Rationalization of Lattice Thermal Expansion for Beta-Blocker Organic Crystals





Abstract
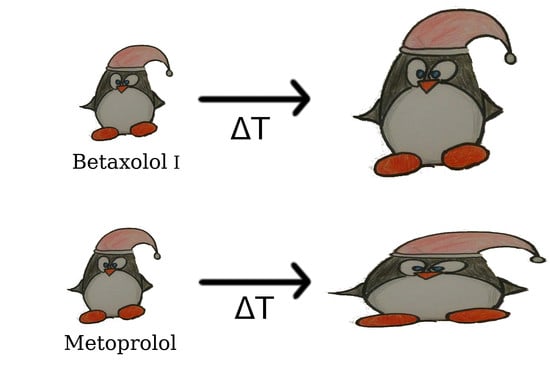
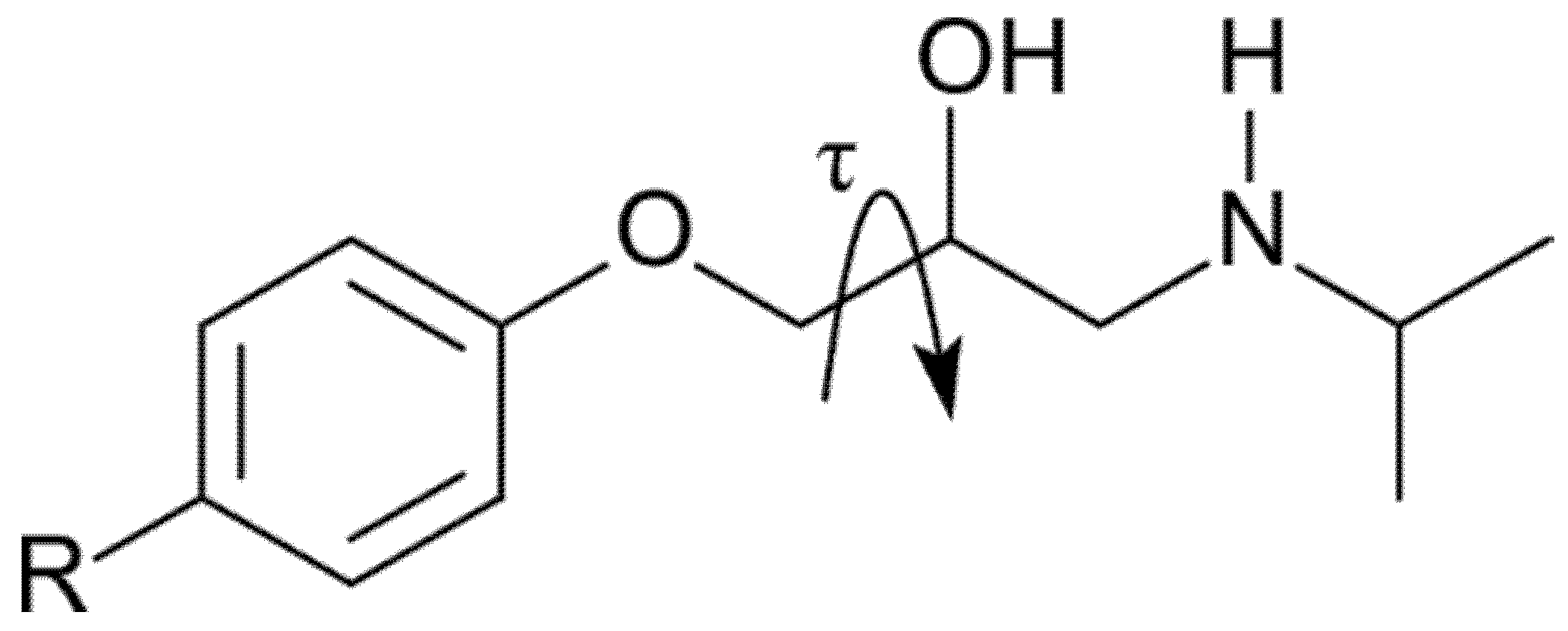
1. Introduction
2. Materials and Methods
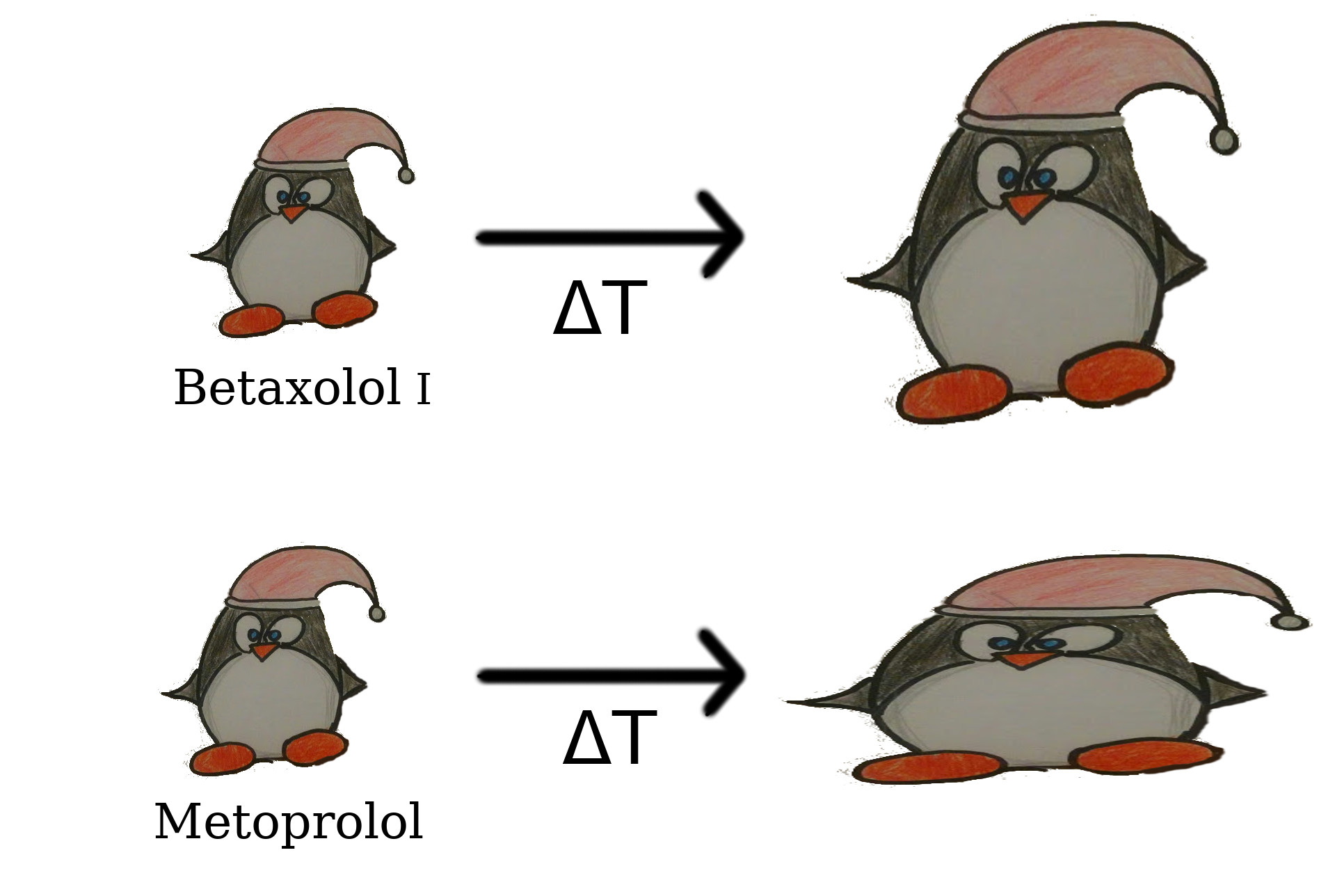
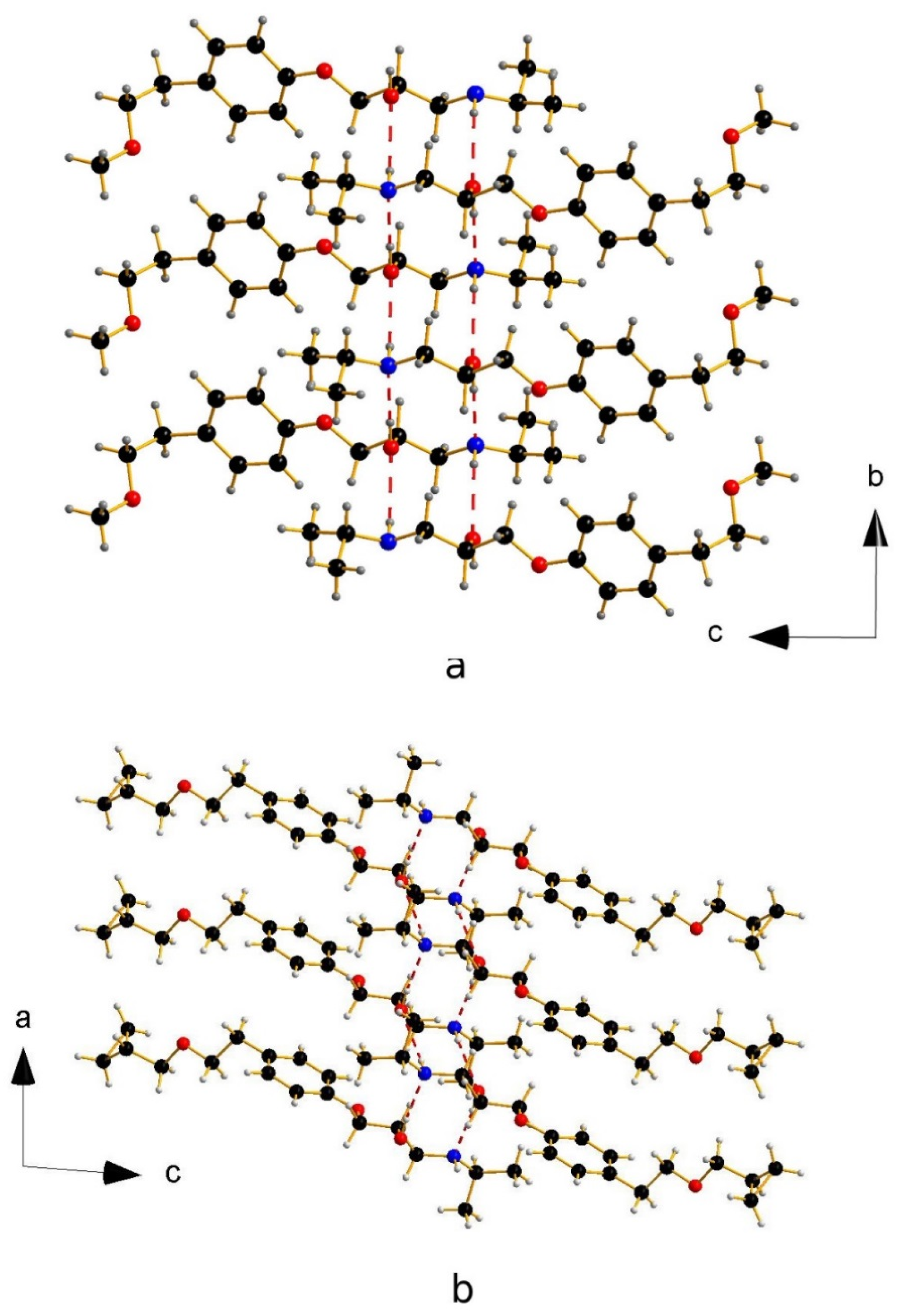
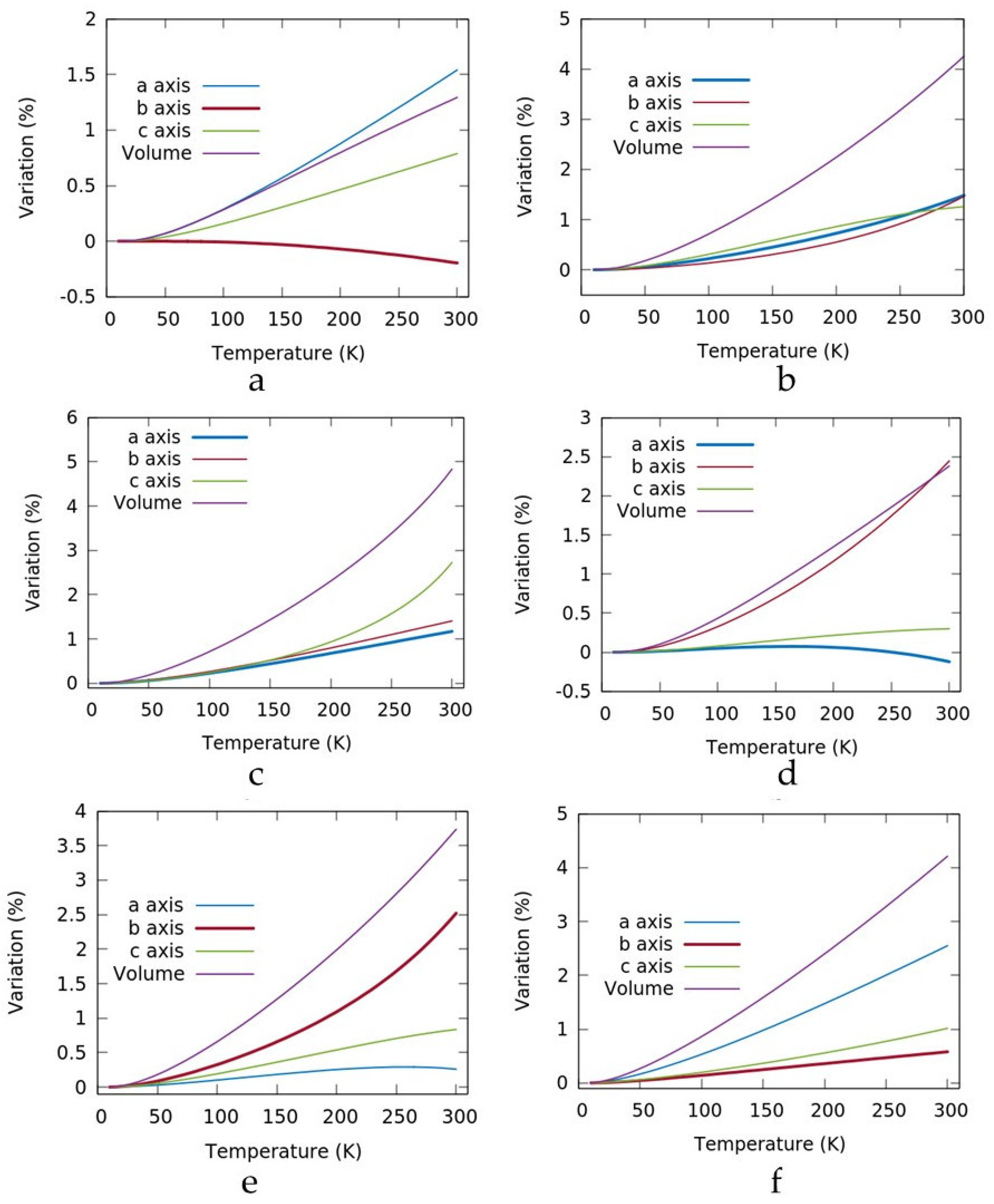
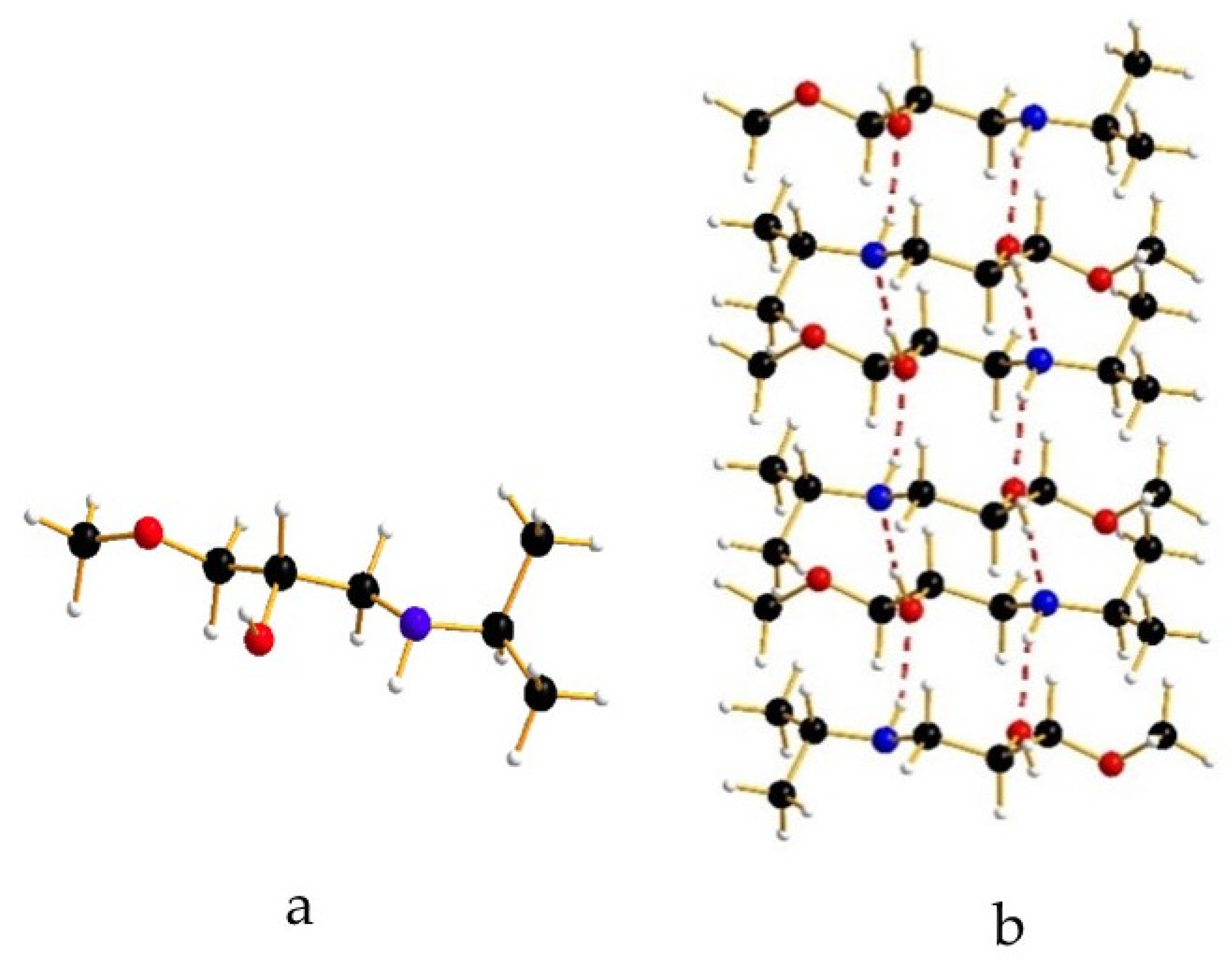
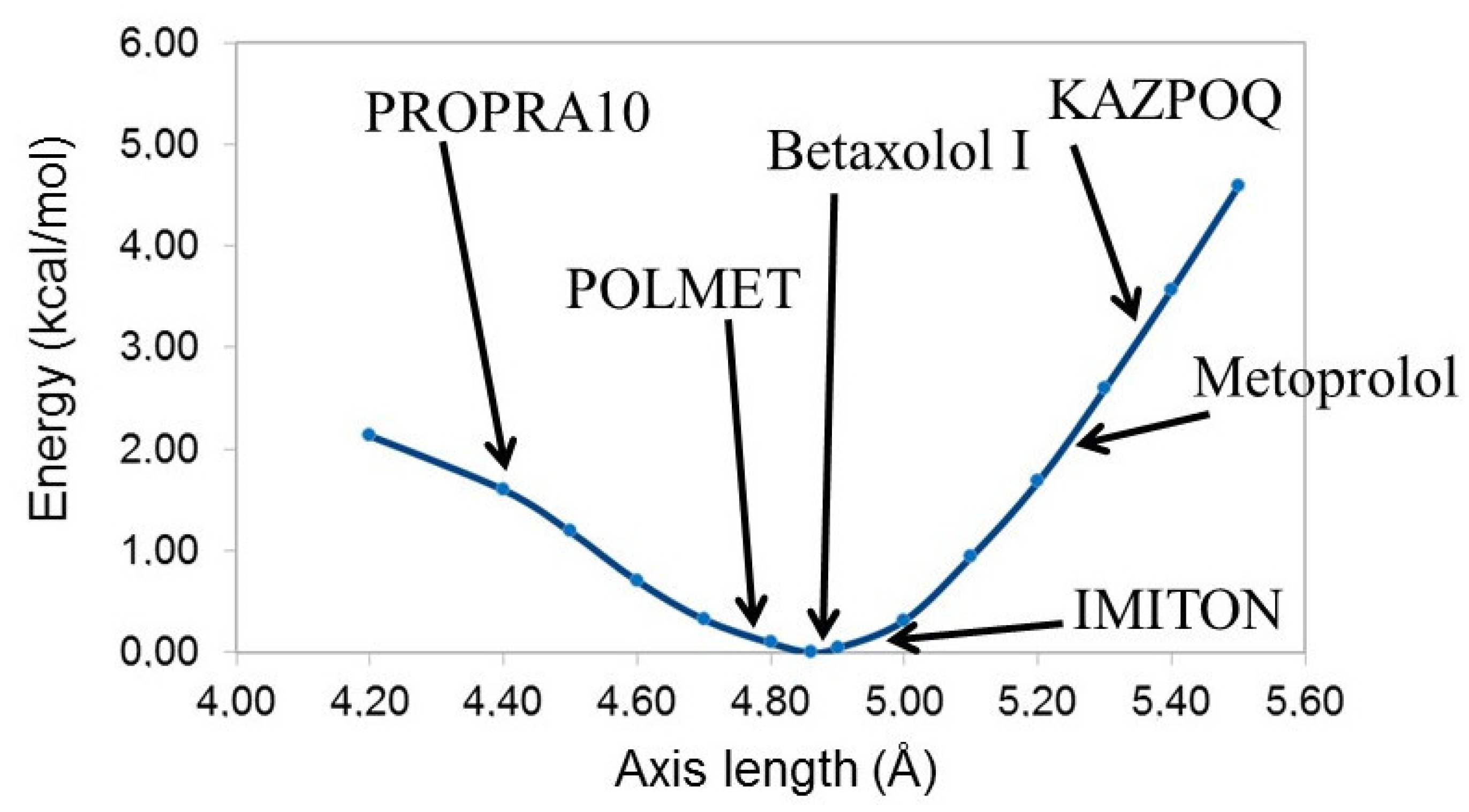
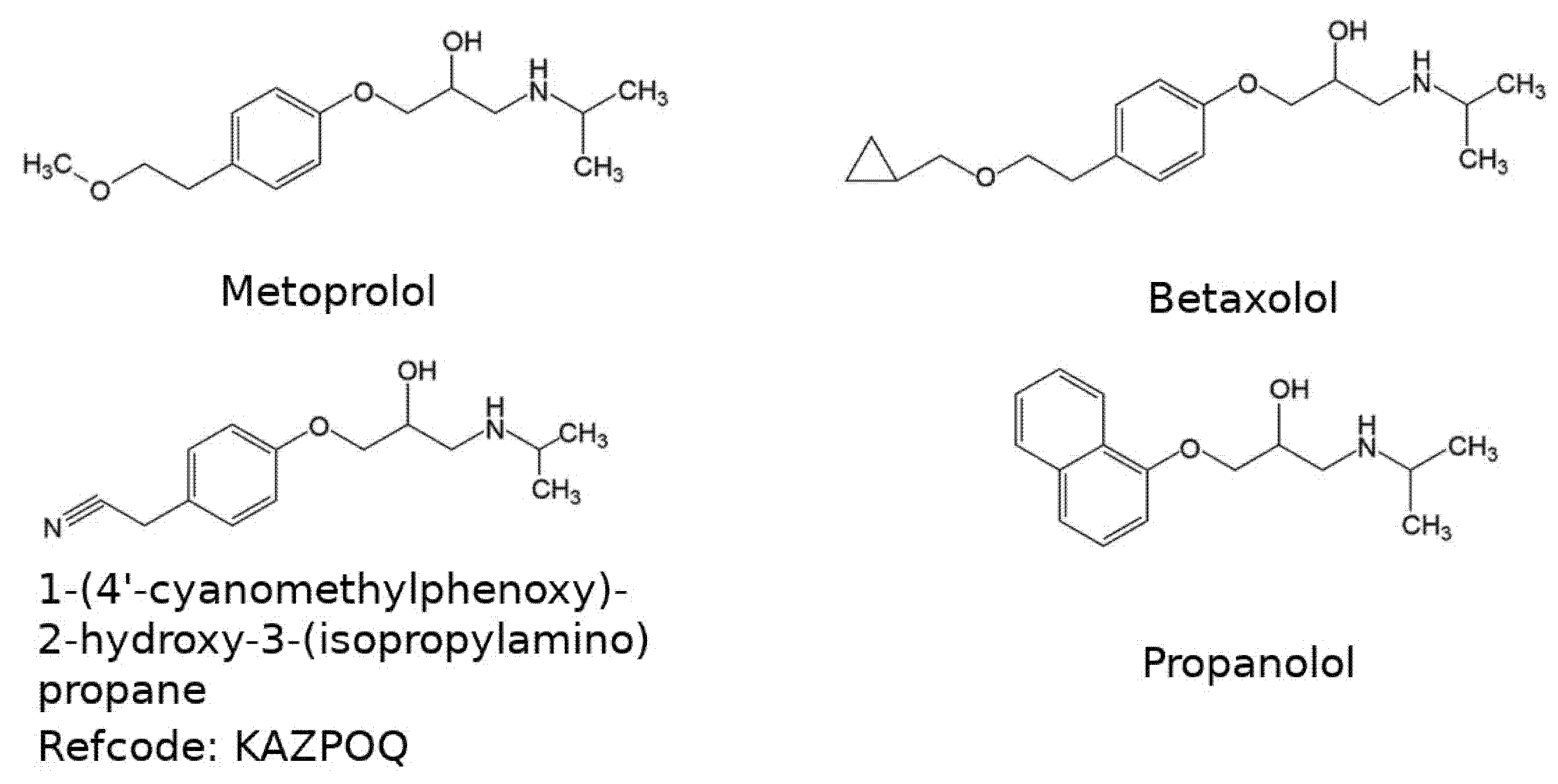
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dunitz, J.D.; Gavezzotti, A. How molecules stick together in organic crystals: weak intermolecular interactions. Chem. Soc. Rev. 2009, 38, 2622–2633. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Maniam, S.; Langford, S.J.; McNeill, C.R. Influence of side-chain length and geometry on the thermal expansion behavior and polymorphism of naphthalene diimide-based thin films. Phys. Rev. Mater. 2019, 3, 013606. [Google Scholar] [CrossRef]
- Abraham, N.S.; Shirts, M.R. Adding Anisotropy to the Standard Quasi-Harmonic Approximation Still Fails in Several Ways to Capture Organic Crystal Thermodynamics. Cryst. Growth Des. 2019, 19, 6911–6924. [Google Scholar] [CrossRef]
- Stephenson, G.A. Anisotropic lattice contraction in pharmaceuticals: The influence of cryo-crystallography on calculated powder diffraction patterns. J. Pharm. Sci. 2006, 95, 821–827. [Google Scholar] [CrossRef]
