Review of the Magnetocaloric Effect in RMnO3 and RMn2O5 Multiferroic Crystals

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
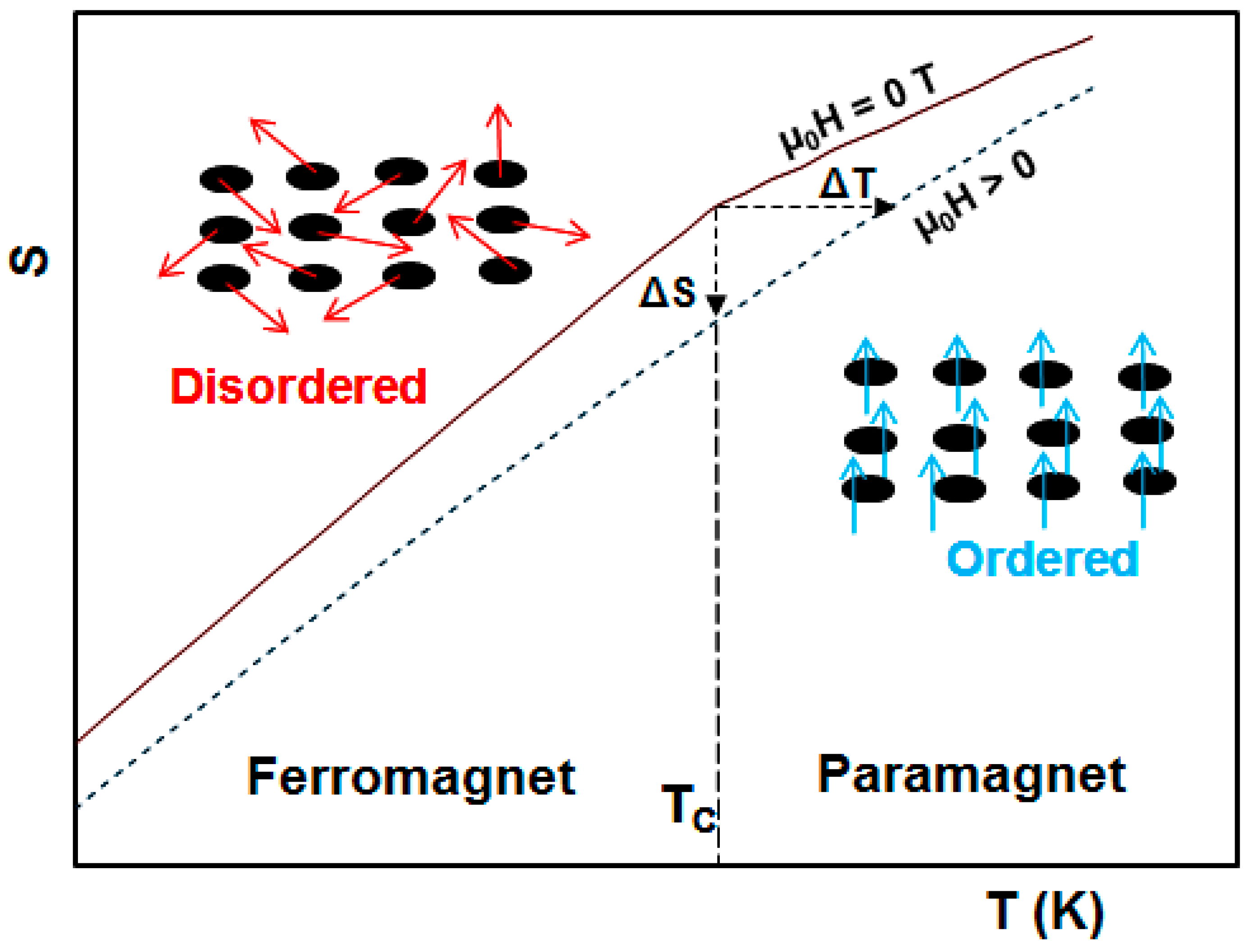
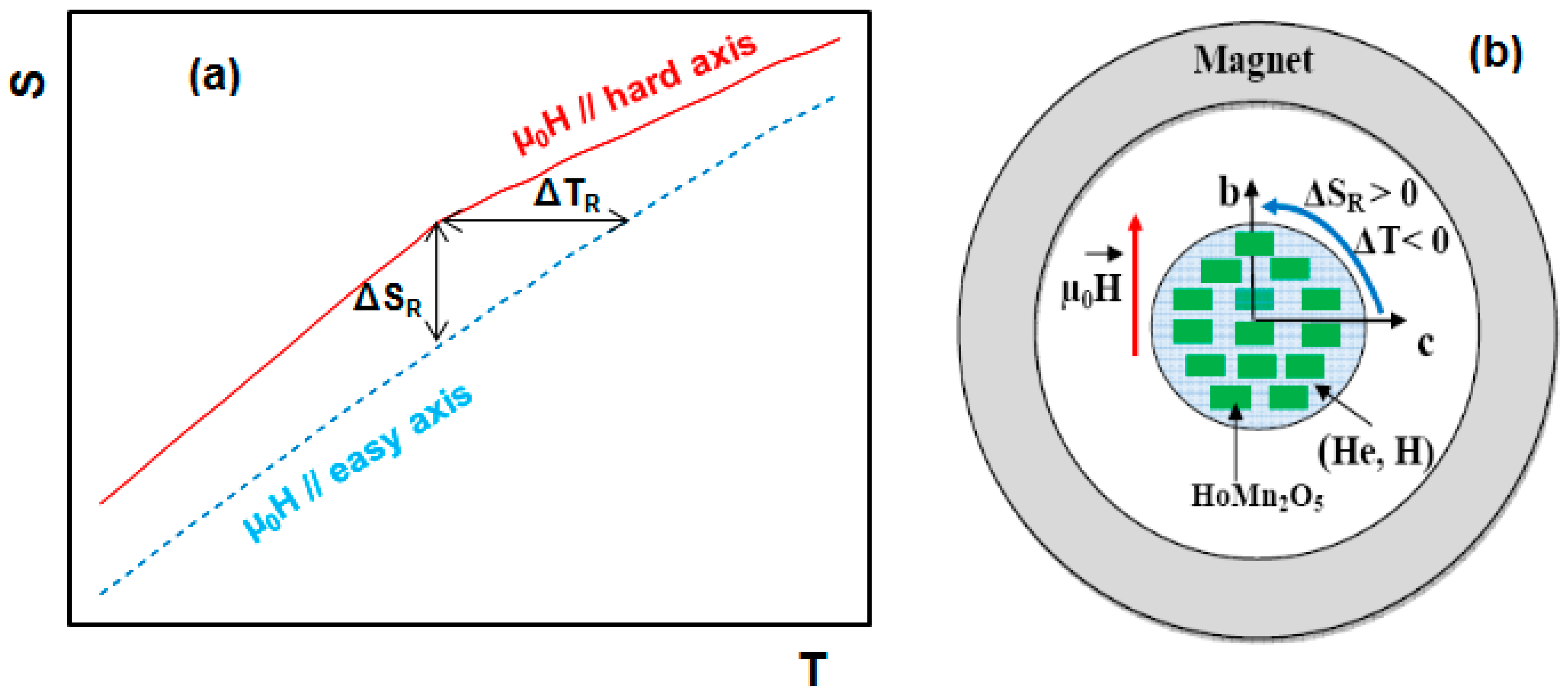
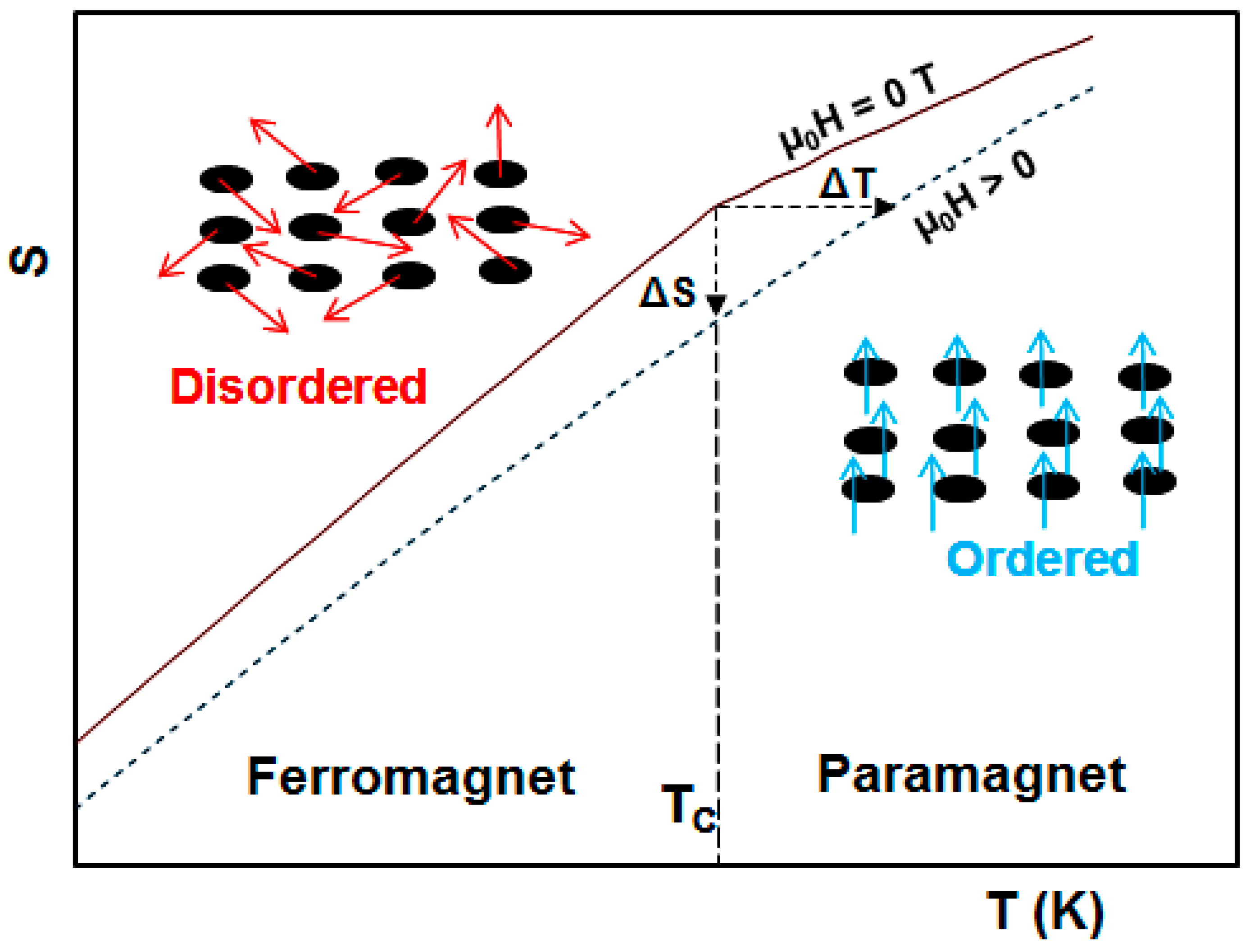
2. Magnetocaloric Effect: Theoretical Aspects
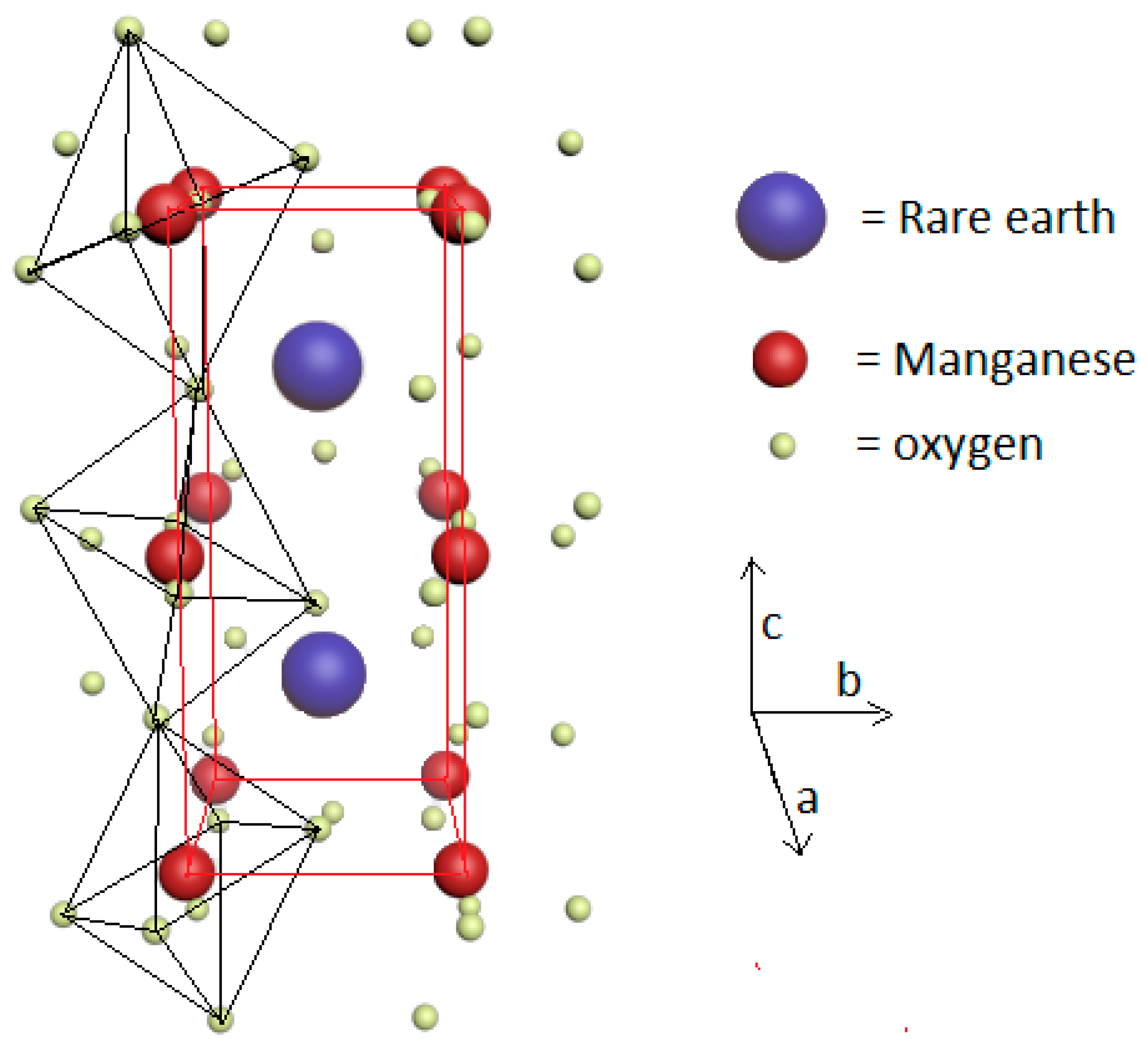
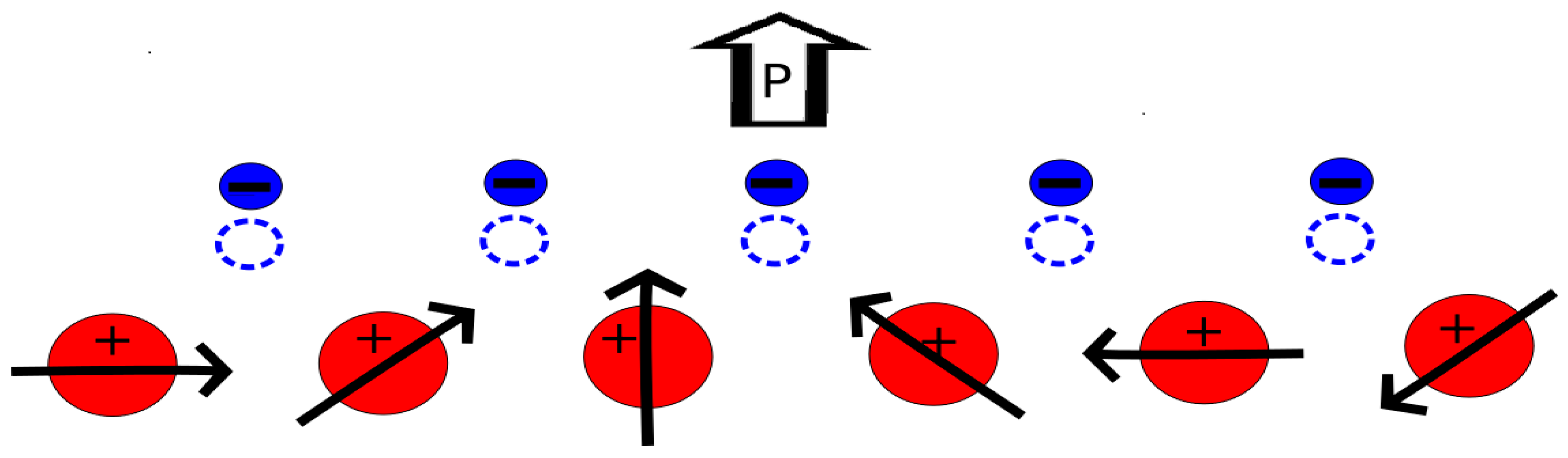
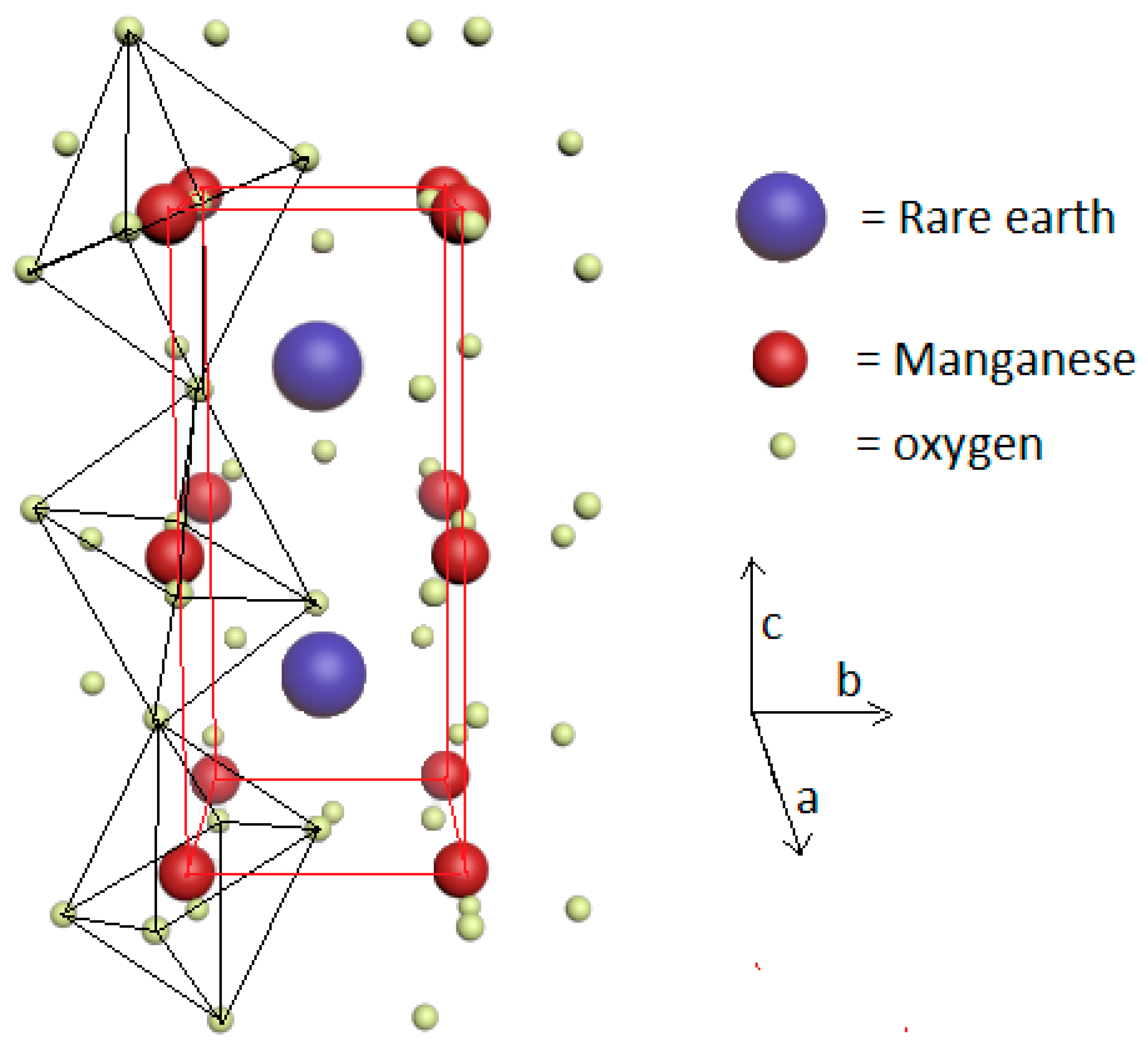
3. Brief Description of the Magnetoelectric Interplay in RMnO3 and RMn2O5 Multiferroics
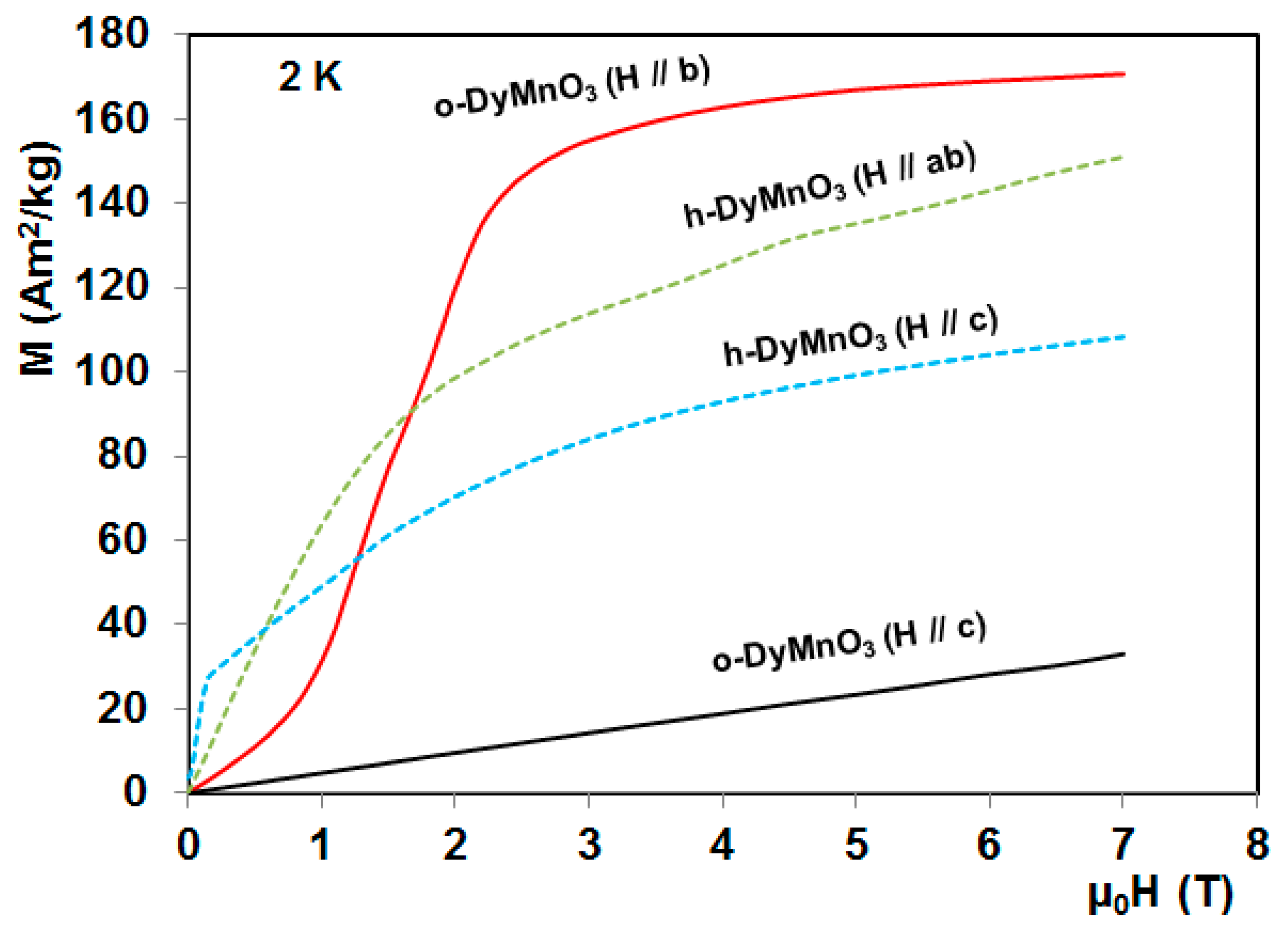
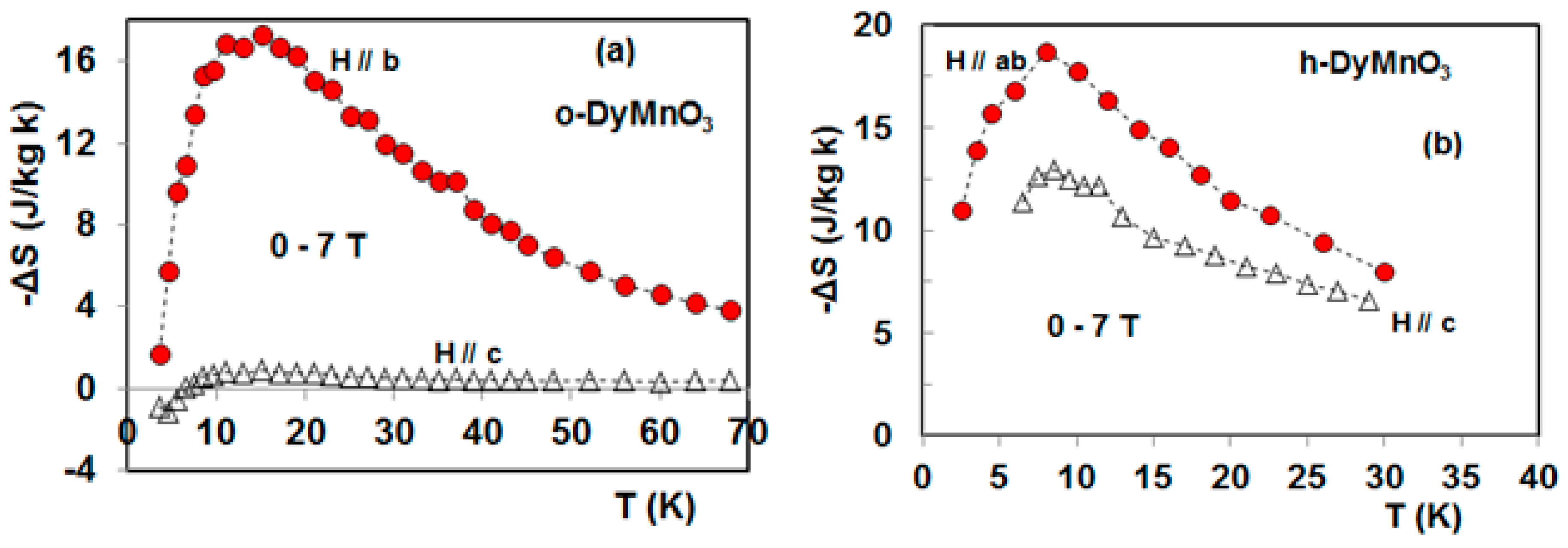
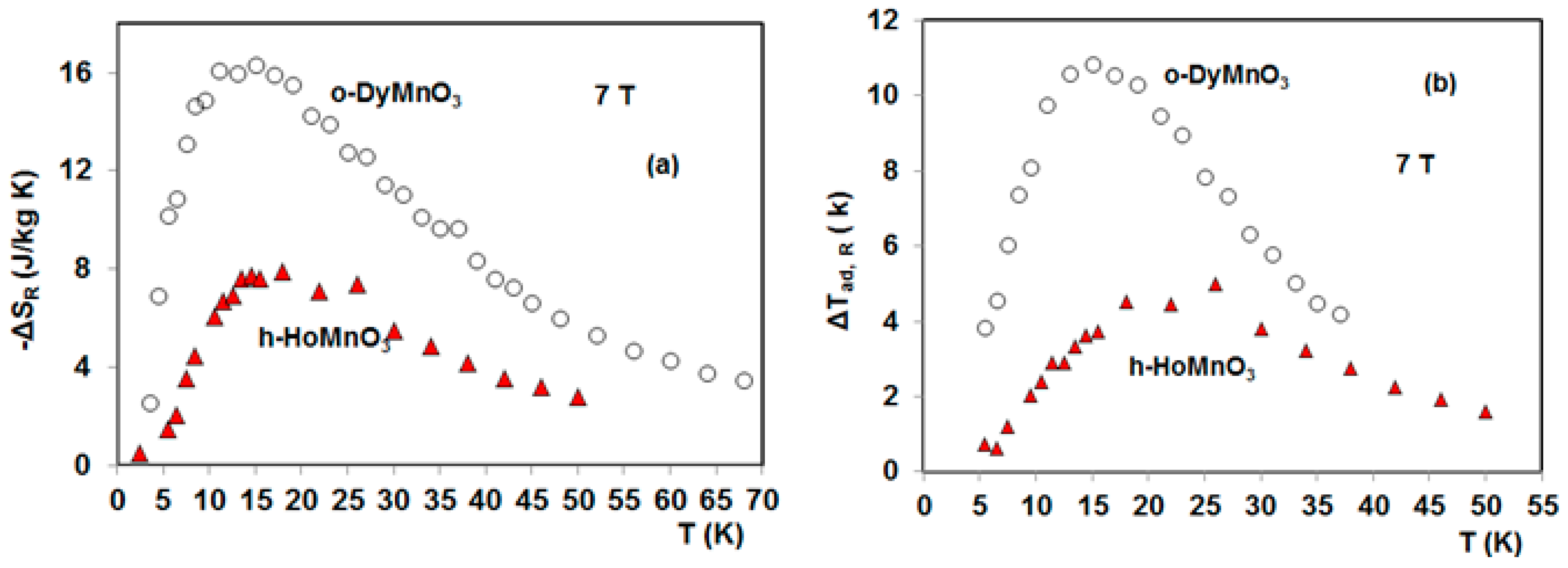
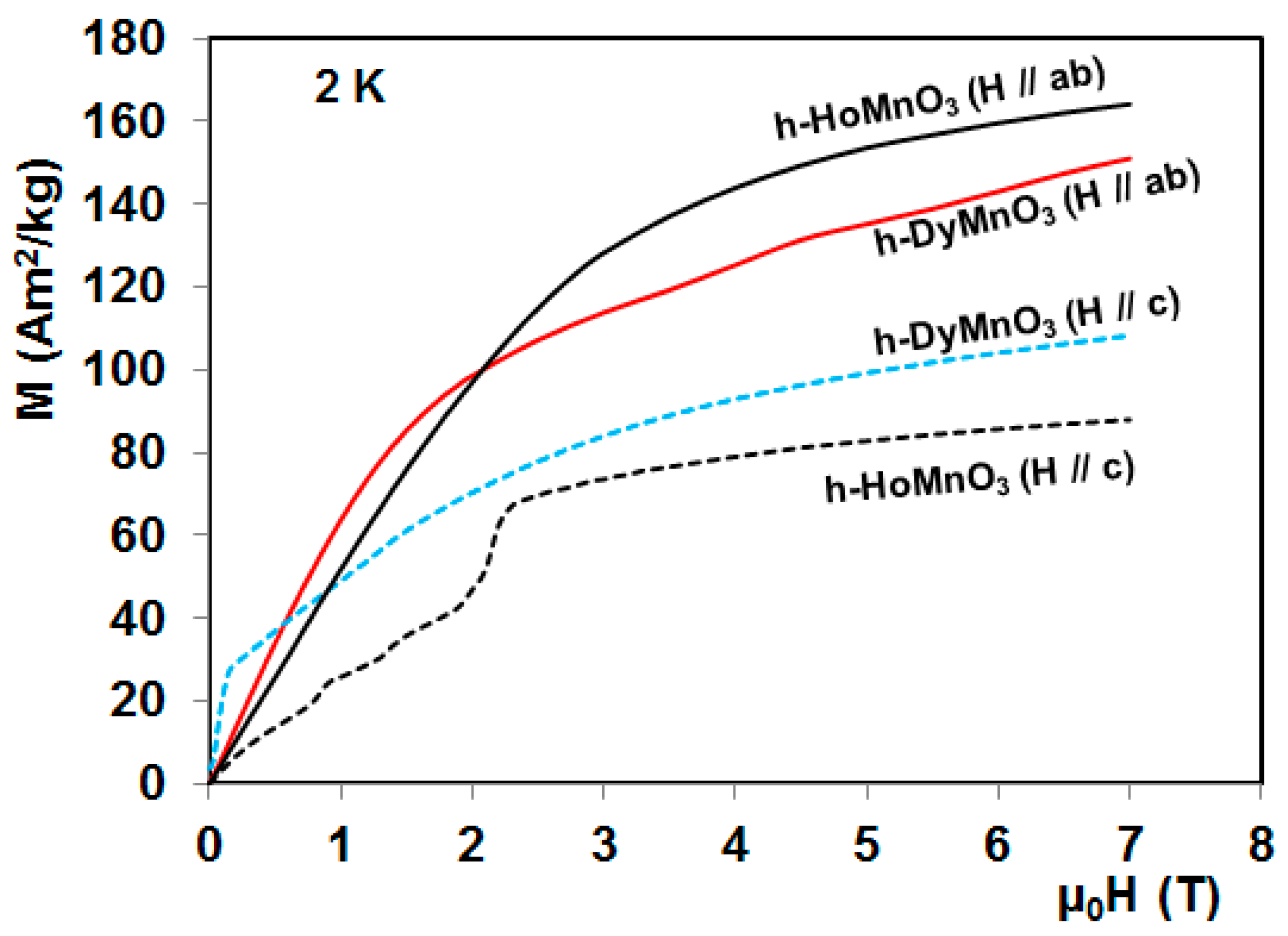
4. Magnetocaloric Properties of RMnO3 Multiferroic Crystals
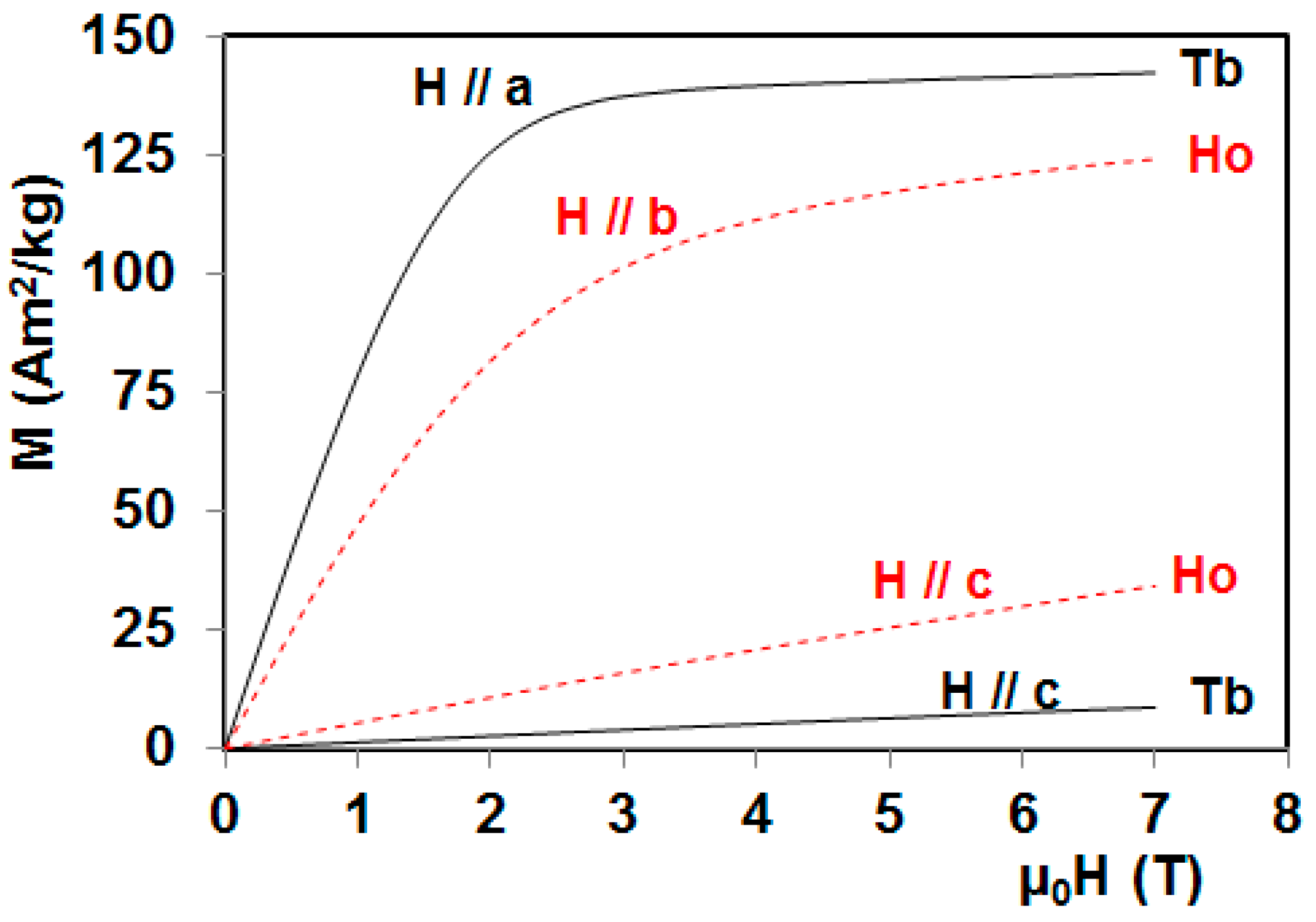
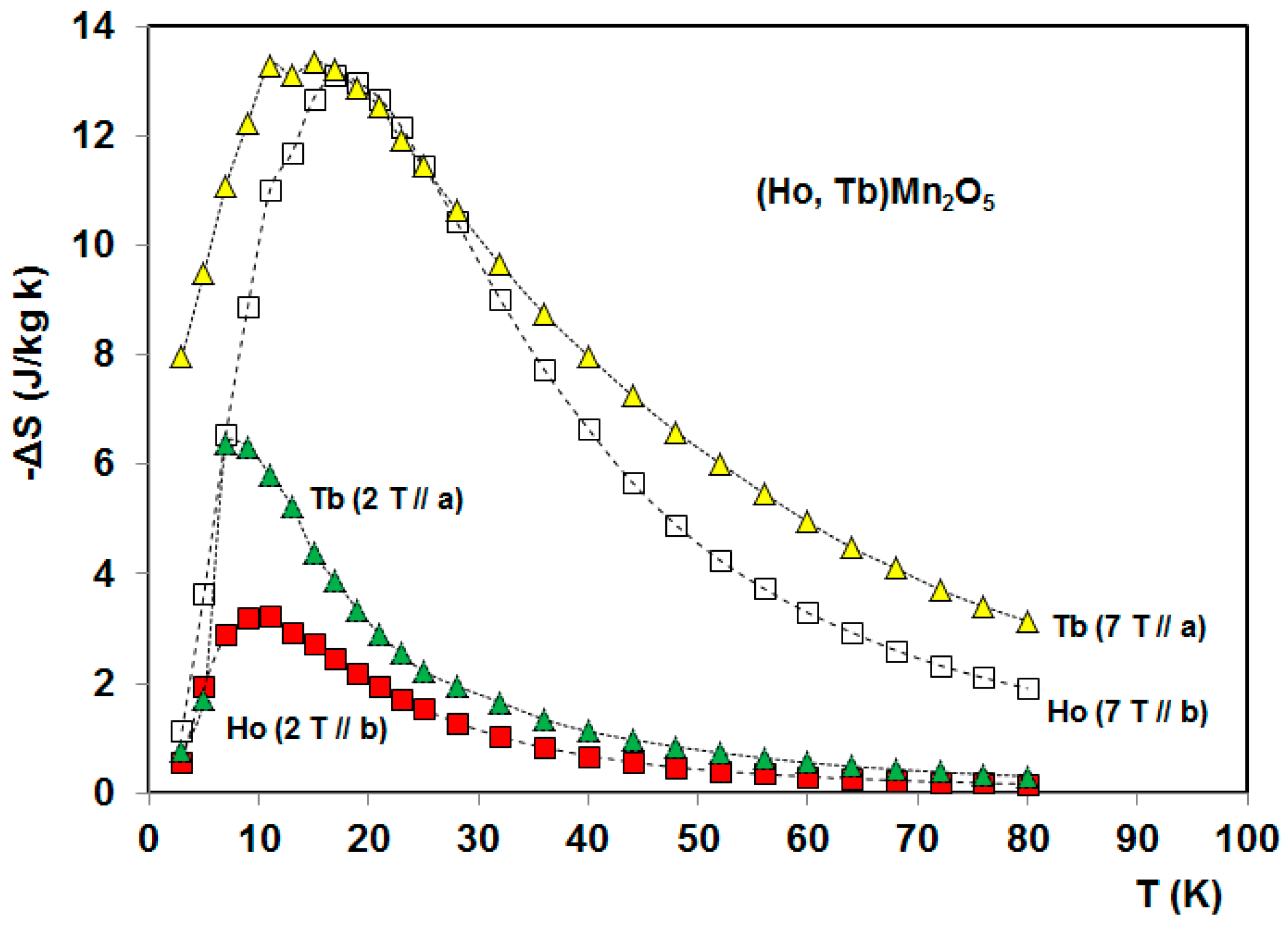
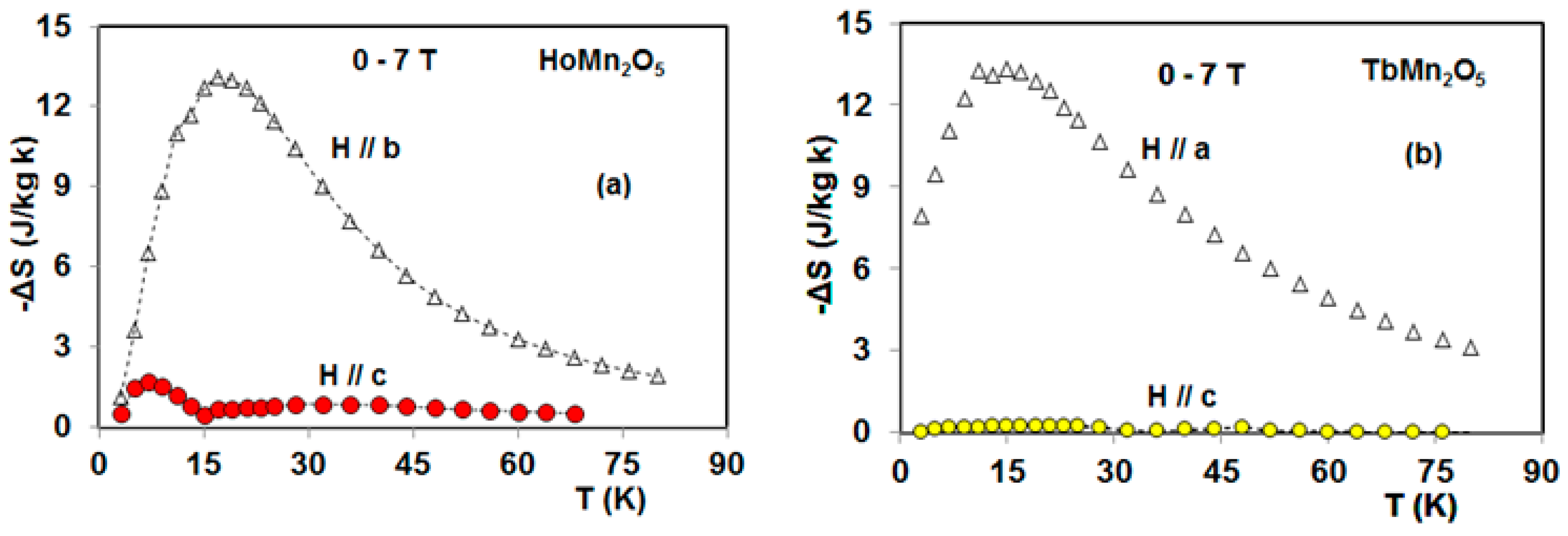
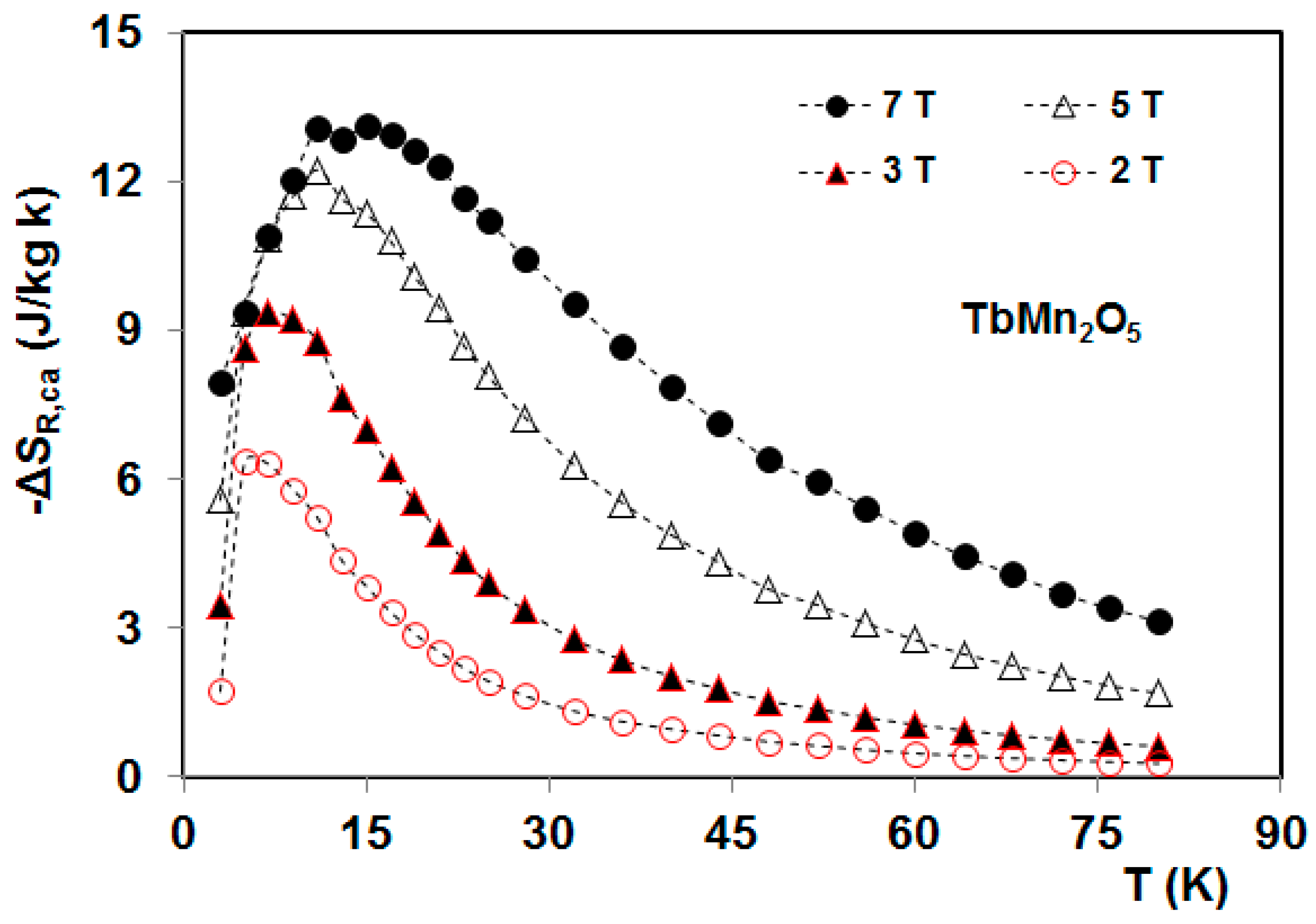
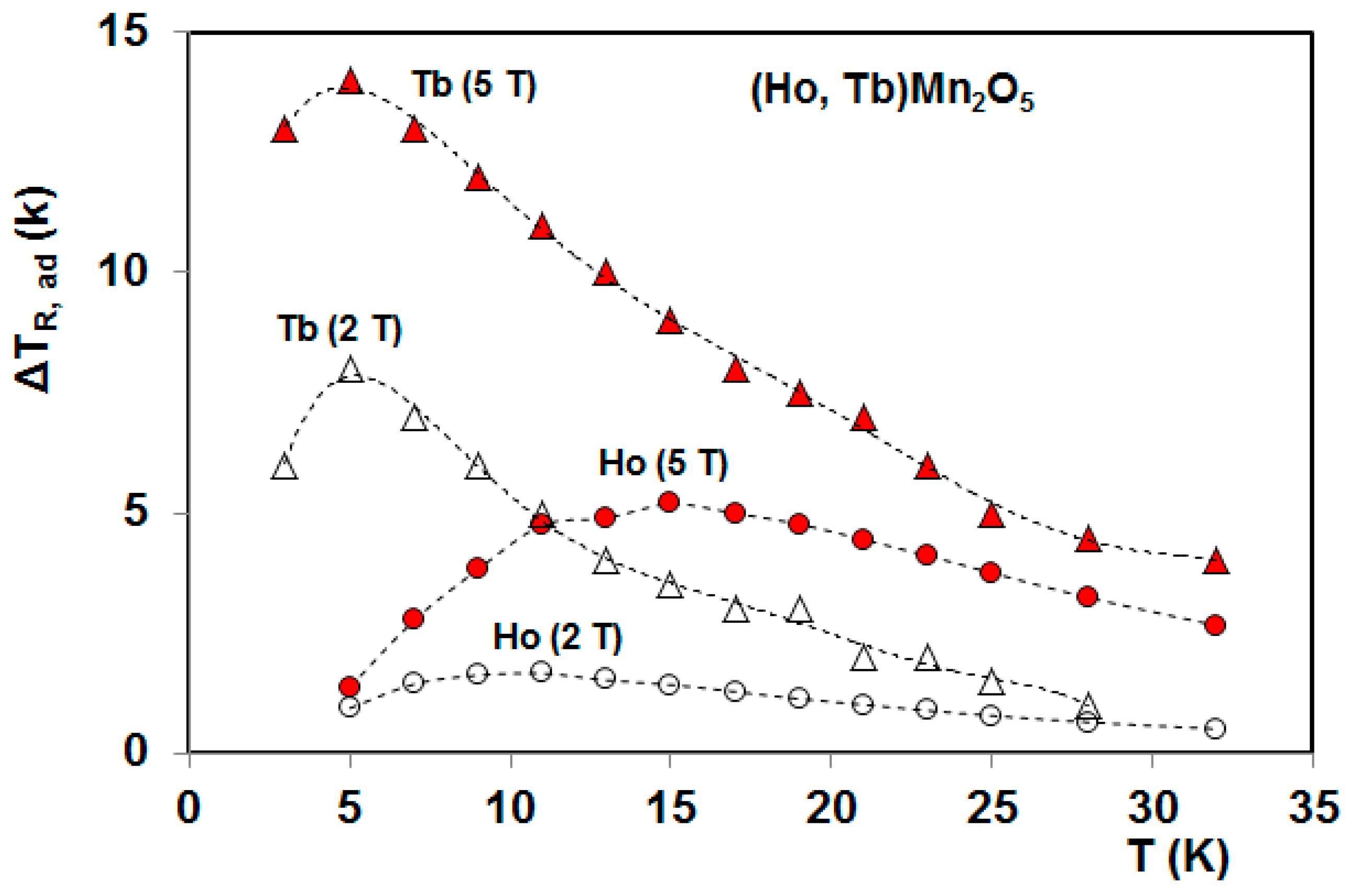
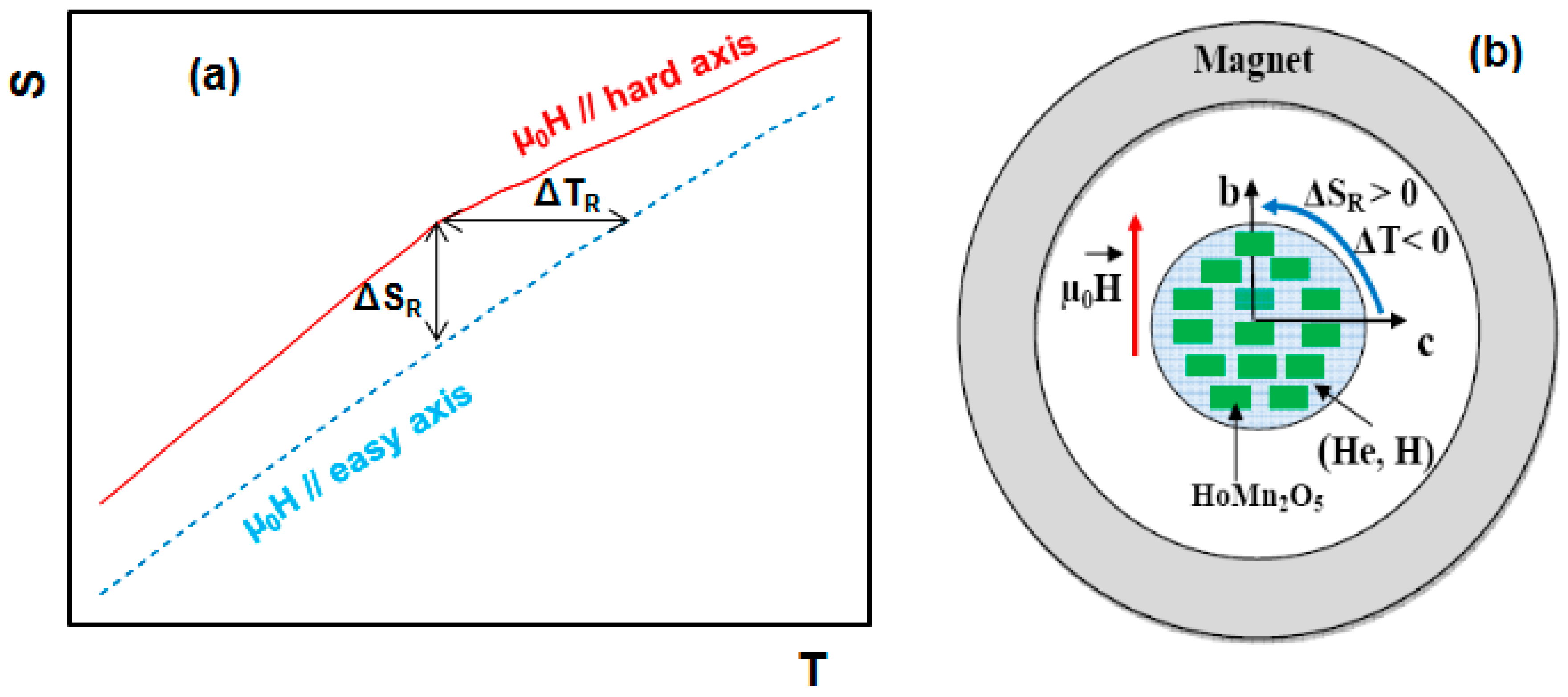
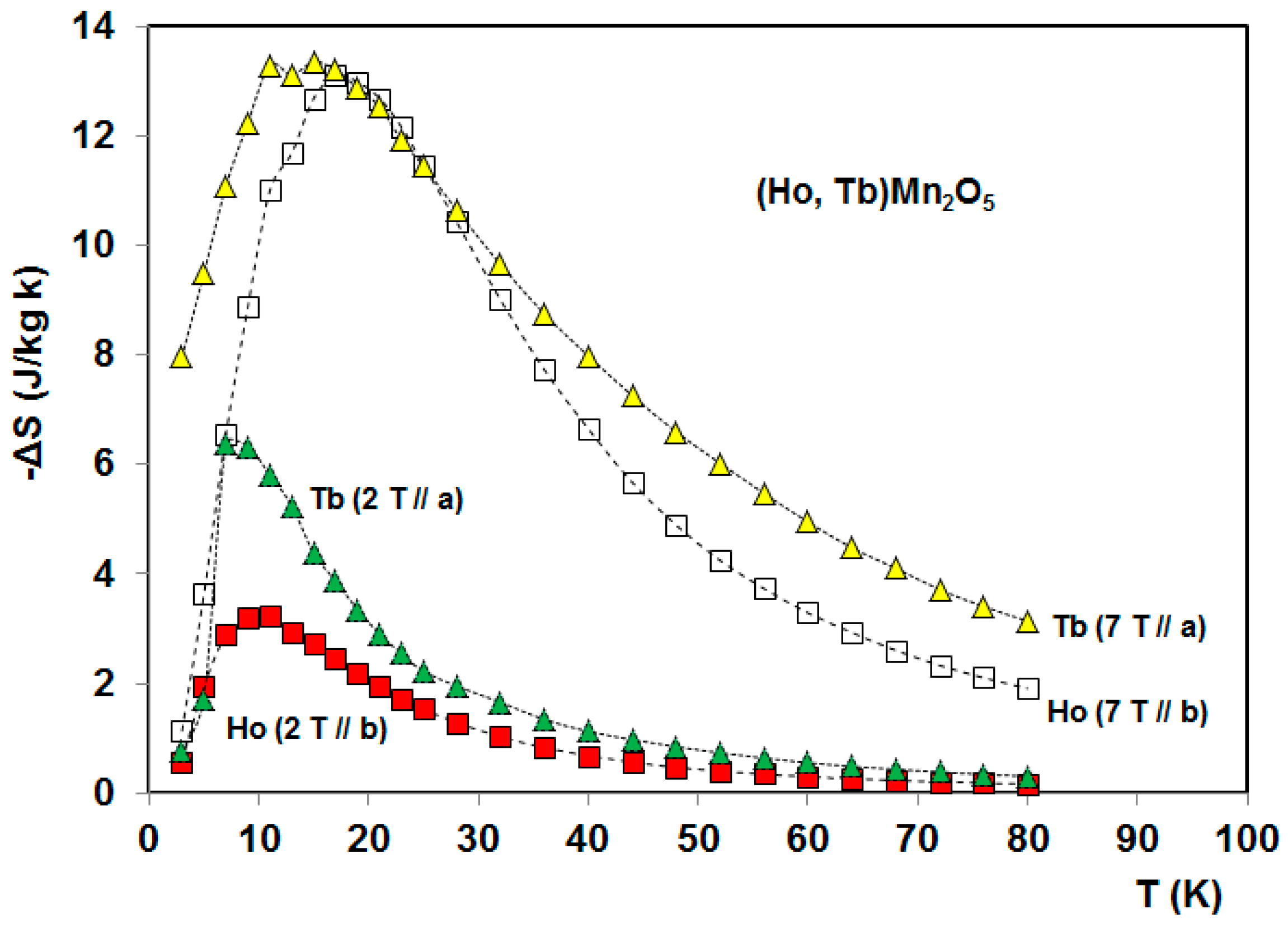
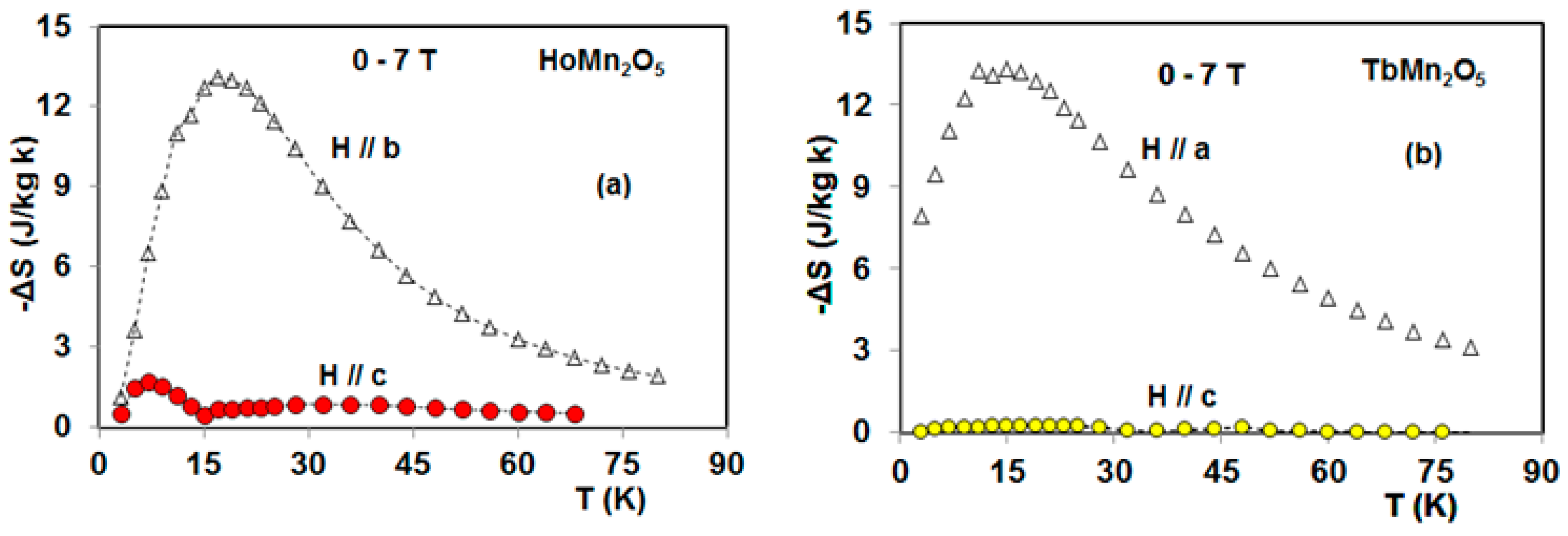
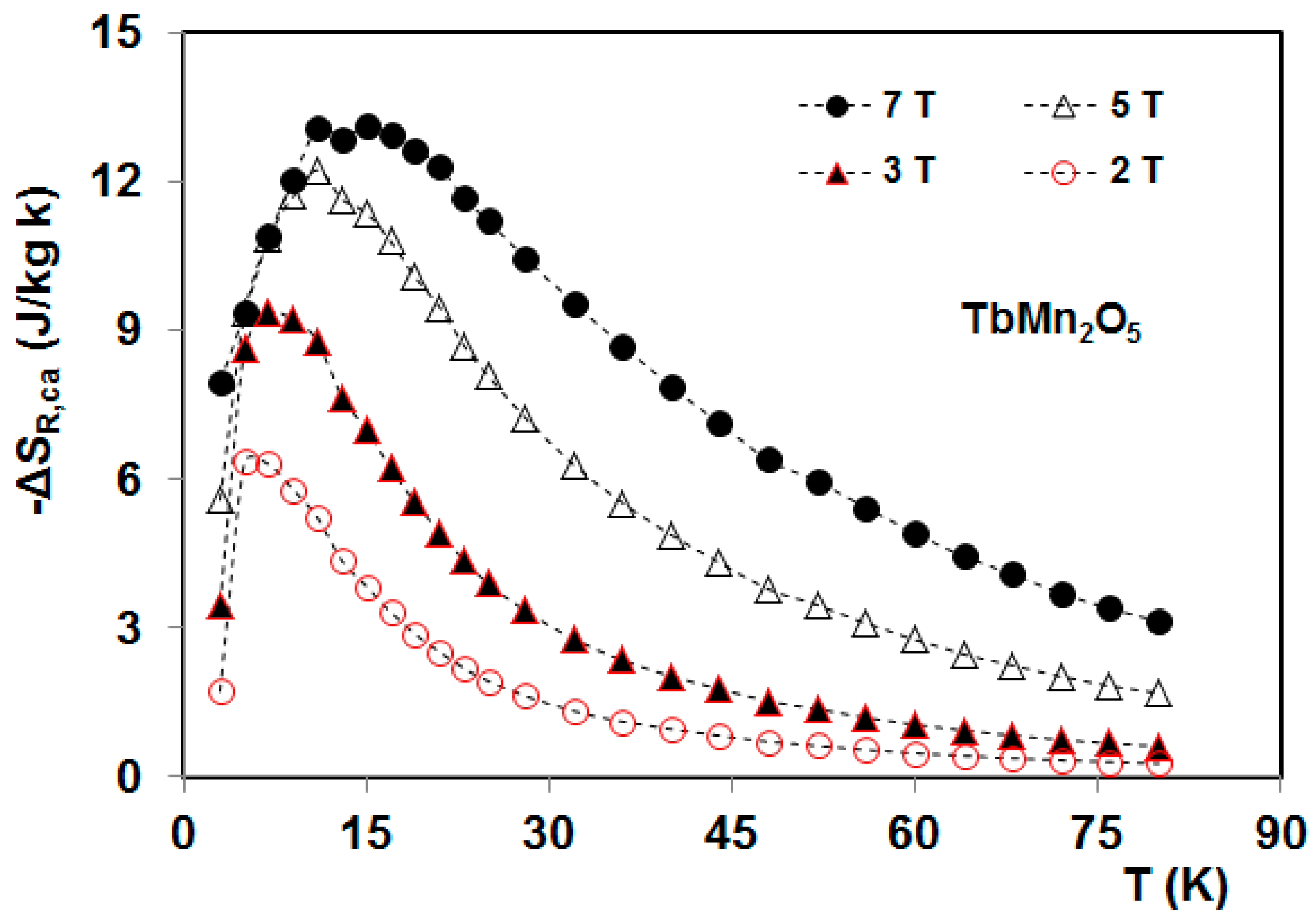
