Investigating Differences and Similarities between Betaxolol Polymorphs

 ,
, 
 ,
,  and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Synthesis and Crystallization
2.2. Single Crystal X-Ray Data Collection and Structure Solution
2.3. Computational Methods
3. Results and Discussion
3.1. Synthesis
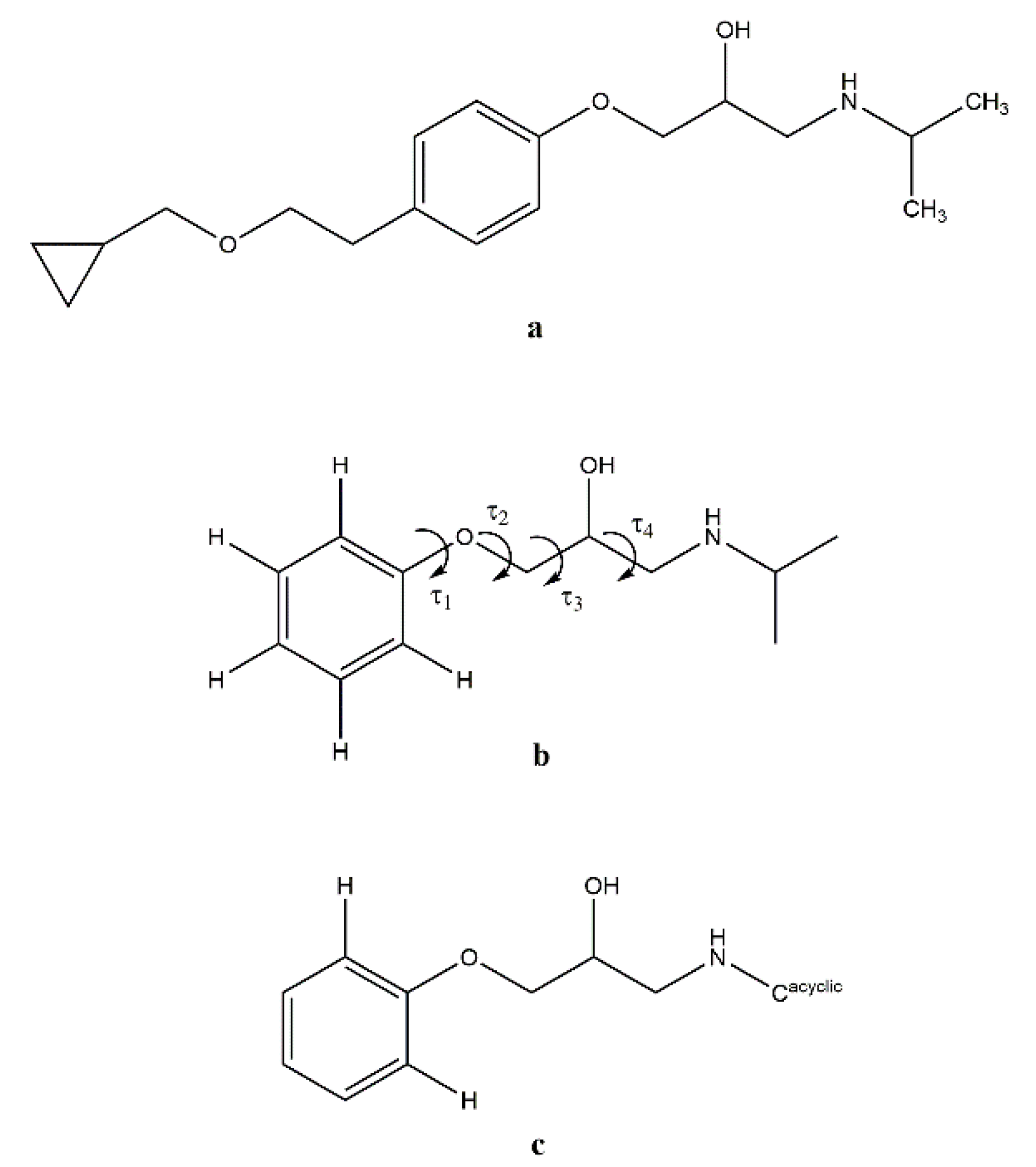
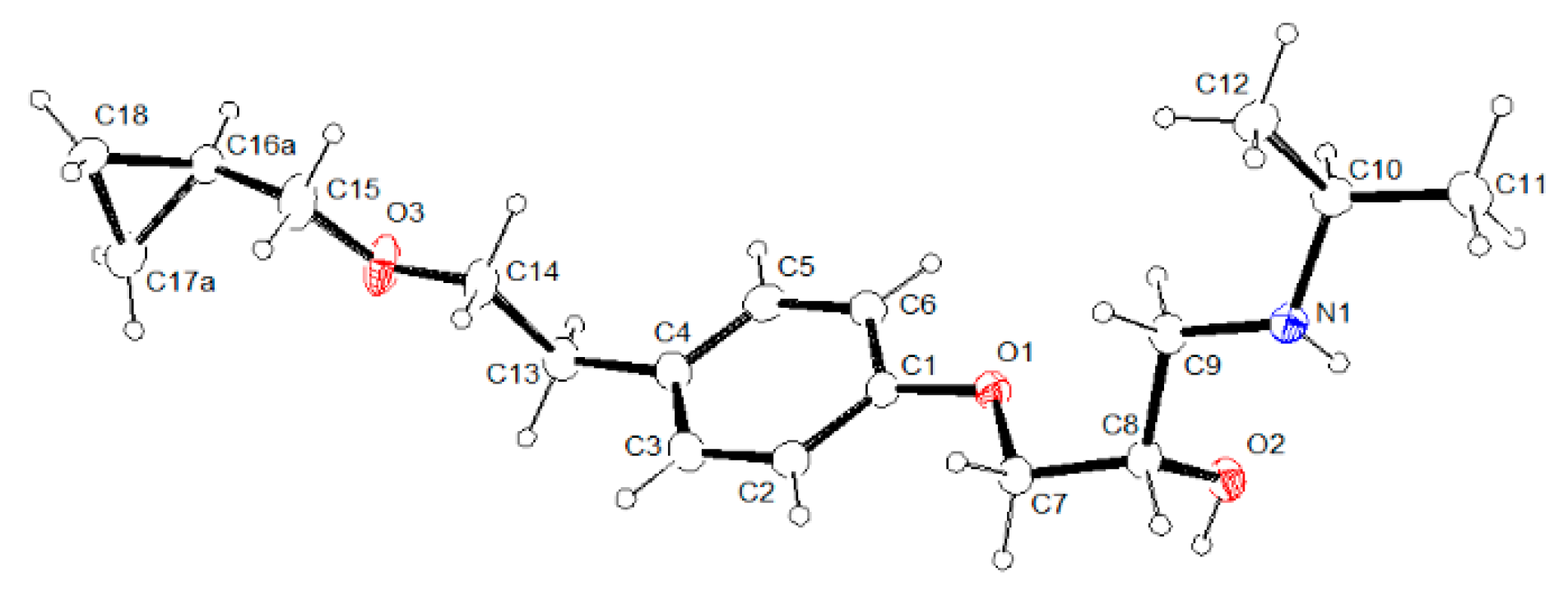
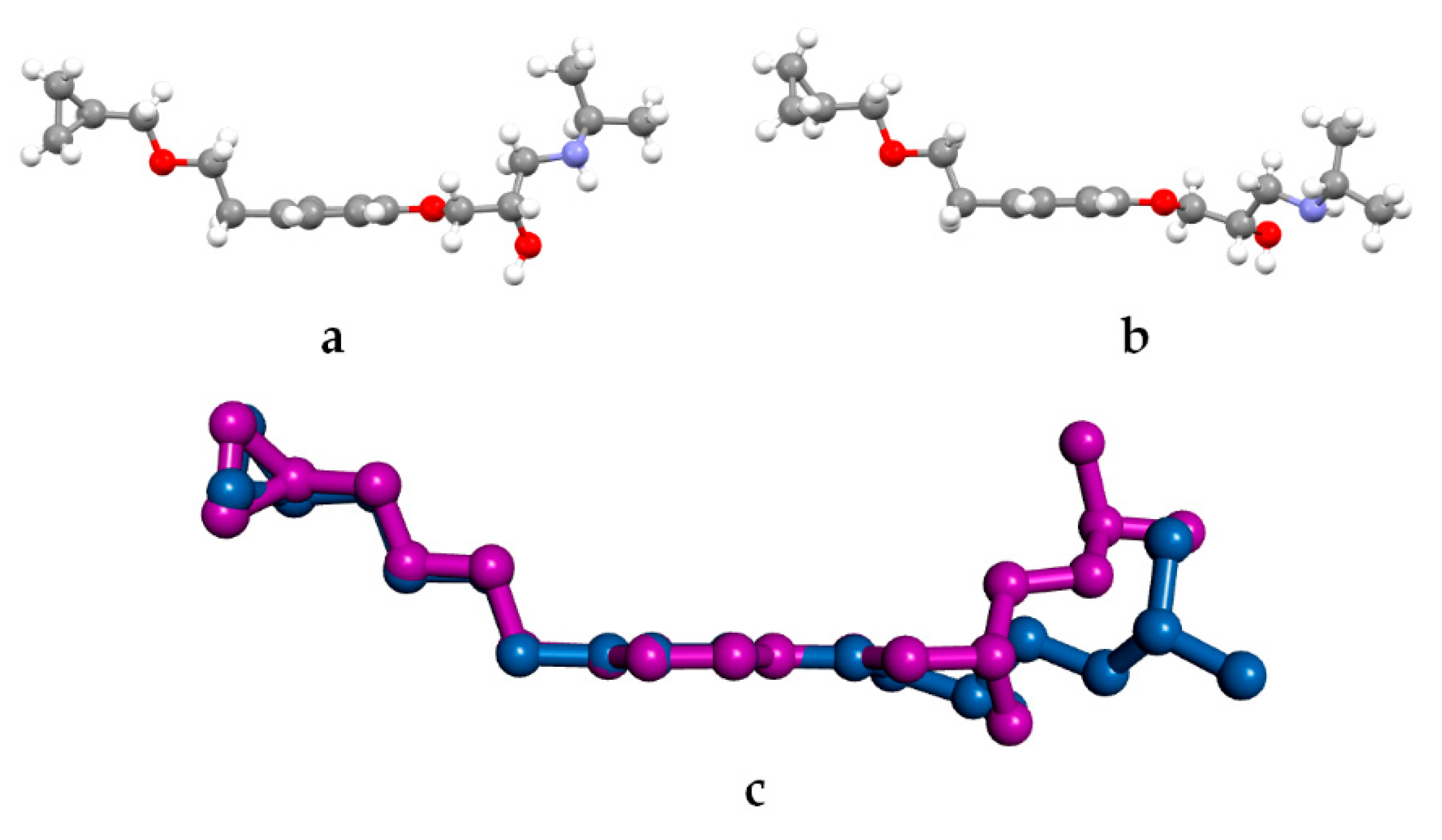
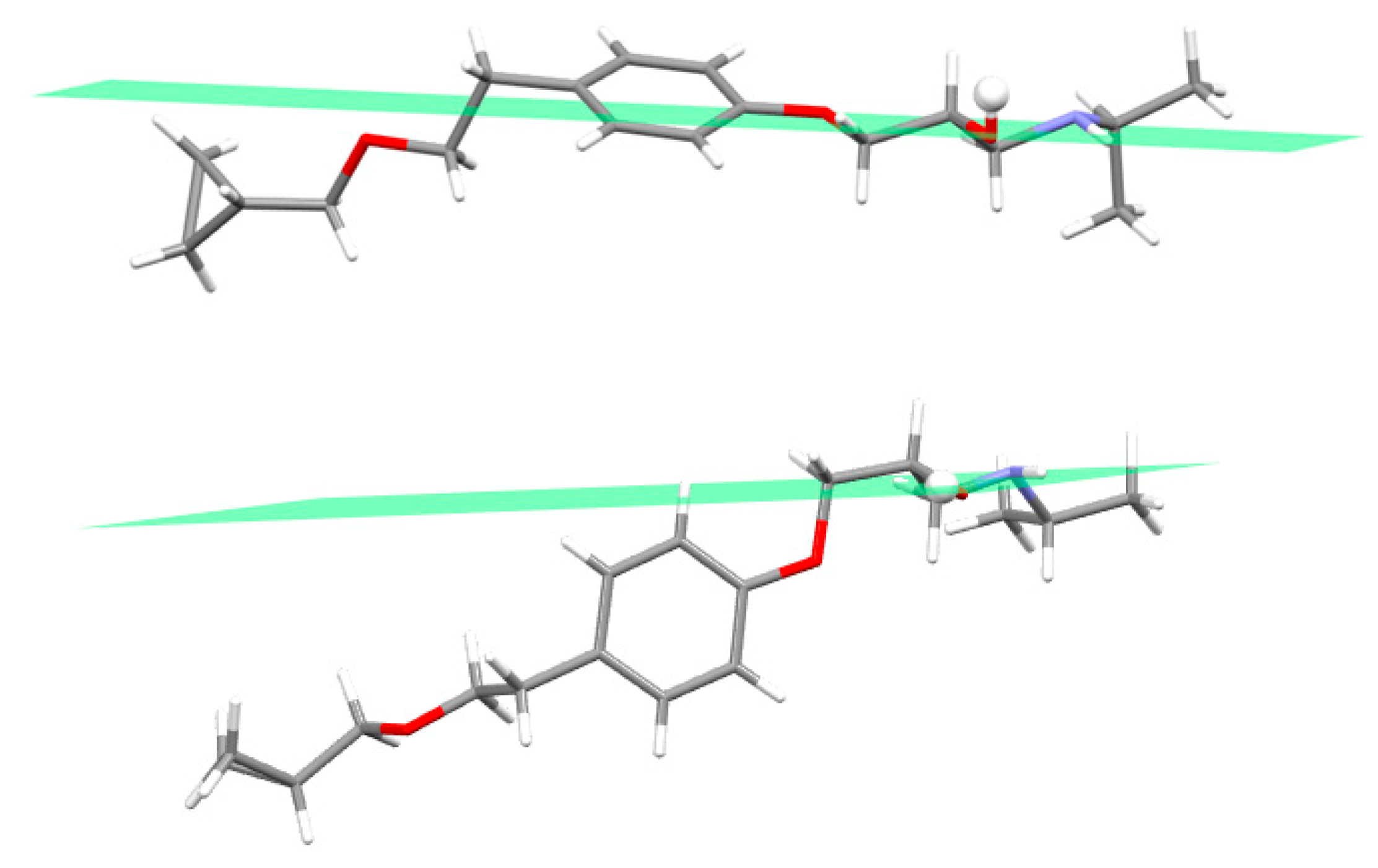
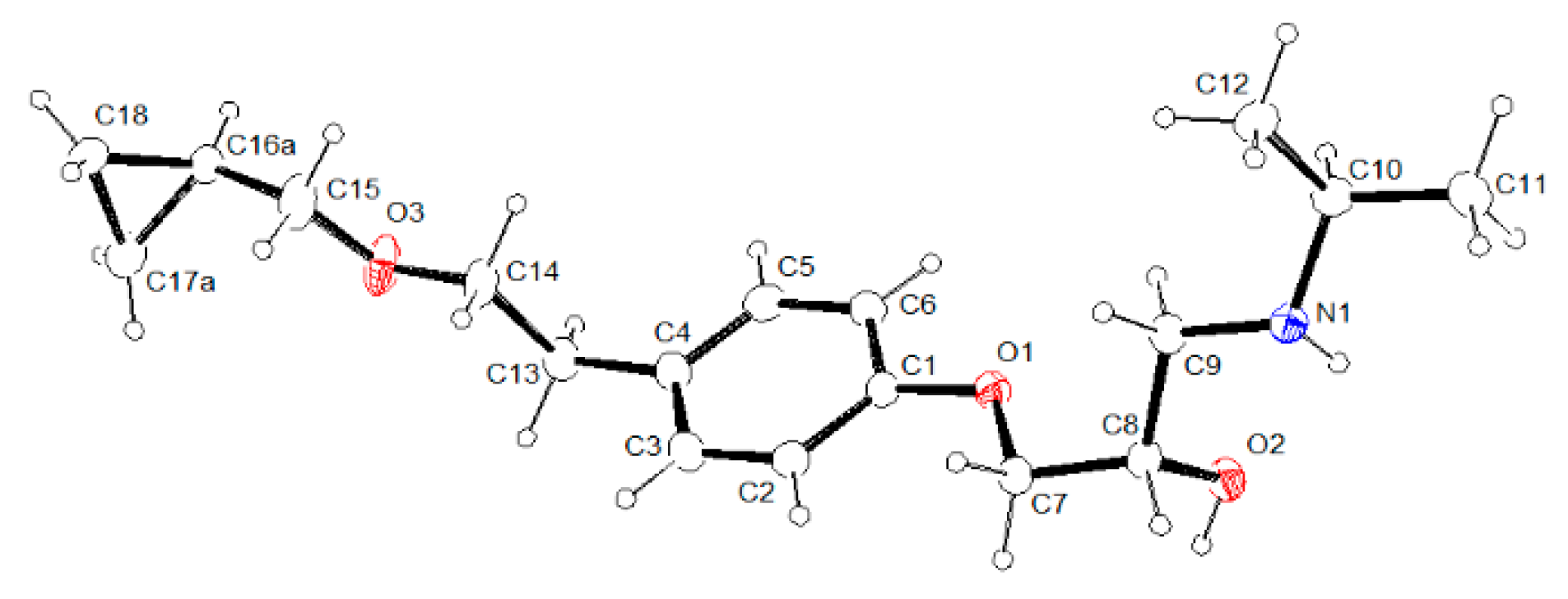
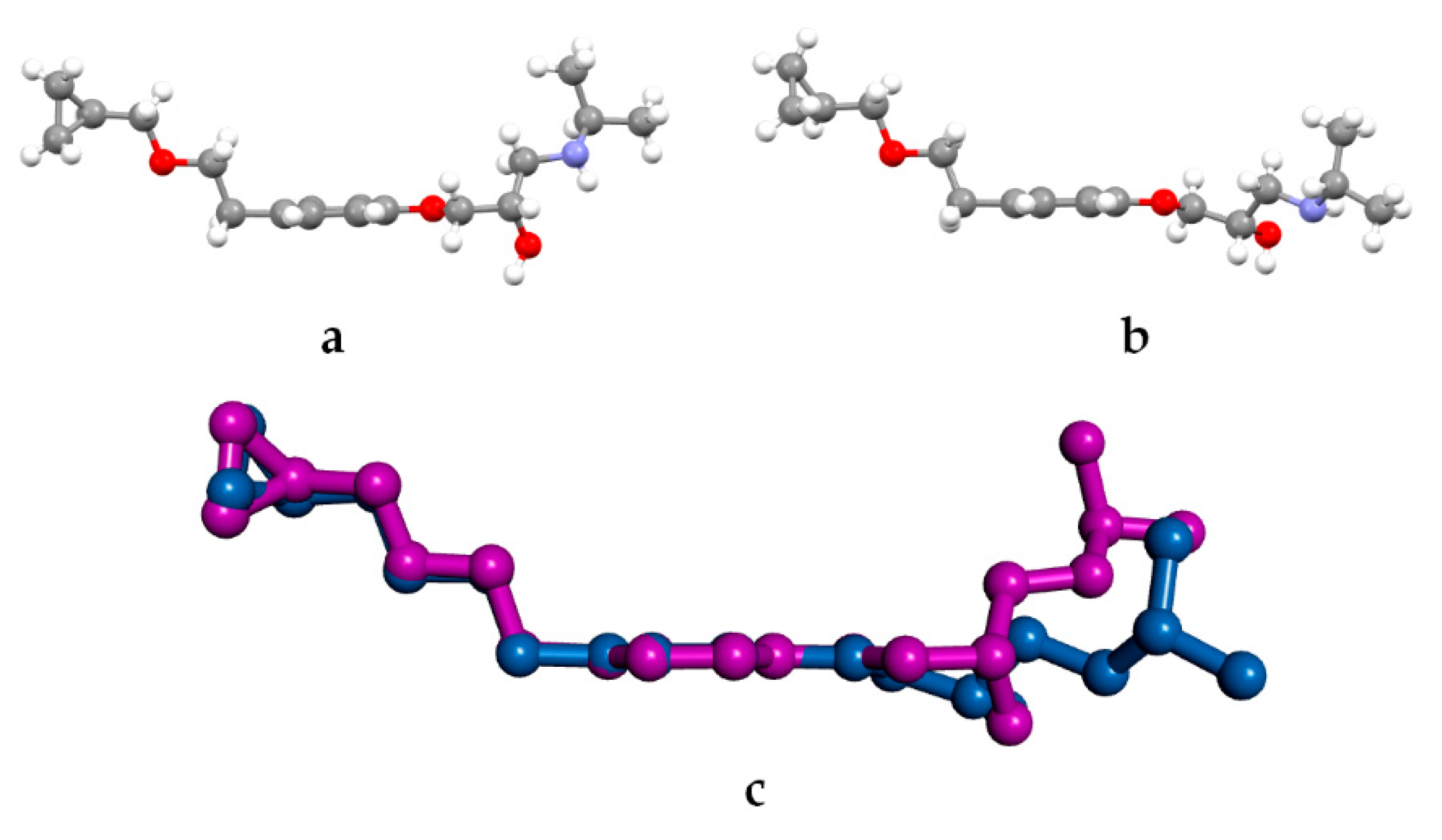
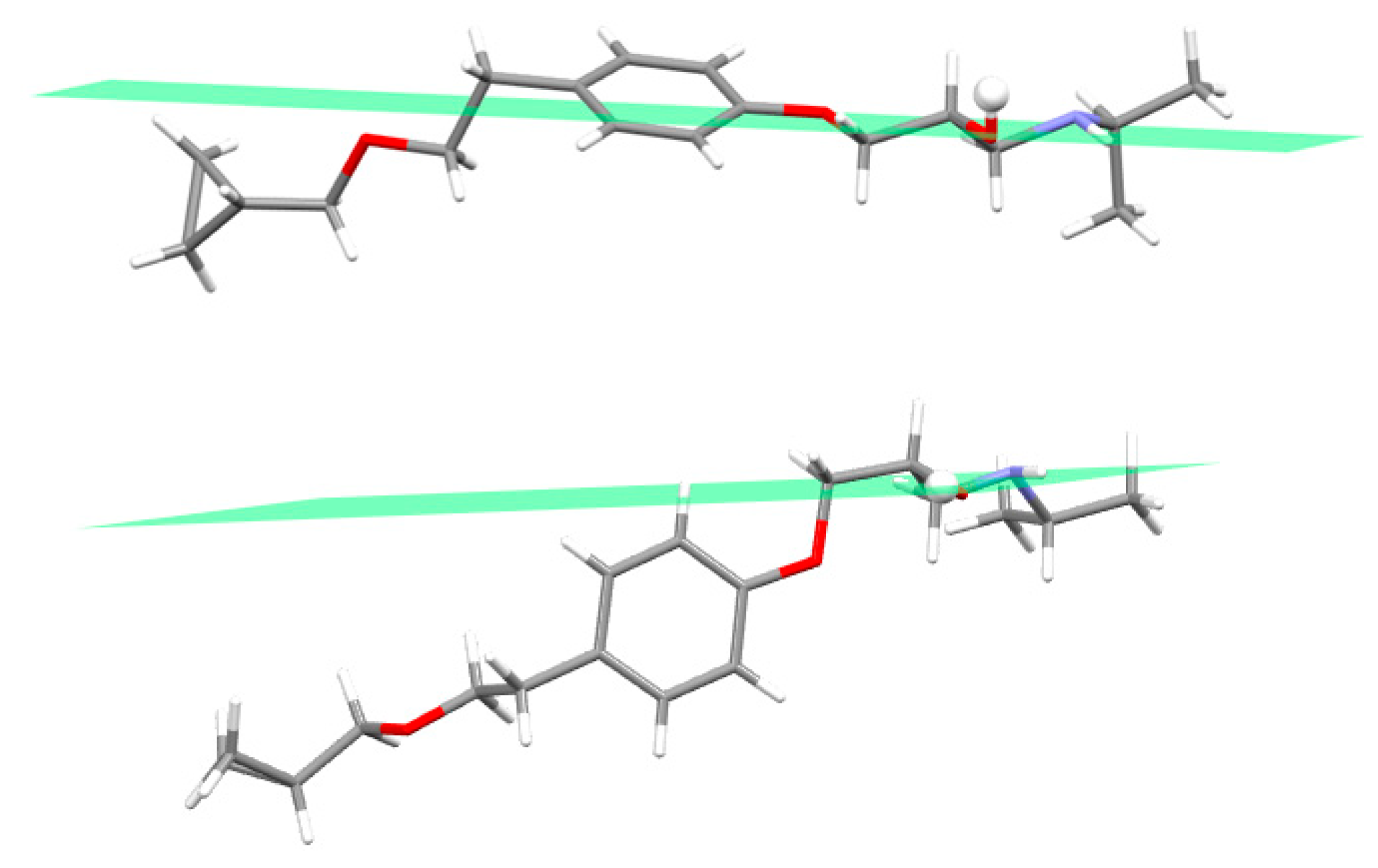
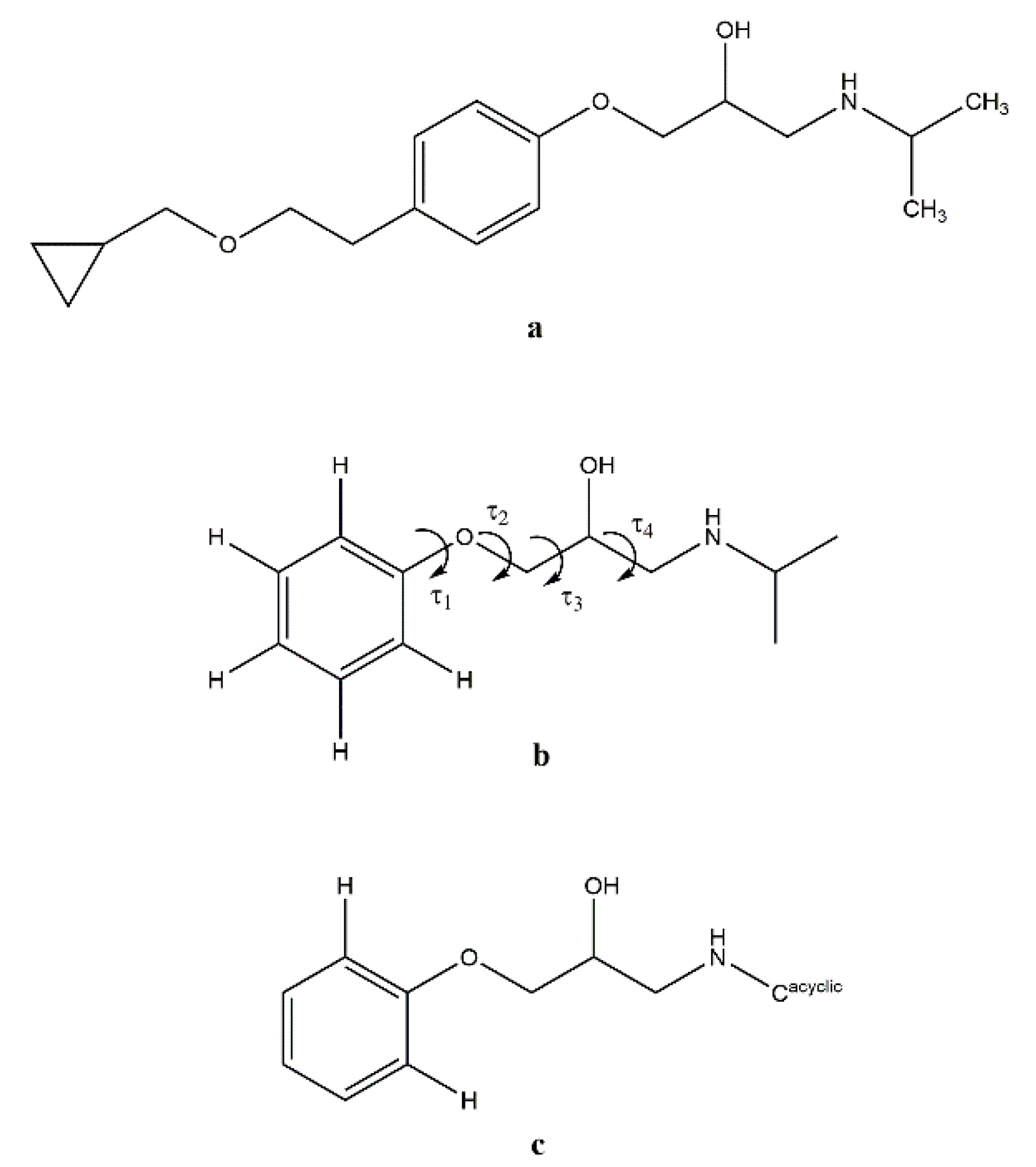
3.2. Molecular Structure from Single-Crystal X-Ray Diffraction and Computational Methods
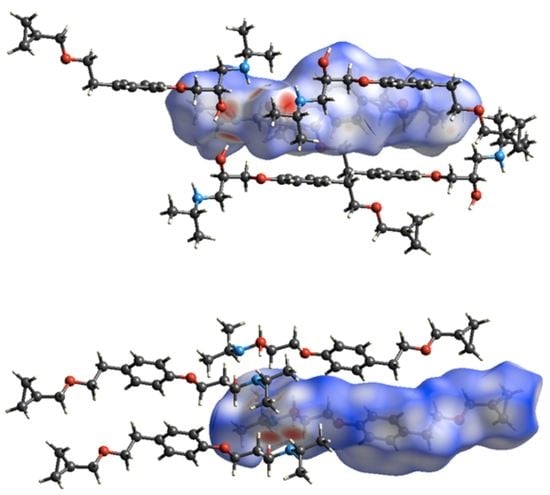
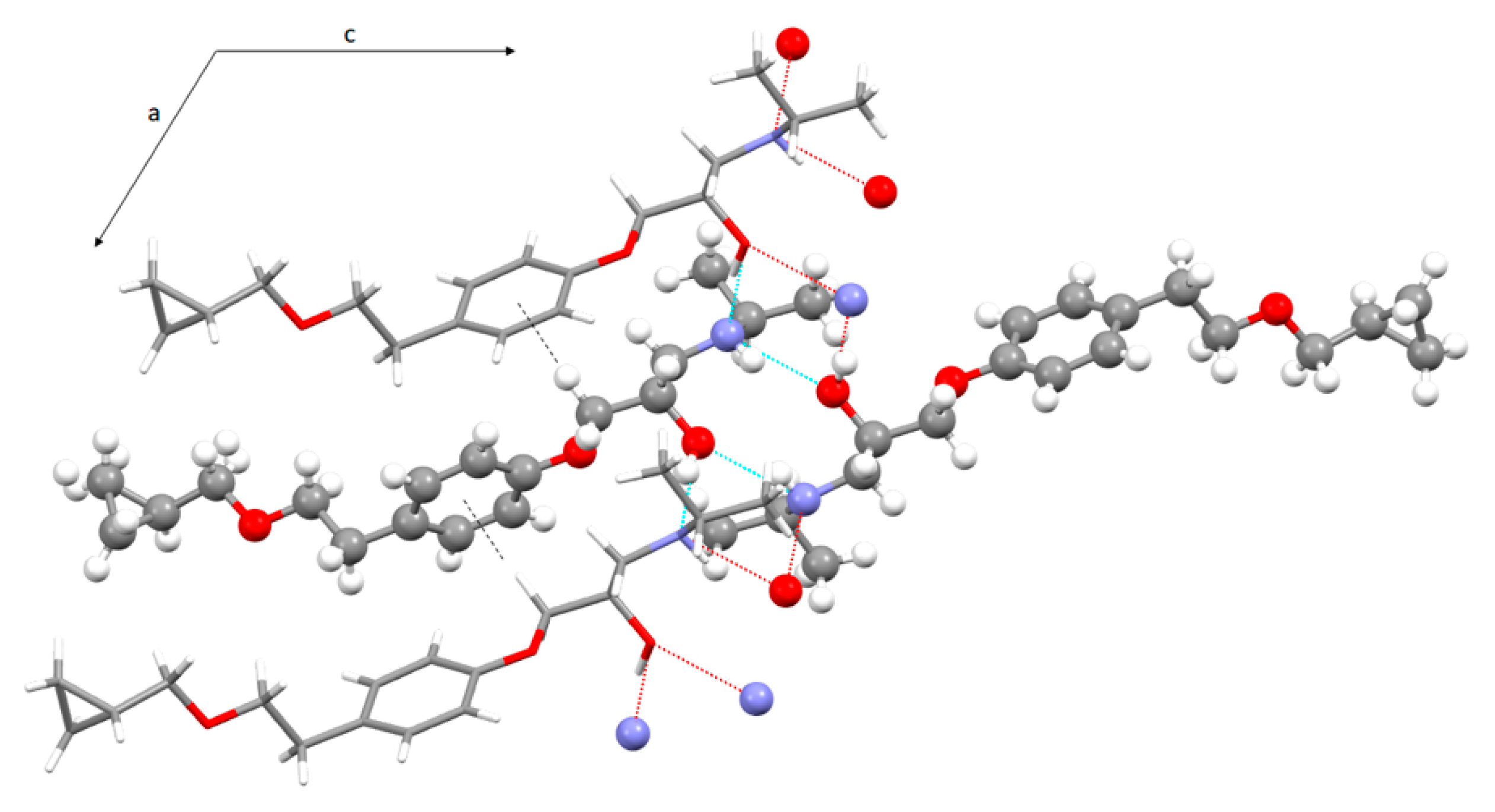
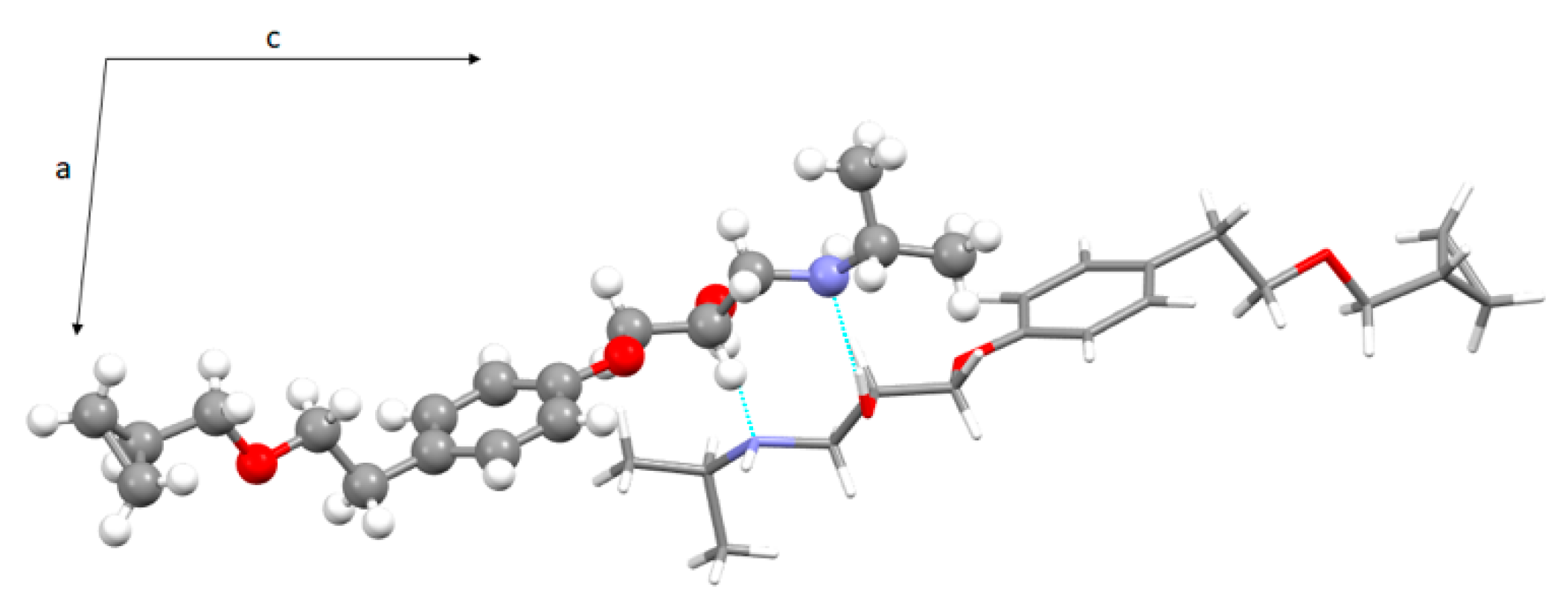
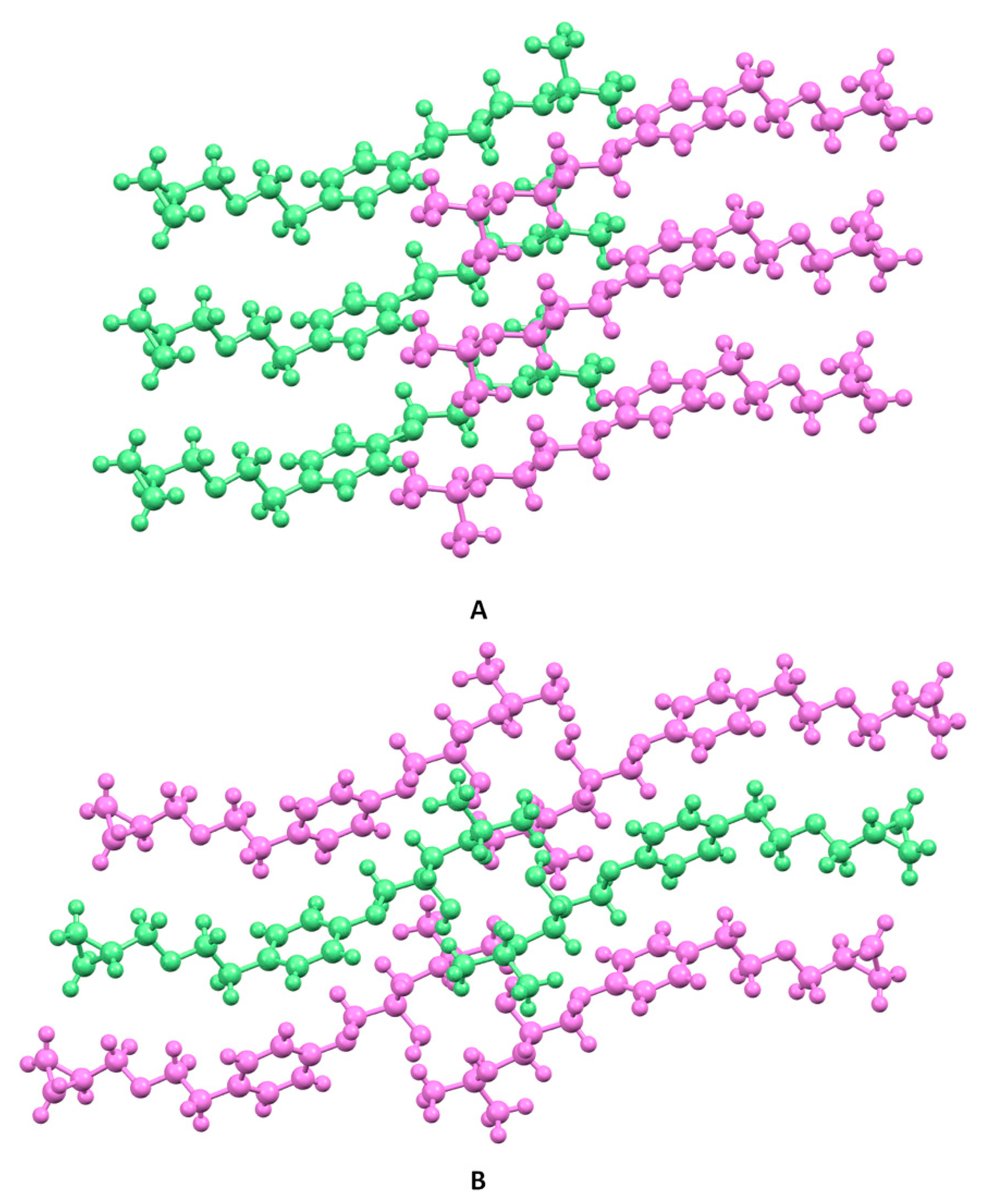
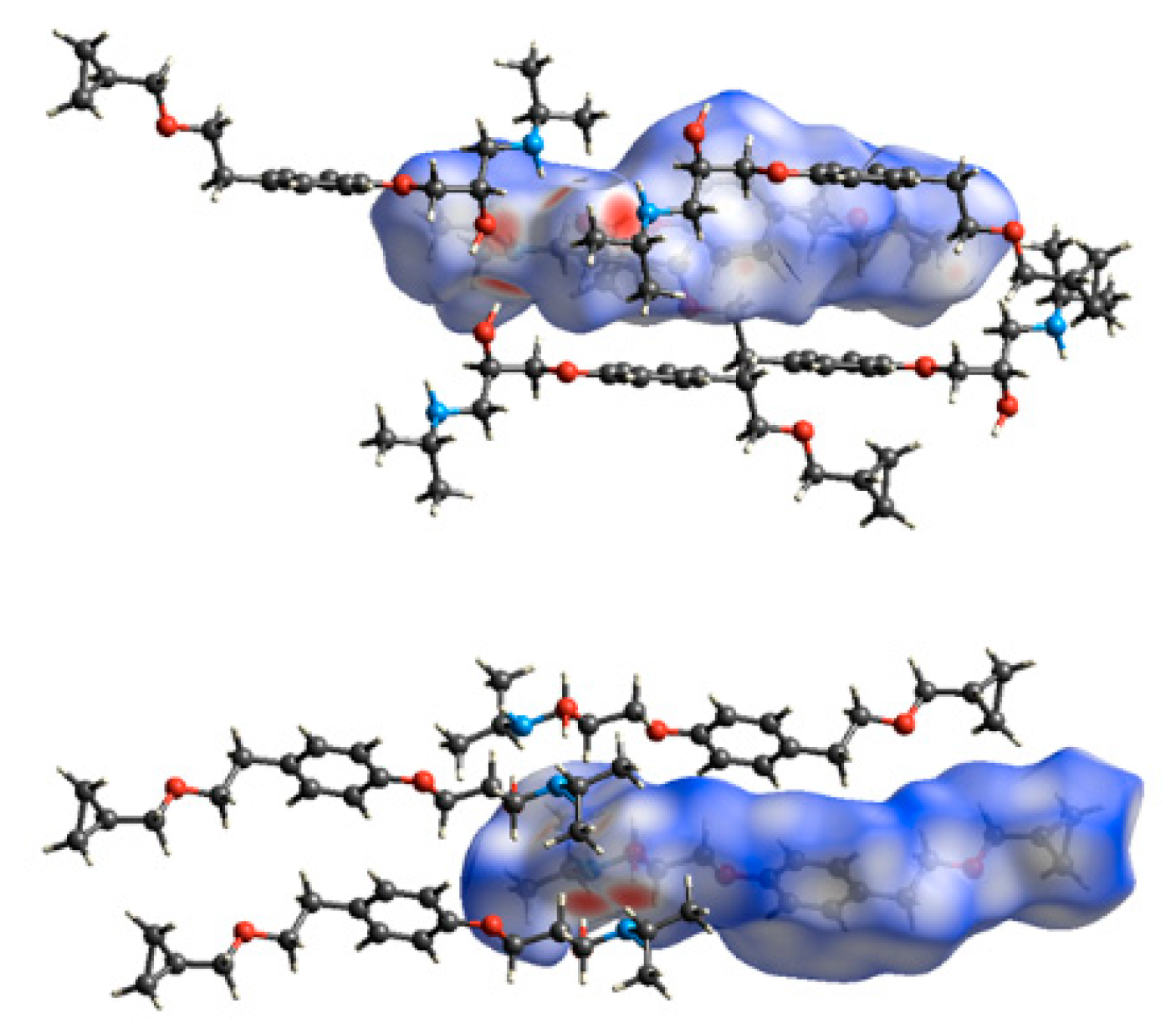
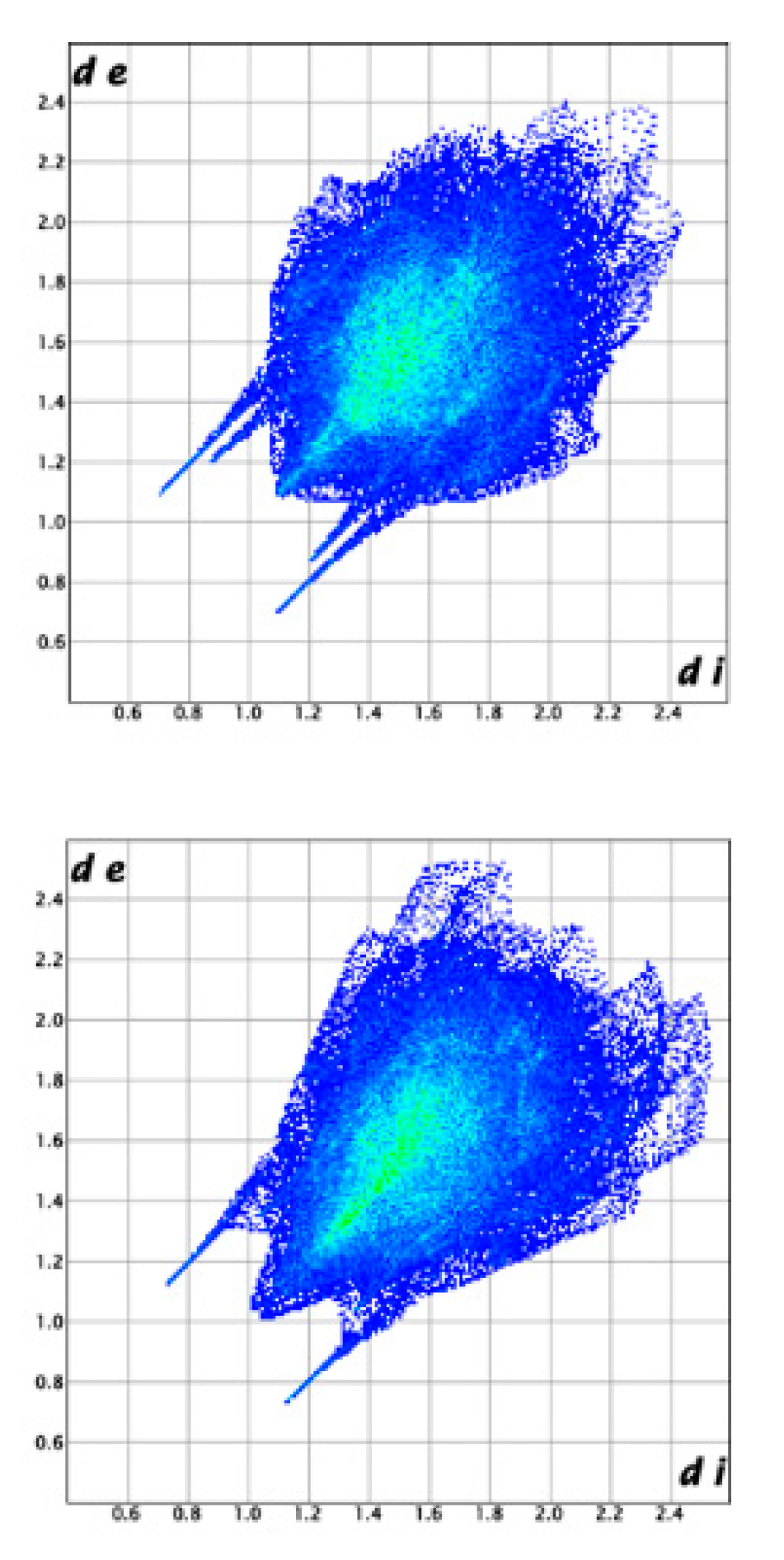
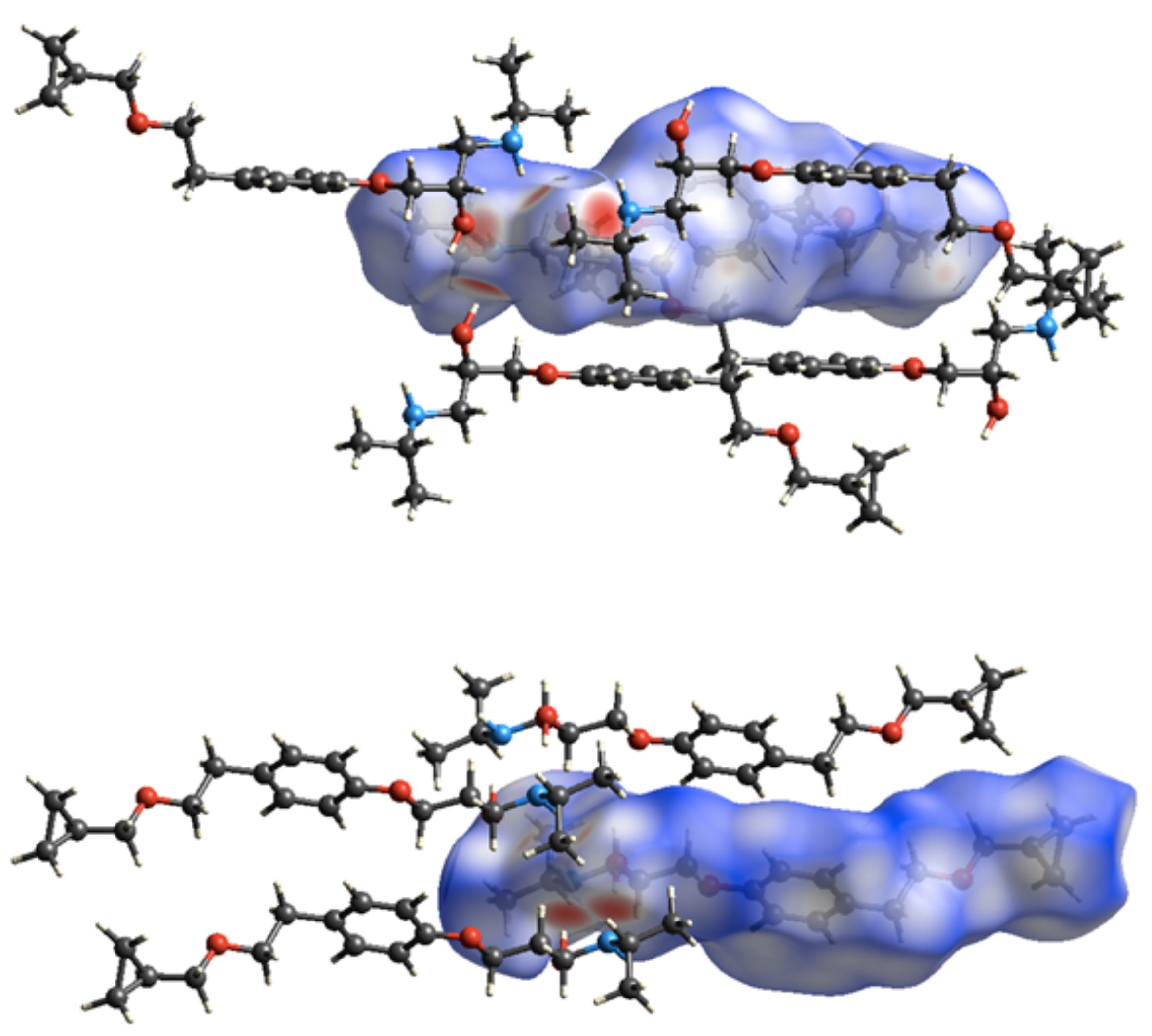
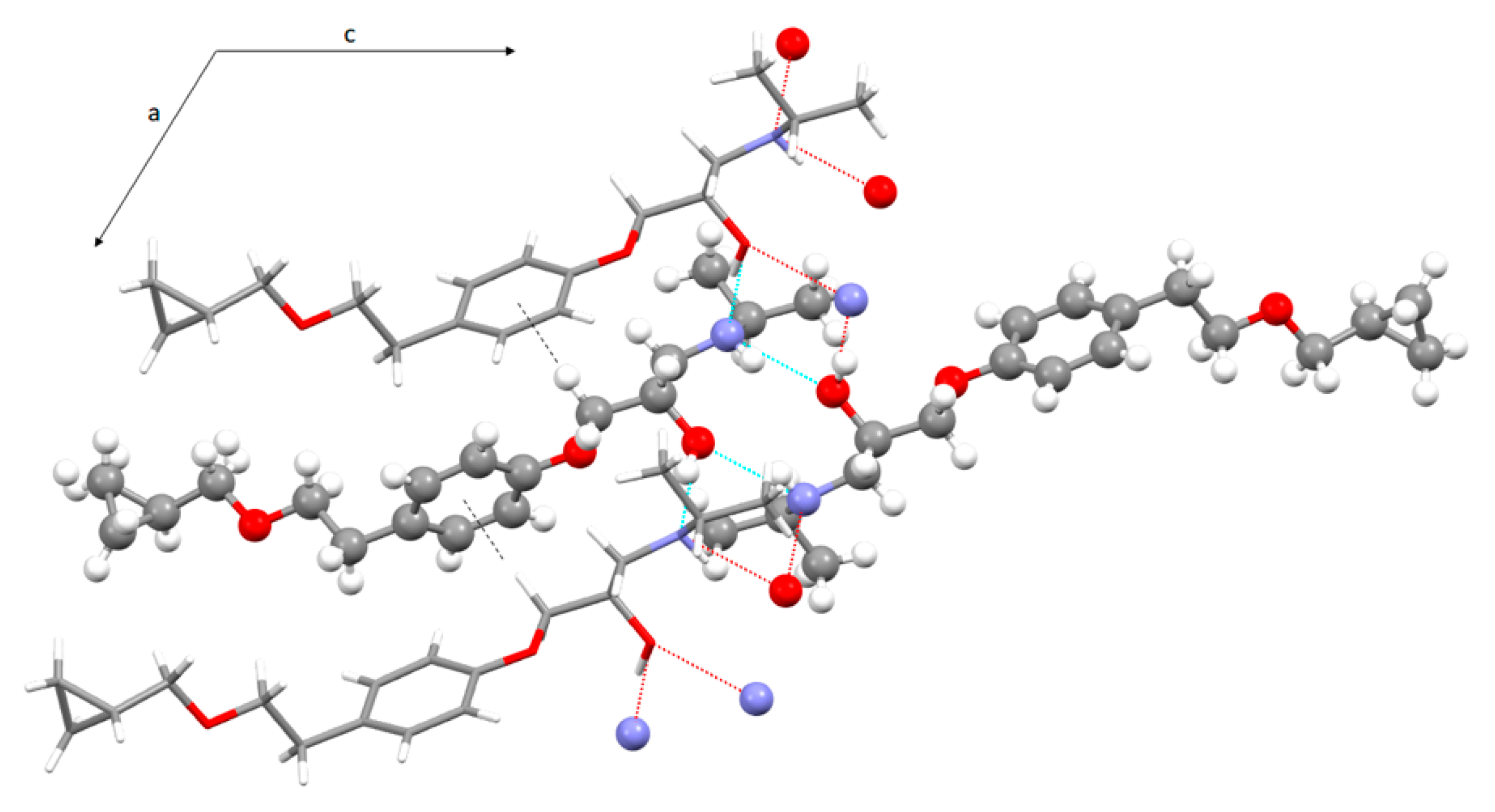
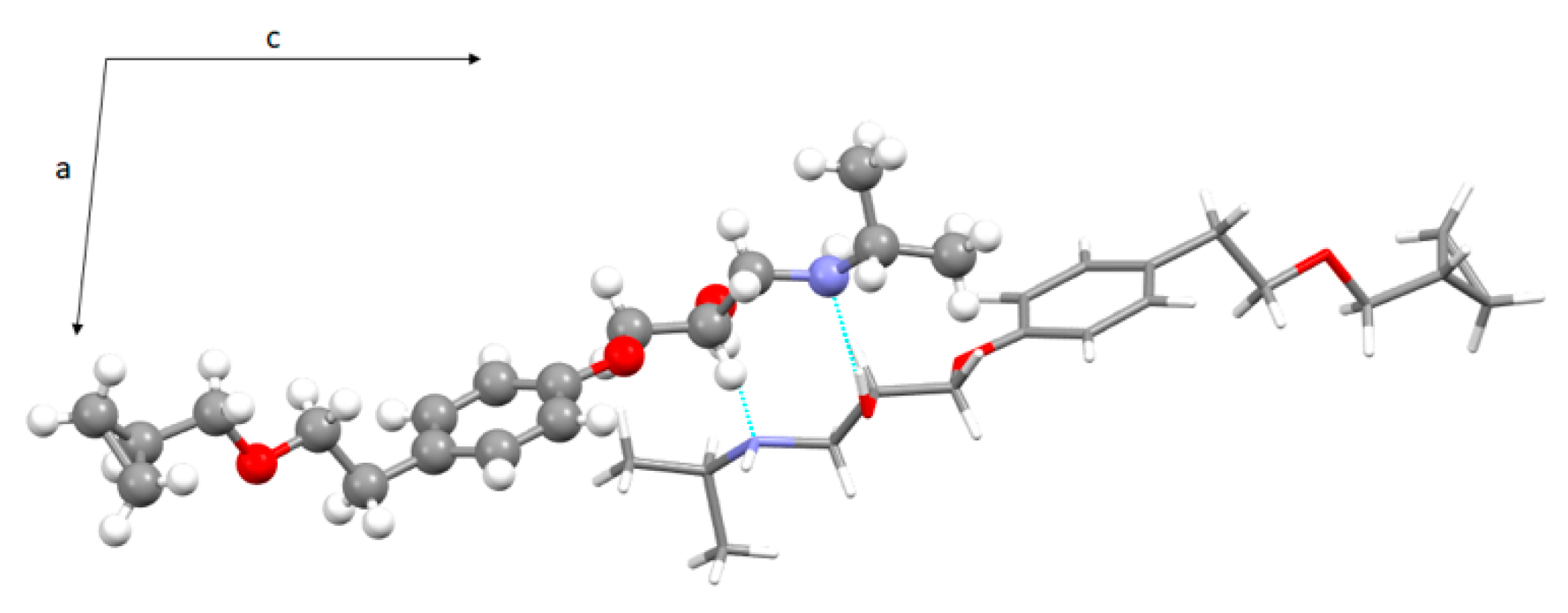
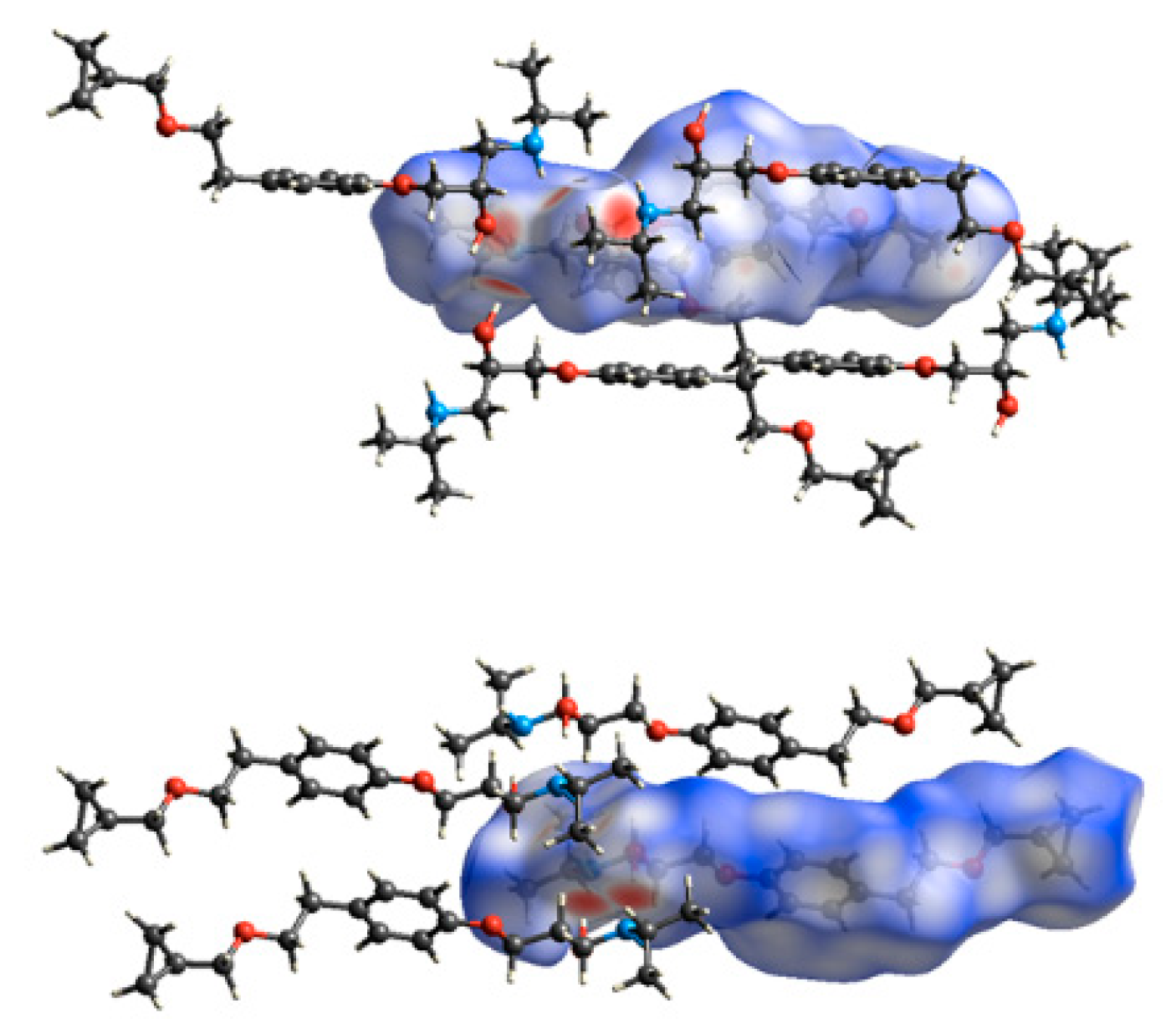
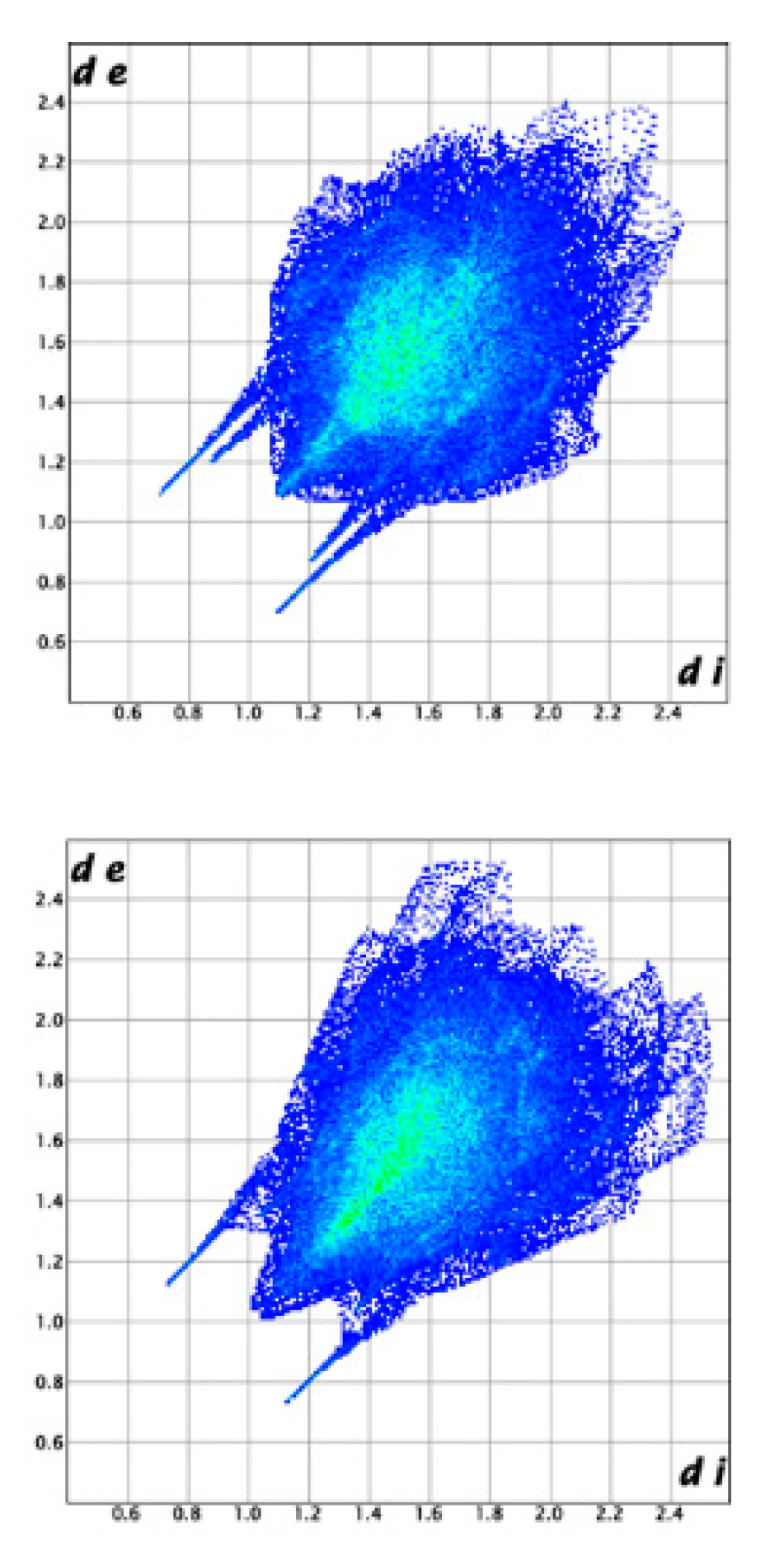
