals grown with magnesium formate (2.3 Å compared to 1.4 Å). These results emphasise the importance of the role of the precipitant used in crystallisation.
4.2. Experimental and Technical Evaluations
Vapour diffusion and microbatch follow different kinetics which can lead to different outcomes [26]. In batch experiments, the drop reaches instant supersaturation when the protein and precipitant are mixed, whereas in vapour diffusion experiments, the drop is undersaturated and the precipitant reservoir will slowly draw water out of the crystallisation drop and increase its saturation. As the conditions in vapour diffusion experiments change over time, there is a self-screening process as the drop saturation increases, whereas there is very little change in drop conditions under paraffin oil [26]. However, in the case of Glulisine with magnesium formate, crystals were produced in many more conditions in microbatch than in vapour diffusion, implying that Glulisine preferred to reach instant supersaturation. In addition, crystals in microbatch appeared faster than in vapour diffusion (seven days compared to two weeks). In contrast, when using NaKT as precipitant, vapour diffusion produced crystals under more conditions compared to microbatch. This highlighted the importance of the precipitant and the crystallisation method used.
The marketed formulation of Apidra consists of a mixture of Glulisine oligomers, not purely hexamers. The proportion of Glulisine monomers increases with dilution of Apidra. At 3.49 mg/mL, the Apidra formulation contains 25% Glulisine monomers; this increases to 37% at 1.74 mg/mL [27]. Large complexes of Glulisine are present at higher concentrations and when diluted, they readily dissociate into monomers which attributes to the rapid activity of Glulisine in vivo [17]. Naomi’s nucleant successfully produced diffracting crystals at metastable conditions of 0.3 M magnesium formate and 1.16 mg/mL Glulisine. However, under identical conditions, the nanoporous gold did not produce crystals. This may be attributed to the difference in pore size between the two materials. Nanoporous gold contains 5 to 10 nm sized pores, whereas Naomi’s nucleant contains pores ranging between 2 to 10 nm. Since it is the ability of the cavities to attract and entrap protein molecules that lead to crystal formation it may be that the pores of the gold foil are too large for Glulisine, which has varying radii molecules due a mixture of monomers and other oligomers.
4.3. Conclusions
The reported study presented the crystallisation of the rapid-acting insulin analogue Glulisine assisted by phase diagrams and nucleants. We have shown that the same protein can have different solubility behaviours depending on the nature of the salt in the precipitating agent. Glulisine with magnesium formate produced crystals faster at lower concentrations but with NaKT the opposite was observed. Interestingly, the microbatch also produced crystals faster (16 h) than in vapour diffusion trials (48 h) with magnesium formate. Glulisine with NaKT followed the expected trends i.e., forming crystals or precipitate at higher concentration and clear drops at lower concentrations of the precipitant. Glulisine was successfully crystallised and the crystals diffracted up to a resolution limit of 1.4 Å.
As Glulisine is a very interesting protein regarding its opposite solubility behaviour in the presence of different salts, it is an excellent candidate for further systematic studies to understand crystallisation. Glulisine will be a unique model protein as it is also of medical interest, unlike current model proteins such as lysozyme, thaumatin etc. which are very commonly studied.
Author Contributions
Conceptualization and supervision N.E.C., methodology Y.L., L.G., H.V.S., formal analysis N.E.C., Y.L., L.G., writing—original draft preparation Y.L., writing—review and editing N.E.C., L.G, R.B.G., G.G.A.