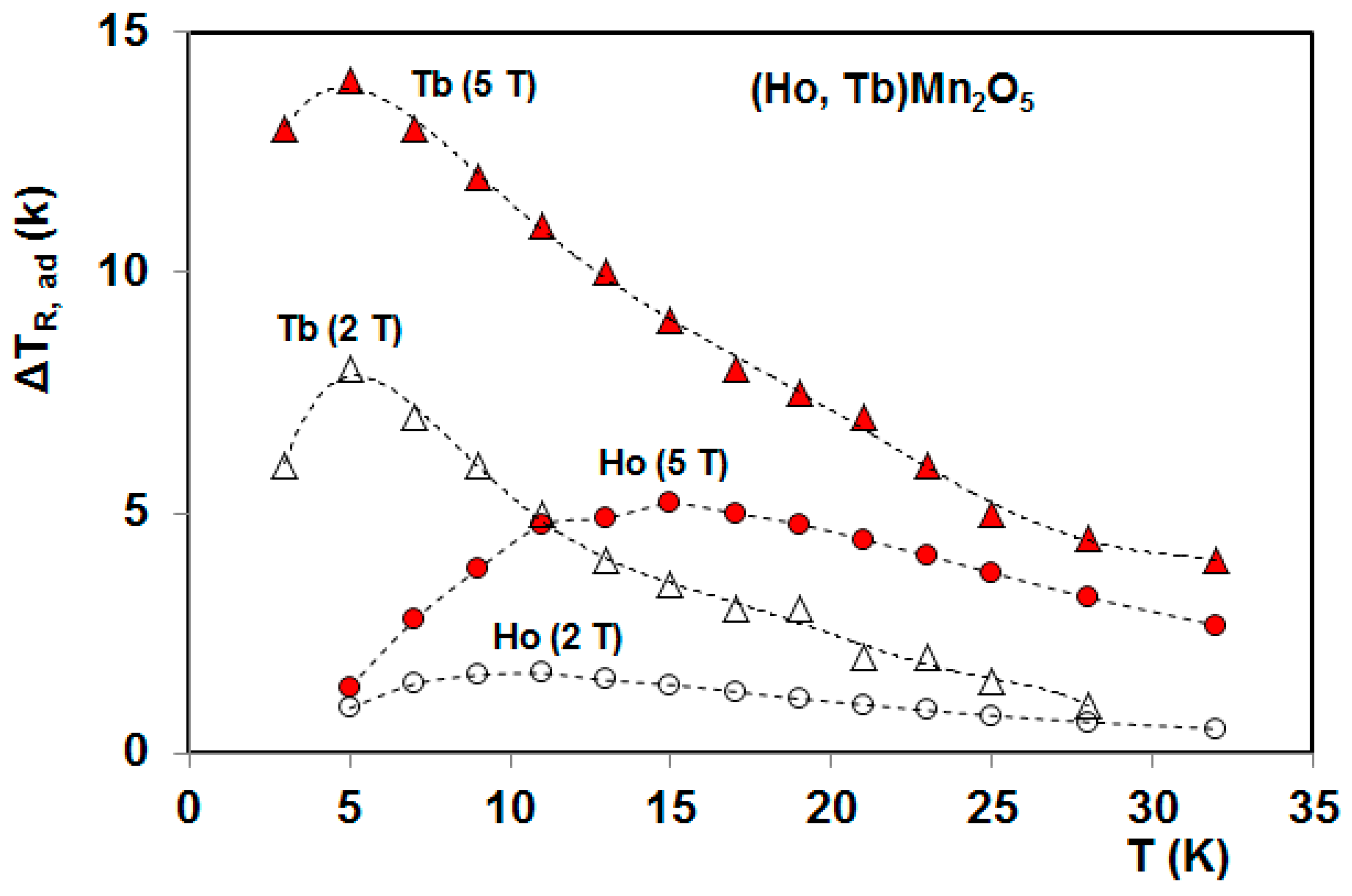
5. Magnetocaloric Properties of RMn2O5 Multiferroic Crystals
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gschneidner, K.A., Jr.; Pecharsky, V.K.; Tsokol, A.O. Recent developments in magnetocaloric materials. Rep. Prog. Phys. 2005, 68, 1479. [Google Scholar] [CrossRef]
- Moya, X.; Kar-Narayan, S.; Mathur, N.D. Caloric materials near ferroic phase transitions. Nat. Mater. 2014, 13, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Pecharsky, V.K.; Gschneidner, K.A., Jr. Giant Magnetocaloric Effect in Gd5Ge2Si2. Phys. Rev. Lett. 1997, 78, 4494. [Google Scholar] [CrossRef]
- Wada, H.; Tanabe, Y. Giant magnetocaloric effect of MnAs1-xSbx. Appl. Phys. Lett. 2001, 79, 3302. [Google Scholar] [CrossRef]
- Balli, M.; Fruchart, D.; Gignoux, D.; Dupuis, C.; Kedous-Lebouc, A.; Zach, R. Giant magnetocaloric effect in Mn1-x (Ti0. 5V0. 5) xAs: Experiments and calculations. J. Appl. Phys. 2008, 103, 103908. [Google Scholar] [CrossRef]
