Crystal Structures and High-Temperature Vibrational Spectra for Synthetic Boron and Aluminum Doped Hydrous Coesite

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Synthesis and Characterization
2.2. Single-Crystal X-ray Diffraction (XRD)
2.3. Vibrational Spectra at Room and High Temperatures
3. Results and Discussion
3.1. Hydration and B/Al Concentrations
3.2. Crystal Structures
3.3. Lattice Vibrations and Grüneisen Parameters γiP
3.4. OH-Stretching Modes at High Temperature
4. Conclusions
- (1)
- We synthesized several hydrous coesite samples with different B and Al compositions at pressures of about 5–7.5 GPa (1273 K) in the multi-anvil press. The concentrations of the B/Al trace elements were measured by fs-LA–ICP-MS, while H2O contents were estimated by FTIR. The B concentrations are more than 400 atomic ppm (B/106Si) with ~350 ppmw H2O, while the Al3+ contents are about 1100–1300 atomic ppm, which were cross checked by both ICP-MS and EPMA. Al-substitution significantly reduces the hydrogen concentration in coesite. Hence, the mechanism controlled by oxygen vacancies (2Si4+ = 2Al3+ + OV) may be dominant for the Al incorporation, which is similar to that in aluminous stishovite, while the B incorporation may prefer the electrostatically coupled substitution (Si4+ = B3+ + H+);
- (2)
- The doped B3+ and Al3+ cations would prefer the Si1 and Si2 tetrahedra, respectively, and the single-crystal structure refinements reveal that B3+ significantly shortens the Si1–O4 and Si1–O5 bond lengths, whereas Al3+ noticeably elongates the Si2–O4 and Si2–O5 distances;
- (3)
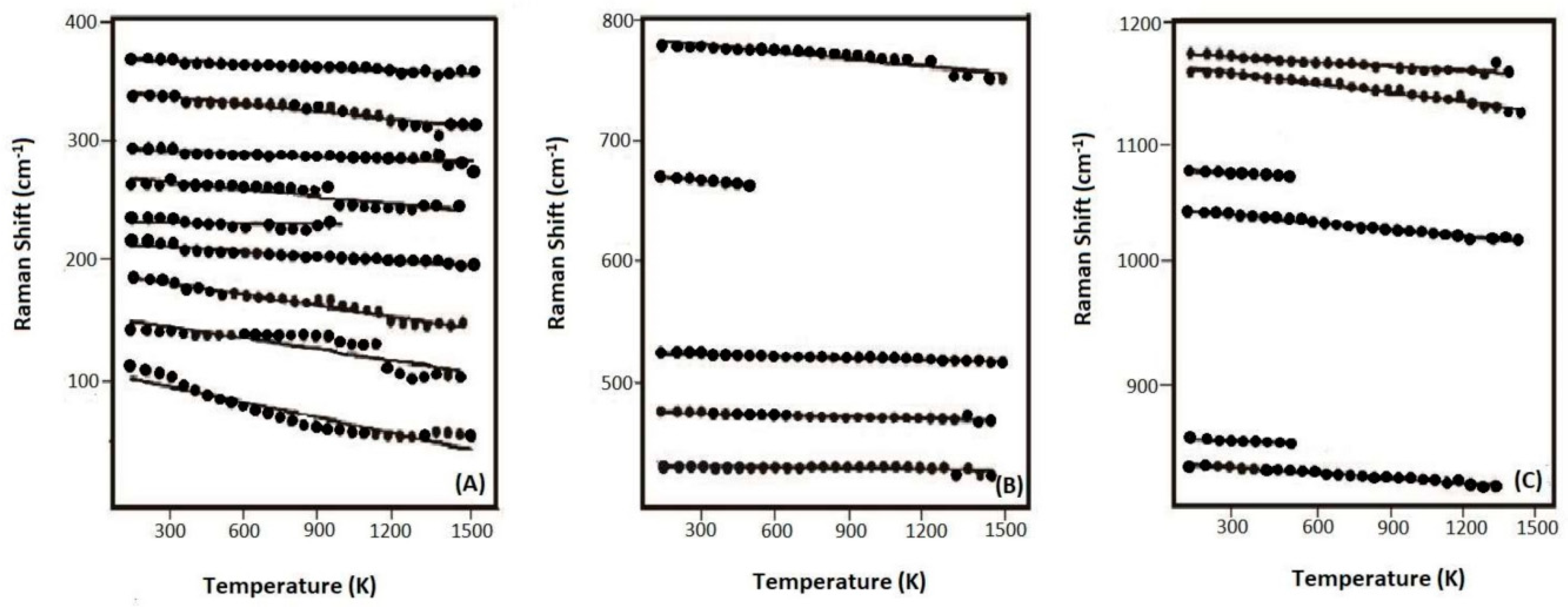
- In-situ high-temperature Raman spectra were collected on these synthetic samples of up to 1500 K (at ambient condition), and no amorphization of phase transition was observed throughout the heating procedures. The derived isobaric mode Grüneisen parameters (γiP) for the external vibrations of SiO4 units (below 350 cm−1) are significantly reduced for the Al-doped samples, as compared with the Al-free ones. Hence, the relaxation of the SiO4 units might be hindered to some extent due to the enlarged tetrahedra units by Al-substitution. On the other hand, the γiP parameters for the internal bending and stretching modes of SiO4 tetrahedra in coesite (above 400 cm−1) are significantly larger than those of most silicate minerals, due to the abnormally small thermal expansion coefficient for coesite;
- (4)