3.3. Crystal Structure from Single Crystal X-Ray Diffraction and Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Prasanthi, N.L.; Sudhir, M.; Jyothi, N.; Sri vajrapriya, V. A review on polymorphism perpetuates pharmaceuticals. Am. J. Adv. Drug Deliv. 2016, 4, 58–63. [Google Scholar]
- Redinha, J.S.; Lopes Jesus, A.J.; Crystal. Crystal Growth of Pharmaceuticals from Melt. In Crystallization and Materials Science of Modern Artificial and Natural Crystals, 1st ed.; Borisenko, E., Kolesnikov, N., Eds.; InTech: Rijeka, Croatia, 2012; Volume 10, pp. 225–248. [Google Scholar]
- Dunitz, J.D.; Bernstein, J. Disappearing polymorphs. Acc. Chem. Res. 1995, 28, 193–200. [Google Scholar] [CrossRef]
- Paoli, P.; Rossi, P.; Chelazzi, L.; Altamura, M.; Fedi, V.; Giannotti, D. Solid State Investigation and Characterization of a Nepadutant Precursor: Polymorphic and Pseudopolymorphic Forms of MEN11282. Cryst. Growth Des. 2016, 16, 5294–5304. [Google Scholar] [CrossRef]
- Rossi, P.; Macedi, E.; Paoli, P.; Bernazzani, L.; Carignani, E.; Borsacchi, S.; Geppi, M. Solid-Solid Transition between Hydrated Racemic Compound and Anhydrous Conglomerate in Na-Ibuprofen: A Combined X-ray Diffraction, Solid-State NMR, Calorimetric, and Computational Study. Cryst. Growth Des. 2014, 14, 2441–2452. [Google Scholar] [CrossRef]