- Balli, M.; Fruchart, D.; Gignoux, D.; Tobola, J.; Hlil, E.K.; Wolfers, P.; Zach, R. Magnetocaloric effect in ternary metal phosphides (Fe 1-xNix)2 P. J. Magn. Magn. Mater. 2007, 316, 358–360. [Google Scholar] [CrossRef]
- Tegus, O.; Brück, E.; Buschow, K.H.J.; De Boer, F.R. Transition-metal-based magnetic refrigerants for room-temperature applications. Nature 2002, 415, 150–152. [Google Scholar] [CrossRef] [PubMed]
- Fujita, A.; Fujieda, S.; Hasegawa, Y.; Fukamichi, K. Itinerant-electron metamagnetic transition and large magnetocaloric effects in La(FexSi1-x)13 compounds and their hydrides. Phys. Rev. B 2003, 67, 104416. [Google Scholar] [CrossRef]
- Hu, F.X.; Shen, B.G.; Sun, J.R.; Wang, G.J.; Cheng, Z.H. Very large magnetic entropy change near room temperature in LaFe11.2Co0.7Si1.1. Appl. Phys. Lett. 2002, 80, 826–828. [Google Scholar] [CrossRef]
- Balli, M.; Fruchart, D.; Gignoux, D. The LaFe11.2Co0.7Si1.1Cx carbides for magnetic refrigeration close to room temperature. Appl. Phys. Lett. 2008, 92, 232505. [Google Scholar] [CrossRef]
- Balli, M.; Fruchart, D.; Gignoux, D. Optimization of La (Fe, Co)13-xSix based compounds for magnetic refrigeration. J. Phys. Condens. Matter 2007, 19, 236230. [Google Scholar] [CrossRef]