Funding
This research was funded by the Engineering and Physical Sciences Research Council (EPSRC), grant No. EP/K503733/1 and the Independent Diabetes Trust (IDDT), grant: ID/TT025272/1
Acknowledgments
The authors thank Rhodri Marc Morgan for assistance with screening the Glulisine crystals.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Khurshid, S.; Saridakis, E.; Govada, L.; Chayen, N.E. Porous nucleating agents for protein crystallization. Nat. Protoc. 2014, 9, 1621. [Google Scholar] [CrossRef]
- Luft, J.; Snell, E.; DeTitta, G. Lessons from high-throughput protein crystallization screening: 10 years of practical experience. Expert Opin. Drug Discov. 2011, 6, 465–480. [Google Scholar] [CrossRef]
- Asherie, N. Protein crystallization and phase diagrams. Methods 2004, 34, 266–272. [Google Scholar] [CrossRef]
- Chayen, N.E. Methods for separating nucleation and growth in protein crystallisation. Prog. Biophys. Mol. Biol. 2005, 88, 329–337. [Google Scholar] [CrossRef]
- Chayen, N.; Akins, J.; Campbell-Smith, S.; Blow, D.M. Solubility of glucose isomerase in ammonium sulphate solutions. J. Cryst. Growth 1988, 90, 112–116. [Google Scholar] [CrossRef]
- Saridakis, E.; Chayen, N.E. Systematic Improvement of Protein Crystals by Determining the Supersolubility Curves of Phase Diagrams. Biophys. J. 2003, 84, 1218–1222. [Google Scholar] [CrossRef] [Green Version]
- Chayen, N.E.; Saridakis, E. Protein crystallization: From purified protein to diffraction-quality crystal. Nat. Methods 2008, 5, 147–153. [Google Scholar] [CrossRef]
- Zhou, R.-B.; Cao, H.-L.; Zhang, C.-Y.; Yin, D.-C. A review on recent advances for nucleants and nucleation in protein crystallization. CrystEngComm 2017, 19, 1143–1155. [Google Scholar] [CrossRef]
- Nanev, C.N.; Saridakis, E.; Chayen, N.E. Protein crystal nucleation in pores. Sci. Rep. 2017, 7, 35821. [Google Scholar] [CrossRef]
- Chayen, N.E.; Saridakis, E.; El-Bahar, R.; Nemirovsky, Y. Porous silicon: An effective nucleation-inducing material for protein crystallization11Edited by R. Huber. J. Mol. Biol. 2001, 312, 591–595. [Google Scholar] [CrossRef]
- Page, A.J.; Sear, R.P. Heterogeneous nucleation in and out of pores. Phys. Rev. Lett. 2006, 97, 65701. [Google Scholar] [CrossRef] [PubMed]
- Chayen, N.E.; Saridakis, E.; Sear, R.P. Experiment and theory for heterogeneous nucleation of protein crystals in a porous medium. Proc. Natl. Acad. Sci. USA 2006, 103, 597–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saridakis, E.; Chayen, N.E. Towards a “universal” nucleant for protein crystallization. Trends Biotechnol. 2009, 27, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Kertis, F.; Khurshid, S.; Okman, O.; Kysar, J.W.; Govada, L.; Chayen, N.; Erlebacher, J. Heterogeneous nucleation of protein crystals using nanoporous gold nucleants. J. Mater. Chem. 2012, 22, 21928–21934. [Google Scholar] [CrossRef]
- Hirsch, I.B. Insulin Analogues. N. Engl. J. Med. 2005, 352, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Sanlioglu, A.D.; Altunbas, H.A.; Balci, M.K.; Griffith, T.S.; Sanlioglu, S. Clinical utility of insulin and insulin analogs. Islets 2013, 5, 67–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garnock-Jones, K.P.; Plosker, G.L. Insulin glulisine: A review of its use in the management of diabetes mellitus. Drugs 2009, 69, 1035–1057. [Google Scholar] [CrossRef] [PubMed]
- Hartman, I. Insulin analogs: Impact on treatment success, satisfaction, quality of life, and adherence. Clin. Med. Res. 2008, 6, 54–67. [Google Scholar] [CrossRef]
- Chayen, N.E.; Helliwell, J.R.; Solomon-Gamsu, H.V.; Govada, L.; Morgan, M.; Gillis, R.B.; Adams, G. Insulin Glulisine. Available online: https://www.rcsb.org/structure/6GV0 (accessed on 26 August 2019).