- Amatori, S.; Ambrosi, G.; Borgogelli, E.; Fanelli, M.; Formica, M.; Fusi, V.; Giorgi, L.; Macedi, E.; Micheloni, M.; Paoli, P.; et al. Modulating the Sensor Response to Halide Using NBD-Based Azamacrocycles. Inorg. Chem. 2014, 53, 4560–4569. [Google Scholar] [CrossRef]
- Crociani, B.; Antonaroli, S.; Burattini, M.; Paoli, P.; Rossi, P. Palladium complexes with a tridentate PNO ligand. Synthesis of η1-allyl complexes and cross-coupling reactions promoted by boron compounds. Dalton Trans. 2010, 39, 3665–3672. [Google Scholar] [CrossRef] [PubMed]
- Rossi, P.; Paoli, P.; Ienco, A.; Biagi, D.; Valleri, M.; Conti, L. A new crystal form of the NSAID dexketoprofen. Acta Cryst. 2019, 75, 783–792. [Google Scholar] [CrossRef]
- Paoli, P.; Rossi, P.; Macedi, E.; Ienco, A.; Chelazzi, L.; Bartolucci, G.L.; Bruni, B. Similar but Different: The Case of Metoprolol Tartrate and Succinate Salts. Cryst. Growth Des. 2016, 16, 789–799. [Google Scholar] [CrossRef]
- Rossi, P.; Paoli, P.; Chelazzi, L.; Conti, L.; Bencini, A. Metroprolol Fumarate: Crystal Structure from Powder X-ray Diffraction Data and Comparison with the Tartrate and Succinate Salts. Cryst. Growth Des. 2018, 18, 7015–7026. [Google Scholar] [CrossRef]
- Rossi, P.; Paoli, P.; Chelazzi, L.; Conti, L.; Bencini, A. The solid-state structure of the β-blocker metoprolol: A combined experimental and in silico investigation. Acta Cryst. 2019, C75, 87–96. [Google Scholar]