- Phan, M.H.; Yu, S.C. Review of the magnetocaloric effect in manganite materials. J. Magn. Magn. Mat 2007, 308, 325–340. [Google Scholar] [CrossRef]
- Balli, M.; Fruchart, D.; Gignoux, D. Effect of Ni substitution on the magnetic and magnetocaloric properties of the Dy (Co1-xNix)2 Laves phase. J. Phys. D Appl. Phys. 2007, 40, 7601. [Google Scholar] [CrossRef]
- Balli, M.; Fruchart, D.; Gignoux, D.; Hlil, E.K.; Miraglia, S.; Wolfers, P. Gd1-xTbx alloys for Ericsson-like magnetic refrigeration cycles. J. Alloys Compd. 2007, 442, 129–131. [Google Scholar] [CrossRef]
- Sari, O.; Balli, M. From conventional to magnetic refrigerator technology. Int. J. Refrig. 2014, 37, 8–15. [Google Scholar] [CrossRef]
- Balli, M.; Sari, O.; Mahmed, C.; Besson, C.; Bonhote, P.; Duc, D.; Forchelet, J. A pre-industrial magnetic cooling system for room temperature application. Appl. Energy 2012, 98, 556–561. [Google Scholar] [CrossRef]
- Zimm, C.; Jastrab, A.; Sternberg, A.; Pecharsky, V.; Gschneidner, K., Jr.; Osborne, M.; Anderson, I. Description and Performance of a Near-Room Temperature Magnetic Refrigerator. Adv. Cryog. Eng. 1998, 43, 1759–1766. [Google Scholar]