- Paoli, P.; Rossi, P.; Macedi, E.; Ienco, A.; Chelazzi, L.; Bartolucci, G.L.; Bruni, B. Similar but different: The case of metoprolol tartrate and succinate salts. Cryst. Growth Des. 2016, 16, 789–799. [Google Scholar] [CrossRef]
- Rossi, P.; Paoli, P.; Chelazzi, L.; Conti, L.; Bencini, A. Metroprolol fumarate: crystal structure from powder X-ray diffraction data and comparison with the tartrate and succinate salts. Cryst. Growth Des. 2018, 18, 7015–7026. [Google Scholar] [CrossRef]
- Benfield, P.; Clissold, S.P.; Brogden, R.N. Metoprolol. Drugs 1986, 31, 376–429. [Google Scholar] [CrossRef]
- Rossi, P.; Paoli, P.; Chelazzi, L.; Conti, L.; Bencini, A. The solid-state structure of the β-blocker metoprolol: A combined experimental and in silico investigation. Acta Cryst. C 2019, 75, 87–96. [Google Scholar] [CrossRef]
- Canotilho, J.; Castro, R.A.E.; Rosado, M.T.S.; Ramos Silva, M.; Matos Beja, A.; Paixão, J.A.; Simões Redinha, J. The structure of betaxolol from single crystal X-ray diffraction and natural bond orbital analysis. J. Mol. Struct. 2008, 891, 437–442. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. Patterns in hydrogen bonding: Functionality and graph set analysis in crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Rossi, P.; Paoli, P.; Milazzo, S.; Chelazzi, L.; Ienco, A.; Conti, L. Investigating differences and similarities between betaxolol polymorphs. Crystals 2019, 9, 509. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The cambridge structural database. Acta Cryst. B 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Akisanya, J.; Parkins, A.W.; Steed, J.W. A synthesis of atenolol using a nitrile hydration catalyst. Org. Process Res. Dev. 1998, 2, 274–276. [Google Scholar] [CrossRef]