- Cierpka-Kmieć, K.; Hering, D. Tachycardia: The hidden cardiovascular risk factor in uncomplicated arterial hypertension. Cardiol. J. 2019, in press. [Google Scholar] [CrossRef]
- Mehvar, R.; Brocks, D.R. Stereospecific Pharmacokinetics and Pharmacodynamics of Beta-Adrenergic Blockers in humans. J. Pharm. Pharmaceut. Sci. 2001, 4, 185–200. [Google Scholar]
- Canotilho, J.; Castro, R.A.E.; Rosado, M.T.S.; Silva, M.R.; Beja, A.M.; Paixão, J.A.; Redinha, J.S. The structure of betaxolol from single crystal X-ray diffraction and natural bond orbital analysis. J. Mol. Struct. 2008, 891, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Maria, T.M.R.; Castro, R.A.E.; Silva, M.R.; Ramos, M.L.; Justino, L.L.G.; Burrows, H.D.; Canolitho, J.; Eusébio, M.E.S. Polymorphism and melt crystallisation of racemic betaxolol, a β-adrenergic antagonist drug. J. Therm. Anal. Calorim. 2013, 111, 2171–2178. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—new features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17, version 17; University of Western Australia: Crawley, Australia, 2017. [Google Scholar]
- CrysAlisPro, Oxford Diffraction, version 2011; Agilent Technologies UK Ltd.: Yarnton, UK, 2011.