- The OH-stretching modes v1, v2a,b, v3, and v4 are observed for all these hydrous samples with the various compositions, and another strong band v6 is also observed for the B-doped ones. Most of these OH vibrational modes shift to higher frequencies at elevated temperatures (except the weak v1 mode around 3600 cm−1), implying that the O...O distances between different SiO4 gets longer during the thermal relaxation of the lattice framework at a high temperature. On the other hand, about 20–40% dehydration of OH groups were observed for these hydrous coesite samples at high temperatures above 1000 K at ambient pressure.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Coes, L. A new dense crystalline silica. Science 1953, 118, 131–132. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, S.W.; Phakey, P.P.; Christie, J.M. Shock processes in porous quartzite: Transmission electron microscope observations and theory. Contrib. Mineral. Petrol. 1976, 59, 41–93. [Google Scholar] [CrossRef]
- Folco, L.; Mugnaioli, E.; Gemelli, M.; Masotta, M.; Campanale, F. Direct quartz-coesite transformation in shocked porous sandstone from Kamil Crater (Egypt). Geology 2018, 9, 739–742. [Google Scholar] [CrossRef]
- Smyth, J.R.; Hatton, C.J. A coesite-sanidine grospydite from the Roberts-Victor Kimberlite. Earth Planet. Sci. Lett. 1977, 34, 284–290. [Google Scholar] [CrossRef]
- Wang, X.M.; Liou, J.G.; Mao, H.K. Coesite-bearing eclogite from the Dabie mountains in central China. Geology 1989, 17, 1085–1088. [Google Scholar] [CrossRef]
- Bi, H.; Song, S.; Dong, J.; Yang, L.; Qi, S.; Allen, M.B. First discovery of coesite in eclogite from East Kunlun, northwest China. Sci. Bull. 2018, 63, 1536–1538. [Google Scholar] [CrossRef]
- Mosenfelder, J.L.; Bohlen, S.R. Kinetics of the coesite to quartz transformation. Earth Planet. Sci. Lett. 1997, 153, 133–147. [Google Scholar] [CrossRef]
- Chopin, C. Coesite and pure pyrope in high-grade blueschists of the Western Alps: A first record and some consequences. Contrib. Mineral. Petrol. 1984, 86, 107–118. [Google Scholar] [CrossRef]
- Angel, R.J.; Mosenfelder, J.L.; Shaw, C.S.J. Anomalous compression and equation of state of coesite. Phys. Earth Planet. Inter. 2001, 124, 71–79. [Google Scholar] [CrossRef]
- Angel, R.J.; Shaw, C.S.J.; Gibbs, G.V. Compression mechanisms of coesite. Phys. Chem. Miner. 2003, 30, 167–176. [Google Scholar] [CrossRef]
- Bourova, E.; Pichet, P.; Petitet, J.-P. Coesite (SiO2) as an extreme case of superheated crystal: An X-ray diffraction study up to 1776 K. Chem. Geol. 2006, 229, 57–63. [Google Scholar] [CrossRef]
- Chen, T.; Gwanmesia, G.D.; Wang, X.; Zou, Y.; Liebermann, R.C.; Michaut, C.; Li, B. Anomalous elastic properties of coesite at high pressure and implications for the upper mantle X-discontinuity. Earth Planet. Sci. Lett. 2015, 412, 42–51. [Google Scholar] [CrossRef]