- Ammon, H.L.; Howe, D.-B.; Erhardt, W.D.; Balsamo, A.; Macchia, B.; Macchia, F.; Keefe, W.E. The crystal structures of dichloroisoproterenol, propranolol and propranolol hydrochloride. Acta Cryst. B 1977, 33, 21–29. [Google Scholar] [CrossRef]
- Bredikhin, A.A.; Savel"ev, D.V.; Bredikhina, Z.A.; Gubaidullin, A.T.; Litvinov, I.A. Crystallization of chiral compounds. 2. Propranolol: Free base and hydrochloride. Russ. Chem. Bull. 2003, 52, 853–861. [Google Scholar] [CrossRef]
- Paoli, P.; Rossi, P.; Chelazzi, L.; Altamura, M.; Fedi, V.; Giannotti, D. Solid state investigation and characterization of a nepadutant precursor: Polymorphic and pseudopolymorphic forms of MEN11282. Cryst. Growth Des. 2016, 16, 5294–5304. [Google Scholar] [CrossRef]
- Rossi, P.; Macedi, E.; Paoli, P.; Bernazzani, L.; Carignani, E.; Borsacchi, S.; Geppi, M. Solid–solid transition between hydrated racemic compound and anhydrous conglomerate in Na-Ibuprofen: A combined X-ray diffraction, solid-state NMR, calorimetric, and computational study. Cryst. Growth Des. 2014, 14, 2441–2452. [Google Scholar] [CrossRef]
- Rossi, P.; Paoli, P.; Ienco, A.; Biagi, D.; Valleri, M.; Conti, L. A new crystal form of the NSAID dexketoprofen. Acta Cryst. C 2019, 75, 783–792. [Google Scholar] [CrossRef]