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; Da Caro, L.; Giacovazzo, C.; Polidori, G.; Spagna, R. SIR2004: An improved tool for crystal structure determination and refinement. J. Appl. Cryst. 2005, 38, 381–388. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT- Integrated space-group and crystal-structure determination. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Nardelli, M. PARST95—An update to PARST: A system of Fortran routines for calculating molecular structure parameters from the results of crystal structure analyses. J. Appl. Cryst. 1995, 28, 659. [Google Scholar] [CrossRef]
- Farrugia, L. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Discovery Studio Visualizer, version 19.1.0.18287; Dassault Systèmes BIOVIA: San Diego, CA, USA, 2019.
- Access Structures. Available online: https://www.ccdc.cam.ac.uk/structures (accessed on 24 September 2019).
- GAUSSIAN09; revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2010.
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Peng, C.; Ayala, P.Y.; Schlegel, H.B.; Frisch, M.J. Using redundant internal coordinates to optimize equilibrium geometries and transition states. J. Comput. Chem. 1996, 17, 49–56. [Google Scholar] [CrossRef]
- Gavezzotti, A. Are Crystal Structures Predictable? Acc. Chem. Res. 1994, 27, 309–314. [Google Scholar] [CrossRef]
- Gavezzotti, A. The Crystal Packing of Organic Molecules: Challenge and Fascination Below 1000 Da. Crystallogr. Rev. 1998, 7, 5–121. [Google Scholar] [CrossRef]
- Canotilho, J.; Castro, R.A.E. The structure of betaxolol studied by infrared spectroscopy and natural bond orbital theory. Spectrochimica Acta Part A. 2010, 76, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Ionescu, C.; Caira, M.R.; Bojita, M.T.; Nassimbeni, L.R.; Mhlongo, W.T. Solid state characterization of metoprolol free base and metoprolol tartrate. FARMACIA 2006, 54, 9–17. [Google Scholar]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond: In Structural Chemistry and Biology, 1st ed.; Oxford Science Publications: Great Britain, UK, 1999. [Google Scholar]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.L. Patterns in hydrogen bonding: Functionality and graph set analysis in crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Ammon, H.L.; Howe, D.-B.; Erhardt, W.D.; Balsamo, A.; Macchia, B.; Macchia, F.; Keefe, W.E. The crystal structures of dichloroisoproterenol, propranolol and propranolol hydrochloride. Acta Cryst. 1977, B33, 21–29. [Google Scholar] [CrossRef]
- Akisanya, J.; Parkins, A.W.; Steed, J.W. A Synthesis of Atenolol Using a Nitrile Hydration Catalyst. Org. Process Res. Dev. 1998, 2, 274–276. [Google Scholar] [CrossRef]