- Chayen, N.E.; Shaw Stewart, P.D.; Blow, D.M. Microbatch crystallization under oil—A new technique allowing many small-volume crystallization trials. J. Cryst. Growth 1992, 122, 176–180. [Google Scholar] [CrossRef]
- Arakawa, T.; Timasheff, S.N. Mechanism of protein salting in and salting out by divalent cation salts: Balance between hydration and salt binding. Biochemistry 1984, 23, 5912–5923. [Google Scholar] [CrossRef]
- Collins, K.D. Ions from the Hofmeister series and osmolytes: Effects on proteins in solution and in the crystallization process. Methods 2004, 34, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Tansel, B.; Sager, J.; Rector, T.; Garland, J.; Strayer, R.F.; Levine, L.; Roberts, M.; Hummerick, M.; Bauer, J. Significance of hydrated radius and hydration shells on ionic permeability during nanofiltration in dead end and cross flow modes. Sep. Purif. Technol. 2006, 51, 40–47. [Google Scholar] [CrossRef]
- Database, P.C.; National Center for Biotechnology Information. PubChem Compound Database. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/119432#section=Top%0A or https://pubchem.ncbi.nlm.nih.gov/compound/5988; (accessed on 16 August 2018).
- Collins, K.D. Charge density-dependent strength of hydration and biological structure. Biophys. J. 1997, 72, 65–76. [Google Scholar] [CrossRef] [Green Version]