- Rossi, P.; Paoli, P.; Chelazzi, L.; Milazzo, S.; Biagi, D.; Valleri, M.; Ienco, A.; Valtancoli, B.; Conti, L. Relationships between anhydrous and solvated species of dexketoprofen trometamol: A solid-state point of view. Cryst. Growth Des. 2020, 20, 226–236. [Google Scholar] [CrossRef]
- Amatori, S.; Ambrosi, G.; Borgogelli, E.; Fanelli, M.; Formica, M.; Fusi, V.; Giorgi, L.; Macedi, E.; Micheloni, M.; Paoli, P.; et al. Modulating the sensor response to halide using NBD-based azamacrocycles. Inorg. Chem. 2014, 53, 4560–4569. [Google Scholar] [CrossRef]
- Crociani, B.; Antonaroli, S.; Burattini, M.; Paoli, P.; Rossi, P. Palladium complexes with a tridentate PNO ligand. Synthesis of η1-allyl complexes and cross-coupling reactions promoted by boron compounds. Dalton Trans. 2010, 39, 3665–3672. [Google Scholar] [CrossRef] [PubMed]
- Grirrane, A.; Pastor, A.; Galindo, A.; del Río, D.; Orlandini, A.; Mealli, C.; Ienco, A.; Caneschi, A.; Sanz, J.F. Supramolecular interactions as determining factors of the geometry of metallic building blocks: Tetracarboxylate dimanganese species. Angew. Chem. Int. Ed. 2005, 44, 3429–3432. [Google Scholar] [CrossRef] [PubMed]
- Grirrane, A.; Pastor, A.; Galindo, A.; Álvarez, E.; Mealli, C.; Ienco, A.; Orlandini, A.; Rosa, P.; Caneschi, A.; Barra, A.-L.; et al. Thiodiacetate–manganese chemistry with N ligands: Unique control of the supramolecular arrangement over the metal coordination mode. Chem. A Eur. J. 2011, 17, 10600–10617. [Google Scholar] [CrossRef] [PubMed]