- Haines, J.; Leger, J.M.; Gorelli, F.; Hanfland, M. Crystalline post-quartz phase in silica at high pressure. Phys. Rev. Lett. 2001, 87, 155503. [Google Scholar] [CrossRef]
- Černok, A.; Ballaran, T.B.; Caracas, R.; Miyajima, N.; Bykova, E.; Prakapenka, V.; Libermann, H.P.; Dubrovinsky, L. Pressure-induced phase transitions in coesite. Am. Miner. 2014, 99, 755–763. [Google Scholar] [CrossRef][Green Version]
- Hu, Q.Y.; Shu, J.-F.; Cadien, A.; Meng, Y.; Yang, W.G.; Sheng, H.W.; Mao, H.-K. Polymorphic phase transition mechanism of compressed coesite. Nat. Commun. 2015, 6, 6630. [Google Scholar] [CrossRef] [PubMed]
- Hemley, R.J. Pressure dependence of Raman spectra of SiO2 polymorphs: α-quartz, coesite, and stishovite. In High Pressure Research in Mineral Physics; Manghnani, M.H., Syono, Y., Eds.; Geophysics Monograph Series 39; AGU: Washington, DC, USA, 1987; pp. 347–359. [Google Scholar]
- Gillet, P.; Le, C.A.; Madon, M. High-temperature Raman spectroscopy of SiO2 and GeO2 polymorphs: Anharmonicity and thermodynamic properties at high-temperatures. J. Geophys. Res. 1990, 95, 21635–21655. [Google Scholar] [CrossRef]
- Williams, Q.; Hemley, R.L.; Kruger, M.B.; Jeanloz, R. High pressure infrared spectra of α-quartz, coesite, stishovite and silica glass. J. Geophys. Res. 1993, 98, 157–170. [Google Scholar] [CrossRef]
- Liu, X.; Ma, Y.; He, Q.; He, M. Some IR features of SiO4 and OH in coesite, and its amorphization and dehydration at ambient pressure. J. Asian Earth Sci. 2017, 148, 315–323. [Google Scholar] [CrossRef]
- He, M.; Yan, W.; Chang, Y.; Liu, K.; Liu, X. Fundamental infrared absorption of α-quartz: An unpolarzied single-crystal absorption infrared spectroscopic study. Vib. Spectrosc. 2019, 101, 52–63. [Google Scholar] [CrossRef]
- Mosenfelder, J.L.; Schertl, H.-P.; Smyth, J.R.; Liou, J.G. Factors in the preservation of coesite: The importance of fluid infiltration. Am. Miner. 2005, 90, 779–789. [Google Scholar] [CrossRef]
- Lathe, C.; Koch-Müller, M.; Wirth, R.; Van Westrenen, W.; Mueller, H.-J.; Schilling, F.; Lauterjung, J. The influence of OH in coesite on the kinetics of the coesite-quartz phase transition. Am. Miner. 2005, 90, 36–43. [Google Scholar] [CrossRef][Green Version]
- Mosenfelder, J.L. Pressure dependence of hydroxyl solubility in coesite. Phys. Chem. Miner. 2000, 27, 610–617. [Google Scholar] [CrossRef]
- Koch-Müller, M.; Fei, Y.; Hauri, E. Location and quantitative analysis of OH in coesite. Phys. Chem. Miner. 2001, 28, 693–705. [Google Scholar] [CrossRef]