- Ning, F.; Anderson, R.J.; Hibbs, D.E.; Groundwater, P.W. A new, one-step synthesis of 1-heteroaryl-2-alkylaminoethanols. Tetrahedron Lett. 2010, 51, 843–845. [Google Scholar] [CrossRef]
- Mereiter, K.; Frohlich, J.; Ilk, R. CCDC 164293: Experimental Crystal Structure Determination; Cambridge Crystallographic Data Centre: Cambridge, UK, 2001. [Google Scholar] [CrossRef]
- Batey, R.A.; MacKay, D.B.; Santhakumar, V. Alkenyl and Aryl Boronates-Mild Nucleophiles for the Stereoselective Formation of Functionalized N-Heterocycles. J. Am. Chem. Soc. 1999, 121, 5075–5076. [Google Scholar] [CrossRef]
- Palchykov, V.A.; Zarovnaya, I.S.; Tretiakov, S.V.; Reshetnyak, A.V.; Omelchenko, I.V.; Shishkin, O.V.; Okovytyy, S.I. Synthesis and characterization of sulfolane-based amino alcohols: A combined experimental and computational study. J. Mol. Struct. 2018, 1157, 149–158. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula | C18H29NO3 |
|---|---|
| MW | 307.42 |
| T (K) | 100 |
| λ (Å) | 0.71073 |
| Crystal system, space group | Monoclinic, I2/a |
| Unit cell dimensions (Å, °) | a = 9.3146(7) b = 19.619(1); β = 103.281(8) c = 19.898(1) |
| Volume (Å3) | 3539.0(4) |
| Z, Dc (mg/cm3) | 8, 1.154 |