- Mosquera, M.E.G.; Gomez-Sal, P.; Diaz, I.; Aguirre, L.M.; Ienco, A.; Manca, G.; Mealli, C. Intriguing I2 reduction in the iodide for chloride ligand substitution at a Ru(II) complex: Role of mixed trihalides in the redox mechanism. Inorg. Chem. 2016, 55, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Canossa, S.; Bacchi, A.; Graiff, C.; Pelagatti, P.; Predieri, G.; Ienco, A.; Manca, G.; Mealli, C. Hierarchy of supramolecular arrangements and building blocks: Inverted paradigm of crystal engineering in the unprecedented metal coordination of methylene blue. Inorg. Chem. 2017, 56, 3512–3516. [Google Scholar] [CrossRef] [PubMed]
- Ienco, A.; Manca, G.; Peruzzini, M.; Mealli, C. Modelling strategies for the covalent functionalization of 2D phosphorene. Dalton Trans. 2018, 47, 17243–17256. [Google Scholar] [CrossRef]
- Klimeš, J.; Michaelides, A. Perspective: Advances and challenges in treating van der Waals dispersion forces in density functional theory. J. Chem. Phys. 2012, 137, 120901. [Google Scholar] [CrossRef]
- Grimme, S.; Hansen, A.; Brandenburg, J.G.; Bannwarth, C. Dispersion-corrected mean-field electronic structure methods. Chem. Rev. 2016, 116, 5105–5154. [Google Scholar] [CrossRef]
- Hoja, J.; Reilly, A.M.; Tkatchenko, A. First-principles modeling of molecular crystals: Structures and stabilities, temperature and pressure. WIREs Comput. Mol. Sci. 2017, 7, e1294. [Google Scholar] [CrossRef]
- Otero-de-la-Roza, A.; Johnson, E.R. A benchmark for non-covalent interactions in solids. J. Chem. Phys. 2012, 137, 054103. [Google Scholar] [CrossRef]