- Barclay, J.; Oseen-Senda, K.; Skrzypkowski, M. Unique feature of liquefaction of hydrogen and natural gas using magnetic refrigeration. In Proceedings of 6th IIF-IIR International Conference on Magnetic Refrigeration, Victoria, BC, Canada, 7–10 September 2014.
- Balli, M.; Jandl, S.; Fournier, P.; Gospodinov, M.M. Anisotropy-enhanced giant reversible rotating magnetocaloric effect in HoMn2O5 single crystals. Appl. Phys. Lett. 2014, 104, 232402. [Google Scholar] [CrossRef]
- Balli, M.; Jandl, S.; Fournier, P.; Dimitrov, D.Z. Giant rotating magnetocaloric effect at low magnetic fields in multiferroic TbMn2O5 single crystals. Appl. Phys. Lett. 2016, 108, 102401. [Google Scholar] [CrossRef]
- Jin, J.L.; Zhang, X.Q.; Li, G.K.; Cheng, Z.H.; Zheng, L.; Lu, Y. Giant anisotropy of magnetocaloric effect in TbMnO3 single crystals. Phys. Rev. B 2011, 83, 184431. [Google Scholar] [CrossRef]
- Balli, M.; Jandl, S.; Fournier, P.; Mansouri, S.; Mukhin, A.; Ivanov, Y.V.; Balbashov, A.M. On the magnetocaloric effect in the multiferroic hexagonal DyMnO3 single crystals. J. Magn. Magn. Mat. 2015, 374, 252–257. [Google Scholar] [CrossRef]
- Jin, J.L.; Zhang, X.Q.; Ge, H.; Cheng, Z.H. Rotating field entropy change in hexagonal TmMnO3 single crystal with anisotropic paramagnetic response. Phys. Rev. B 2012, 85, 214426. [Google Scholar] [CrossRef]
- Balli, M.; Mansouri, S.; Jandl, S.; Fournier, P.; Dimitrov, D.Z. Large rotating magnetocaloric effect in the orthorhombic DyMnO3 single crystal. Solid. Stat. Commun. 2016, 239, 9–13. [Google Scholar] [CrossRef]
- Li, L.; Namiki, T.; Huo, D.; Qian, Z.; Nishimura, K. Two successive magnetic transitions induced large refrigerant capacity in HoPdIn compound. Appl. Phys. Lett. 2013, 103, 222405. [Google Scholar]
- Midya, A.; Das, S.N.; Mandal, P.; Pandya, S.; Ganesan, V. Anisotropic magnetic properties and giant magnetocaloric effect in antiferromagnetic RMnO3 crystals (R = Dy, Tb, Ho, and Yb). Phys. Rev. B 2011, 84, 235127. [Google Scholar] [CrossRef]