- Koch-Müller, M.; Dera, P.; Fei, Y.; Reno, B.; Sobolev, N.; Hauri, E.; Wysoczanski, R. OH− in synthetic and natural coesite. Am. Miner. 2003, 88, 1436–1445. [Google Scholar] [CrossRef]
- Deon, F.; Koch-Müller, M.; Hövelmann, J.; Rhede, D.; Thomas, S.-M. Coupled boron and hydrogen incorporation in coesite. Eur. J. Mineral. 2009, 21, 9–16. [Google Scholar] [CrossRef]
- Rossman, G.R.; Smyth, J.R. Hydroxyl contents of accessory minerals in mantle eclogites and related rocks. Am. Miner. 1990, 75, 775–780. [Google Scholar]
- Zhang, J.F.; Shi, F.; Xu, H.J.; Wang, L.; Feng, S.Y.; Liu, W.L.; Wang, Y.F.; Green, H.W. Petrofabric and strength of SiO2 near the quartz-coesite phase boundary. J. Metamorph. Geol. 2013, 31, 83–92. [Google Scholar] [CrossRef]
- Luo, T.; Ni, Q.; Hu, Z.; Zhang, W.; Shi, Q.; Günther, D.; Liu, Y.; Zong, K.; Hu, S. Comparison of signal intensities and elemental fractionation in 257 nm femtosecond LA-ICP-MS using He and Ar as carrier gases. J. Anal. At. Spectrom. 2017, 32, 2217–2225. [Google Scholar] [CrossRef]
- Li, Z.; Hu, Z.; Liu, Y.; Gao, S.; Li, M.; Zong, K.; Chen, H.; Hu, S. Accurate determination of elements in silicate glass by nanosecond and femtosecond laser ablation ICP-MS at high spatial resolution. Chem. Geol. 2015, 400, 11–23. [Google Scholar] [CrossRef]
- Jochum, K.P.; Willbold, M.; Raczek, I.; Stoll, B.; Herwig, K. Chemical characterization of the USGS reference glasses GSA-1G, GSC-1G, GSD-1G, GSE-1G, BCR-2G, BHVO-2G and BIR-1G using EPMA, ID-TIMS, ID-ICP-MS and LA-ICP-MS. Geostand. Geoanal. Res. 2005, 29, 285–302. [Google Scholar] [CrossRef]
- Wang, X.; Xu, X.; Ye, Y.; Wang, C.; Liu, D.; Shi, X.; Wang, S.; Zhu, X. In-situ high-temperature XRD and FTIR for calcite, dolomite and magnesite: Anharmonic contribution to the thermodynamic properties. J. Earth Sci. 2019, 30, 964–976. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Blake, A.J.; Champness, N.R.; Schroder, M. Olex: New software for visualization and analysis of extended crystal structures. J. Appl. Crystallogr. 2003, 36, 1283–1284. [Google Scholar] [CrossRef]
- Cromer, D.T.; Mann, J. X-ray scattering factors computed from numerical Hartree-Fock wave functions. Acta Crystallogr. 1968, A24, 321–325. [Google Scholar] [CrossRef]
- Tokonami, M. Atomic scattering factor for O2−. Acta Crystallogr. 1965, 19, 486. [Google Scholar] [CrossRef]
- Pawley, A.R.; McMillan, P.F.; Holloway, J.R. Hydrogen in stishovite, with implications for mantle water content. Science 1993, 261, 1024–1026. [Google Scholar] [CrossRef]