| μ (mm−1) | 0.077 |
| Rwp (%) | 6.87 |
| GOF’s | 1.056 |
| H-Bonds & CH-π Interactions | Distance (Å) | Distance (Å) | Angle (°) |
|---|---|---|---|
| X-H…Y | X…Y | H…Y | X-H…Y |
| O2-H2o...N1 2 | 2.786(3) | 1.84(3) | 167(3) |
| N1-H1n…O2 3 | 3.038(3) | 2.22(3) | 157(2) |
| C-H…π | H…(C6)centroid (Å) | CH…(C6)centroid (°) | |
| C7-H7a…(C1-C6) 4centroid | 2.78(3) | 132(2) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, P.; Paoli, P.; Milazzo, S.; Chelazzi, L.; Ienco, A.; Conti, L. Investigating Differences and Similarities between Betaxolol Polymorphs. Crystals 2019, 9, 509. https://doi.org/10.3390/cryst9100509
Rossi P, Paoli P, Milazzo S, Chelazzi L, Ienco A, Conti L. Investigating Differences and Similarities between Betaxolol Polymorphs. Crystals. 2019; 9(10):509. https://doi.org/10.3390/cryst9100509
Chicago/Turabian StyleRossi, Patrizia, Paola Paoli, Stella Milazzo, Laura Chelazzi, Andrea Ienco, and Luca Conti. 2019. "Investigating Differences and Similarities between Betaxolol Polymorphs" Crystals 9, no. 10: 509. https://doi.org/10.3390/cryst9100509
APA StyleRossi, P., Paoli, P., Milazzo, S., Chelazzi, L., Ienco, A., & Conti, L. (2019). Investigating Differences and Similarities between Betaxolol Polymorphs. Crystals, 9(10), 509. https://doi.org/10.3390/cryst9100509