- Balli, M.; Roberge, B.; Vermette, J.; Jandl, S.; Fournier, P.; Gospodinov, M.M. Magnetocaloric properties of the hexagonal HoMnO3 single crystal revisited. Physica B 2015, 478, 77–83. [Google Scholar] [CrossRef]
- Balli, M.; Roberge, B.; Jandl, S.; Fournier, P.; Palstra, T.T.M.; Nugroho, A.A. Observation of large refrigerant capacity in the HoVO3 vanadate single crystal. J. Appl. Phys. 2015, 118, 073903. [Google Scholar] [CrossRef]
- Matsumoto, K.; Kondo, T.; Yoshioka, S.; Kamiya, K.; Numazawa, T. Magnetic refrigerator for hydrogen liquefaction. J. Phys. Conf. Ser. 2009, 150, 012028. [Google Scholar] [CrossRef]
- Numazawa, T.; Kamiya, K.; Utaki, T.; Matsumoto, K. Magnetic refrigerator for hydrogen liquefaction. Cryogenics 2014, 62, 185–192. [Google Scholar] [CrossRef]
- Noda, Y.; Kimura, H.; Fukunaga, M.; Kobayashi, S.; Kagomiya, I.; Kohn, K. Magnetic and ferroelectric properties of multiferroic RMn2O5. J. Phys. Condens. Matter 2008, 20, 434206. [Google Scholar] [CrossRef]
- Blake, G.R.; Chapon, L.C.; Radaelli, P.G.; Park, S.; Hur, N.; Cheong, S.W.; Rodriguez-Carvajal, J. Spin structure and magnetic frustration in multiferroic RMn2O5 (R = Tb, Ho, Dy). Phys. Rev. B 2005, 71, 214402. [Google Scholar] [CrossRef]