- Reilly, A.M.; Tkatchenko, A. Understanding the role of vibrations, exact exchange, and many-body van der waals interactions in the cohesive properties of molecular crystals. J. Chem. Phys. 2013, 139, 024705. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, J.G.; Grimme, S. Organic crystal polymorphism: A benchmark for dispersion corrected mean field electronic structure methods. Acta Cryst. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, J.G.; Maas, T.; Grimme, S. Benchmarking DFT and semiempirical methods on structures and lattice energies for ten ice polymorphs. J. Chem. Phys. 2015, 142, 124104. [Google Scholar] [CrossRef] [PubMed]
- Cutini, M.; Civalleri, B.; Corno, M.; Orlando, R.; Brandenburg, J.G.; Maschio, L.; Ugliengo, P. Assessment of different quantum mechanical methods for the prediction of structure and cohesive energy of molecular crystals. J. Chem. Theory Comput. 2016, 12, 3340–3352. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, J.G.; Potticary, J.; Sparkes, H.A.; Price, S.L.; Hall, S.R. Thermal expansion of carbamazepine: Systematic crystallographic measurements challenge quantum chemical calculations. J. Phys. Chem. Lett. 2017, 8, 4319–4324. [Google Scholar] [CrossRef] [PubMed]
- Sure, R.; Grimme, S. Corrected small basis set hartree-fock method for large systems. J. Comput. Chem. 2013, 34, 1672–1685. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL. WIREs Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Erba, A. On combining temperature and pressure effects on structural properties of crystals with standard ab initio techniques. J. Chem. Phys. 2014, 141, 124115. [Google Scholar] [CrossRef]