- Chayen, N.E. Comparative Studies of Protein Crystallization by Vapour-Diffusion and Microbatch Techniques. Acta Crystallogr. Sect. D 1998, 54, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Nagel, N.; Graewert, M.A.; Gao, M.; Heyse, W.; Jeffries, C.M.; Svergun, D.; Berchtold, H. The quaternary structure of insulin glargine and glulisine under formulation conditions. Biophys. Chem. 2019, 253, 106226. [Google Scholar] [CrossRef]
Figure 1.
Working phase diagrams for Glulisine crystallisation with magnesium formate as precipitant. (A) vapour diffusion, (B) microbatch.
Figure 1.
Working phase diagrams for Glulisine crystallisation with magnesium formate as precipitant. (A) vapour diffusion, (B) microbatch.
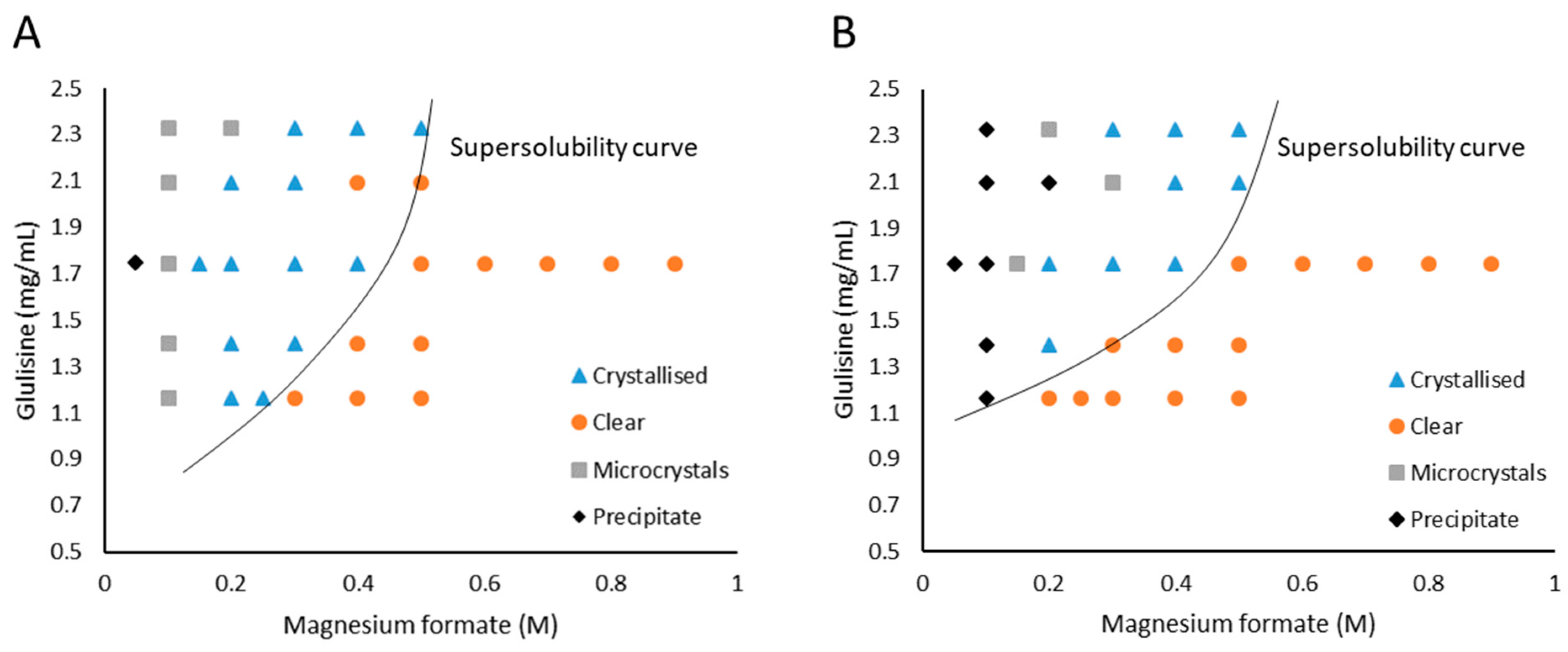
Figure 2.
(A) Glulisine crystals produced in metastable conditions at 1.16 mg/mL Glulisine and 0.3 M magnesium formate with Naomi’s nucleant. (B) Crystals obtained at supersaturated conditions at 1.16 mg/mL Glulisine and 0.2 M magnesium formate with no nucleants. (C) Diffraction of up to a resolution limit of 1.4 Å of the crystals in (A).
Figure 2.