- Litasov, K.D.; Kagi, H.; Shatskiy, A.; Ohtani, E.; Lakshtanov, D.L.; Bass, J.D.; Ito, E. High hydrogen solubility in Al-rich stishovite and water transport in the lower mantle. Earth Planet. Sci. Lett. 2007, 262, 620–634. [Google Scholar] [CrossRef]
- Shimizu, N.; Hart, S.R. Applications of the ion microprobe to geochemistry and cosmochemistry. Annu. Rev. Earth Planet. Sci. 1982, 10, 483–526. [Google Scholar] [CrossRef]
- Araki, T.; Zoltai, T. Refinement of a coesite structure. Z. Krist. 1969, 129, 381–387. [Google Scholar] [CrossRef]
- Levien, L.; Prewitt, C.T. High-pressure crystal structure and compressibility of coesite. Am. Miner. 1981, 66, 324–333. [Google Scholar]
- Smyth, J.R.; Artioli, G.; Smith, J.V.; Kvick, A. Crystal structure of coesite, a high-pressure form of SiO2, at 15 and 298 K from single-crystal neutron and X-ray diffraction data: Test of bonding models. J. Phys. Chem. 1987, 91, 988–992. [Google Scholar] [CrossRef]
- Sasaki, S.; Chen, H.K.; Prewitt, C.T.; Nakajima, Y. Re-examination of “P21/a coesite”. Z. Krist. 1983, 164, 67–77. [Google Scholar] [CrossRef]
- Ikuta, D.; Kawame, N.; Banno, S.; Hirajima, T.; Ito, K.; Rakovan, J.F.; Downs, R.T.; Tamada, O. First in situ X-ray diffraction identification of coesite and retrograde quartz on a glass thin section of an ultrahigh-pressure metamorphic rock and their crystal structure details. Am. Miner. 2007, 92, 57–63. [Google Scholar] [CrossRef]
- Downs, R.T.; Bartelmehs, K.L.; Gibbs, G.V.; Boisen, M.B. Interactive software for calculating and displaying X-ray or neutron power diffractometer patterns of crystalline materials. Am. Miner. 1993, 78, 1104–1107. [Google Scholar]
- Richet, P. Superheating, melting and vitrification through decompression of high-pressure minerals. Nature. 1988, 331, 56–58. [Google Scholar] [CrossRef]
- Liu, L.G.; Mernagh, T.P.; Hibberson, W.O. Raman spectra of high-pressure polymorphs of SiO2 at various temperatures. Phys. Chem. Miner. 1997, 24, 396–402. [Google Scholar] [CrossRef]
- Kieffer, S.W. Thermodynamics and lattice vibrations of minerals: Lattice dynamics and an approximation for minerals with application to simple substances and framework silicates. Rev. Geophys. 1979, 17, 35–39. [Google Scholar] [CrossRef]
- Gillet, P.; Daniel, I.; Guyot, F. Anharmonic properties of Mg2SiO4-forsterite measured from the volume dependence of the Raman spectrum. Eur. J. Mineral. 1997, 9, 255–262. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, Z.; Smyth, J.R.; Liu, J.; Xia, Q. Water effects on the anharmonic properties of forsterite. Am. Miner. 2015, 100, 2185–2190. [Google Scholar] [CrossRef]