- Erba, A.; Maul, J.; Itou, M.; Dovesi, R.; Sakurai, Y. Anharmonic thermal oscillations of the electron momentum distribution in lithium fluoride. Phys. Rev. Lett. 2015, 115, 117402. [Google Scholar] [CrossRef]
- Erba, A.; Shahrokhi, M.; Moradian, R.; Dovesi, R. On how differently the quasi-harmonic approximation works for two isostructural crystals: Thermal properties of periclase and lime. J. Chem. Phys. 2015, 142, 044114. [Google Scholar] [CrossRef]
- Erba, A.; Maul, J.; De La Pierre, M.; Dovesi, R. Structural and elastic anisotropy of crystals at high pressures and temperatures from quantum mechanical methods: The case of Mg2SiO4 forsterite. J. Chem. Phys. 2015, 142, 204502. [Google Scholar] [CrossRef] [PubMed]
- Ponti, G.; Palombi, F.; Abate, D.; Ambrosino, F.; Aprea, G.; Bastianelli, T.; Beone, F.; Bertini, R.; Bracco, G.; Caporicci, M.; et al. The role of medium size facilities in the HPC ecosystem: The case of the new CRESCO4 cluster integrated in the ENEAGRID infrastructure. In Proceedings of the 2014 International Conference on High Performance Computing Simulation (HPCS), Bologna, Italy, 21–25 July 2014; pp. 1030–1033. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | b | C | ||
|---|---|---|---|---|
| Metoprolol | 16.2368 | 5.2503 | 21.6807 | 10K |