(A) Glulisine crystals produced in metastable conditions at 1.16 mg/mL Glulisine and 0.3 M magnesium formate with Naomi’s nucleant. (B) Crystals obtained at supersaturated conditions at 1.16 mg/mL Glulisine and 0.2 M magnesium formate with no nucleants. (C) Diffraction of up to a resolution limit of 1.4 Å of the crystals in (A).
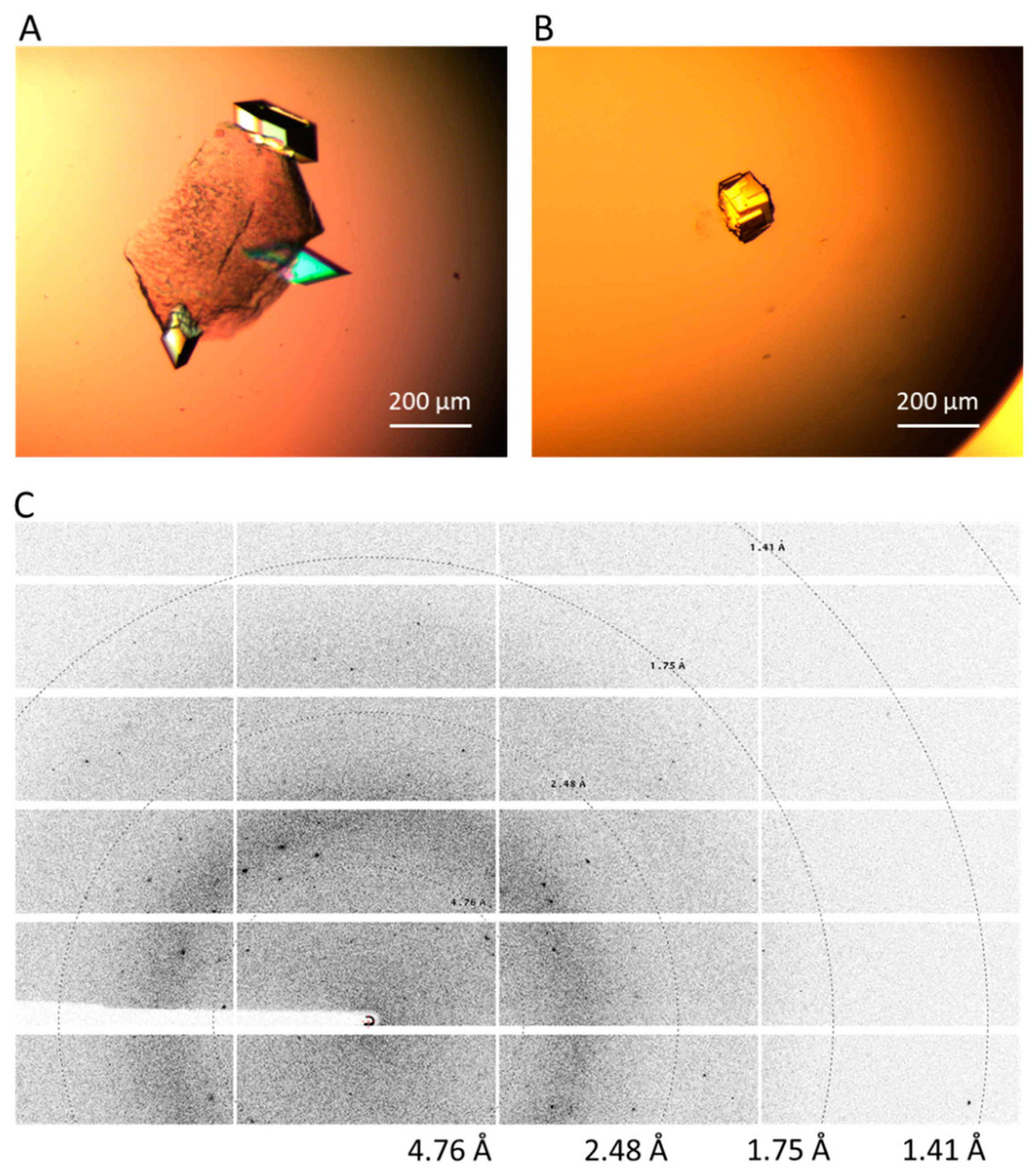
Figure 3.
Working phase diagram for Glulisine crystallisation with sodium potassium tartrate tetrahydrate (NaKT) as precipitant using vapour diffusion.
Figure 3.
Working phase diagram for Glulisine crystallisation with sodium potassium tartrate tetrahydrate (NaKT) as precipitant using vapour diffusion.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, Y.; Govada, L.; Solomon, H.V.; Gillis, R.B.; Adams, G.G.; Chayen, N.E. Analysis of Glulisine Crystallisation Utilising Phase Diagrams and Nucleants. Crystals 2019, 9, 462. https://doi.org/10.3390/cryst9090462
AMA Style
Li Y, Govada L, Solomon HV, Gillis RB, Adams GG, Chayen NE. Analysis of Glulisine Crystallisation Utilising Phase Diagrams and Nucleants. Crystals. 2019; 9(9):462. https://doi.org/10.3390/cryst9090462
Chicago/Turabian StyleLi, Yanmin, Lata Govada, Hodaya V. Solomon, Richard B. Gillis, Gary G. Adams, and Naomi E. Chayen. 2019. "Analysis of Glulisine Crystallisation Utilising Phase Diagrams and Nucleants" Crystals 9, no. 9: 462. https://doi.org/10.3390/cryst9090462
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.