- Gillet, P.; Fiquet, G.; Malezieux, J.M.; Geiger, C.A. High-pressure and high-temperature Raman spectroscopy of end-member garnets: Pyrope, grossular and andradite. Eur. J. Mineral. 1992, 4, 651–664. [Google Scholar] [CrossRef]
- Zucker, R.; Shim, S.-H. In situ Raman spectroscopy of MgSiO3 enstatite up to 1550 K. Am. Miner. 2009, 94, 1638–1646. [Google Scholar] [CrossRef]
- Kroll, H.; Kirfel, A.; Heinemann, R.; Barbier, B. Volume thermal expansion and related thermophysical parameters in the Mg, Fe olivine solid-solution series. Eur. J. Mineral. 2012, 24, 935–956. [Google Scholar] [CrossRef]
- Du, W.; Clark, S.M.; Walker, D. Thermo-compression of pyrope-grossular garnet solid solutions: Non-linear compositional dependence. Am. Miner. 2015, 100, 215–222. [Google Scholar] [CrossRef]
- Jackson, J.M.; Palko, J.W.; Andrault, D.; Sinogeikin, S.V.; Lakshtanov, D.L.; Wang, J.; Bass, J.D.; Zha, C.-S. Thermal expansion of natural orthoenstatite up to 1472 K. Eur. J. Mineral. 2003, 97, 6842–6866. [Google Scholar]
- Keppler, H.; Bagdassarov, N.S. High-temperature FTIR spectra of H2O in rhyolite melt to 1300 °C. Am. Miner. 1993, 78, 1324–1327. [Google Scholar]
- Grzechnik, A.; McMillan, P.F. Temperature dependence of the OH− absorption in SiO2 glass and melt to 1975 K. Am. Miner. 1998, 83, 331–338. [Google Scholar] [CrossRef]
- Moore, G.; Chizmshya, A.; McMillan, P.F. Calibration of a reflectance FTIR method for determination of dissolved CO2 concentration in rhyolitic glasses. Geochim. Gosmochim. Acta 2000, 64, 3571–3579. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run No. Starting Materials (wt.%) | Pressure (GPa) | Temperature (K) | Time (h) | B (ppmw) | B/106Si | Al (ppmw) | Al/106Si | H2O (ppmw) | H/106Si |
|---|---|---|---|---|---|---|---|---|---|
| R503: SiO2 + 1 μL H2O | 7.5 | 1273 | 12 | — | — | — | — | 32.3 ± 14.2 | 215 ± 95 |
| R663: SiO2 (96) + H3BO3 (4) + 1 μL H2O | 5 | 1273 | 10 | 74.6 ± 3.5 | 445 ± 21 | — | — | 51.0 ± 19.8 | 340 ± 132 |
| R694: SiO2 (94) + H3BO3 (3) + Al(OH)3 (3) + 1 μL H2O | 5 | 1273 | 10 | 73.4 ± 9.0 | 437 ± 54 | 115.6 ± 52.0 | 343 ± 154 | 22.8 ± 11.7 | 152 ± 78 |
| R712: SiO2 (94) + H3BO3 (3) + Al(OH)3 (3) + 1 μL H2O | 7.5 | 1273 | 9 | 39.7 ± 8.2 | 237 ± 49 | 139.8 ± 31.7 | 415 ± 94 | 24.1 ± 10.9 | 161 ± 73 |
| R749: SiO2 (96) + Al(OH)3 (4) + 1 μL H2O | 5 | 1273 | 9 | — | — | 445.8 ± 86.5 a 392.7 ± 50.8 b | 1323 ± 257 a 1166 ± 151 b | 7.2 ± 2.9 | 48 ± 19 |
| R503 | R663 | R694 | R712 | R749 | |
|---|---|---|---|---|---|
| a (Å) | 7.1458 (5) | 7.1332 (9) | 7.1355 (5) | 7.1426 (13) | 7.1437(7) |
| b (Å) | 12.3922 (10) | 12.3886 (5) | 12.3678 (5) | 12.3698 (8) | 12.3964 (6) |