| 16.4869 | 5.2400 | 21.8515 | 300K | |
| 0.54 | −0.20 | 0.79 | % | |
| Betaxolol I | 4.8626 | 9.6063 | 19.0185 | 10K |
| 4.9347 | 9.7471 | 19.2575 | 300K | |
| 1.48 | 1.46 | 1.26 | % | |
| POLMET | 4.7727 | 9.7570 | 17.4059 | 10K |
| 4.8286 | 9.8942 | 17.8805 | 300K | |
| 1.17 | 1.41 | 2.72 | % | |
| KAZPOQ | 5.3453 | 7.5992 | 16.2182 | 10K |
| 5.3388 | 7.7851 | 16.2669 | 300K | |
| −0.12 | 2.45 | 0.30 | % | |
| PROPRA10 | 11.4123 | 4.4098 | 26.5950 | 10K |
| 11.4418 | 4.5209 | 26.8169 | 300K | |
| 0.26 | 2.52 | 0.83 | % | |
| IMITON | 11.8377 | 4.9171 | 12.9885 | 10K |
| 12.1394 | 4.9455 | 13.0292 | 300K | |
| 2.54 | 0.58 | 1.01 | % |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paoli, P.; Milazzo, S.; Rossi, P.; Ienco, A. Rationalization of Lattice Thermal Expansion for Beta-Blocker Organic Crystals. Crystals 2020, 10, 350. https://doi.org/10.3390/cryst10050350
Paoli P, Milazzo S, Rossi P, Ienco A. Rationalization of Lattice Thermal Expansion for Beta-Blocker Organic Crystals. Crystals. 2020; 10(5):350. https://doi.org/10.3390/cryst10050350
Chicago/Turabian StylePaoli, Paola, Stella Milazzo, Patrizia Rossi, and Andrea Ienco. 2020. "Rationalization of Lattice Thermal Expansion for Beta-Blocker Organic Crystals" Crystals 10, no. 5: 350. https://doi.org/10.3390/cryst10050350
APA StylePaoli, P., Milazzo, S., Rossi, P., & Ienco, A. (2020). Rationalization of Lattice Thermal Expansion for Beta-Blocker Organic Crystals. Crystals, 10(5), 350. https://doi.org/10.3390/cryst10050350