| c (Å) | 7.1778 (8) | 7.1828 (16) | 7.1763 (8) | 7.1788 (16) | 7.1858 (12) |
| β (°) | 120.293 (11) | 120.31 (2) | 120.358 (10) | 120.37 (2) | 120.292 (15) |
| V (Å3) | 548.82 (10) | 548.01 (18) | 546.47 (9) | 547.24 (19) | 549.46 (13) |
| No. total refl. | 1028 | 777 | 2117 | 911 | 1346 |
| No. unique total | 366 | 436 | 523 | 467 | 487 |
| No. unique I > 4σ | 348 | 427 | 511 | 446 | 461 |
| Goof | 1.084 | 1.059 | 1.021 | 1.051 | 1.041 |
| R1 for all (%) | 2.45 | 2.89 | 2.91 | 2.67 | 2.83 |
| R1 for I > 4σ (%) | 2.33 | 2.84 | 2.86 | 2.57 | 2.72 |
| Rint (%) | 1.33 | 0.62 | 1.04 | 1.23 | 1.47 |
| R503 | R663 | R694 | R712 | R749 | ||
|---|---|---|---|---|---|---|
| Si1 | x | 0.14027 (12) | 0.14048 (13) | 0.14044 (9) | 0.14049 (11) | 0.14050 (11) |
| y | 0.10826 (6) | 0.10833 (5) | 0.10835 (4) | 0.10832 (5) | 0.10829 (5) | |
| z | 0.07226 (10) | 0.07240 (15) | 0.07242 (9) | 0.07232 (11) | 0.07242 (11) | |
| Si2 | x | 0.50653 (12) | 0.50655 (13) | 0.50668 (9) | 0.50682 (11) | 0.50675 (11) |
| y | 0.15808 (6) | 0.15794 (5) | 0.15796 (8) | 0.15798 (5) | 0.15795 (5) | |
| z | 0.54061 (10) | 0.54028 (15) | 0.54069 (8) | 0.54073 (11) | 0.54079 (10) | |
| O2 | x | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| y | 0.1162 (2) | 0.1164 (2) | 0.1164 (15) | 0.1163 (2) | 0.11656 (17) | |
| z | 0.75 | 0.75 | 0.75 | 0.75 | 0.75 | |
| O3 | x | 0.7340 (3) | 0.7333 (3) | 0.7336 (2) | 0.7334 (3) | 0.7335 (3) |
| y | 0.12289 (16) | 0.12287 (15) | 0.12290 (12) | 0.12284 (15) | 0.12280 (13) | |
| z | 0.5603 (3) | 0.5593 (4) | 0.5594 (3) | 0.5594 (3) | 0.5595 (3) | |
| O4 | x | 0.3115 (3) | 0.3103 (4) | 0.3109 (2) | 0.3112 (3) | 0.3107 (3) |
| y | 0.10385 (17) | 0.10397 (15) | 0.10386 (11) | 0.10372 (15) | 0.10377 (12) | |
| z | 0.3282 (3) | 0.3278 (4) | 0.3278 (2) | 0.3282 (3) | 0.3276 (3) | |
| O5 | x | 0.0172 (3) | 0.0168 (3) | 0.0174 (2) | 0.0175 (3) | 0.0175 (3) |
| y | 0.21182 (17) | 0.21177 (15) | 0.21174 (11) | 0.21173 (15) | 0.21168 (13) | |
| z | 0.4784 (3) | 0.4787 (4) | 0.4786 (2) | 0.4786 (3) | 0.4786 (3) |
| R503 | R663 | R694 | R712 | R749 | |
|---|---|---|---|---|---|
| Si1–O1 | 1.5966 (8) | 1.5968 (7) | 1.5949 (5) | 1.5953 (7) | 1.5980 (6) |
| Si1–O3 | 1.6134 (16) | 1.6130 (20) | 1.6105 (15) | 1.6120 (20) | 1.6134 (17) |
| Si1–O4 | 1.6146 (19) | 1.6090 (30) | 1.6088 (16) | 1.6120 (20) | 1.6106 (19) |
| Si1–O5 | 1.6230 (20) | 1.6184 (19) | 1.6191(14) | 1.6205 (19) | 1.6229 (16) |
| <Si1–O> | 1.6119 (22) | 1.6093 (19) | 1.6083 (13) | 1.6100 (16) | 1.6112 (16) |
| Poly. Vol. | 2.1485 (25) | 2.1382 (30) | 2.1344 (24) | 2.1415 (21) | 2.1457 (20) |
| Si2–O2 | 1.6130 (12) | 1.6147 (12) | 1.6109 (7) | 1.6121 (11) | 1.6121 (9) |
| Si2–O3 | 1.6194 (18) | 1.6125 (19) | 1.6150 (14) | 1.6150 (20) | 1.6160 (16) |
| Si2–O4 | 1.6050 (20) | 1.6060 (30) | 1.6049 (15) | 1.6046 (19) | 1.6111 (18) |
| Si2–O5 | 1.6200 (20) | 1.6213 (19) | 1.6191 (14) | 1.6194 (19) | 1.6283 (17) |
| <Si2–O> | 1.6144 (23) | 1.6136 (22) | 1.6125 (14) | 1.6128 (12) | 1.6169 (6) |
| Poly. Vol. | 2.1579 (24) | 2.1563 (30) | 2.1515 (18) | 2.1522 (22) | 2.1645 (20) |
| O1–Si1–O3 | 110.37 (9) | 110.28 (9) | 110.35 (6) | 110.41 (8) | 110.32 (7) |
| O1–Si1–O4 | 109.41 (8) | 109.34 (8) | 109.33 (5) | 109.28 (8) | 109.32 (6) |
| O1–Si1–O5 | 109.94 (8) | 110.03 (16) | 109.83 (6) | 109.75 (8) | 109.80 (7) |
| O4–Si1–O3 | 110.26 (11) | 110.51 (12) | 110.34 (9) | 110.30 (11) | 110.39 (10) |
| O4–Si1–O5 | 108.86 (10) | 108.81 (12) | 108.91 (8) | 108.92 (10) | 108.92 (9) |
| O3–Si1–O5 | 107.97 (10) | 107.84 (11) | 108.05 (7) | 108.14 (10) | 108.05 (9) |
| O2–Si2–O4 | 109.30 (11) | 109.41 (10) | 109.37 (7) | 109.30 (10) | 109.48 (8) |
| O2–Si2–O5 | 110.35 (11) | 110.22 (12) | 110.21 (8) | 110.22 (12) | 110.17 (10) |
| O3–Si2–O2 | 109.42 (8) | 109.56 (10) | 109.75 (7) | 109.75 (9) | 109.55 (8) |
| O3–Si2–O4 | 108.80 (10) | 108.93 (12) | 108.71 (8) | 108.69 (11) | 108.72 (10) |
| O3–Si2–O5 | 109.47 (10) | 109.35 (10) | 109.45 (7) | 109.54 (10) | 109.55 (8) |
| O5–Si2–O4 | 109.45 (11) | 109.35 (11) | 109.25 (7) | 109.31 (10) | 109.23 (9) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miao, Y.; Pang, Y.; Ye, Y.; Smyth, J.R.; Zhang, J.; Liu, D.; Wang, X.; Zhu, X. Crystal Structures and High-Temperature Vibrational Spectra for Synthetic Boron and Aluminum Doped Hydrous Coesite. Crystals 2019, 9, 642. https://doi.org/10.3390/cryst9120642
Miao Y, Pang Y, Ye Y, Smyth JR, Zhang J, Liu D, Wang X, Zhu X. Crystal Structures and High-Temperature Vibrational Spectra for Synthetic Boron and Aluminum Doped Hydrous Coesite. Crystals. 2019; 9(12):642. https://doi.org/10.3390/cryst9120642
Chicago/Turabian StyleMiao, Yunfan, Youwei Pang, Yu Ye, Joseph R. Smyth, Junfeng Zhang, Dan Liu, Xiang Wang, and Xi Zhu. 2019. "Crystal Structures and High-Temperature Vibrational Spectra for Synthetic Boron and Aluminum Doped Hydrous Coesite" Crystals 9, no. 12: 642. https://doi.org/10.3390/cryst9120642
APA StyleMiao, Y., Pang, Y., Ye, Y., Smyth, J. R., Zhang, J., Liu, D., Wang, X., & Zhu, X. (2019). Crystal Structures and High-Temperature Vibrational Spectra for Synthetic Boron and Aluminum Doped Hydrous Coesite. Crystals, 9(12), 642. https://doi.org/10.3390/cryst9120642