- Hur, N.; Park, S.; Sharma, P.A.; Ahn, J.S.; Guha, S.; Cheong, S.W. Electric polarization reversal and memory in a multiferroic material induced by magnetic fields. Nature 2004, 429, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Hur, N.; Park, S.; Sharma, P.A.; Guha, S.; Cheong, S.W. Colossal Magnetodielectric Effects in DyMn2O5. Phys. Rev. Lett. 2004, 93, 107207. [Google Scholar] [CrossRef] [PubMed]
- Mihailova, B.; Gospodinov, M.M.; Güttler, B.; Yen, F.; Litvinchuk, A.P.; Iliev, M.N. Temperature-dependent Raman spectra of HoMn2O5 and TbMn2O5. Phys. Rev. B 2005, 71, 172301. [Google Scholar] [CrossRef]
- Tzankov, D.; Skumryev, V.; Aroyo, M.; Puźniak, R.; Kuz’min, M.D.; Mikhov, M. Magnetic anisotropy of multiferroic HoMn2O5 single crystal. Solid. Stat. Commun. 2008, 147, 212–216. [Google Scholar] [CrossRef]
- Kimura, T.; Goto, T.; Shintani, H.; Ishizaka, K.; Arima, T.; Tokura, Y. Magnetic control of ferroelectric polarization. Nature 2003, 426, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Lottermoser, T.; Lonkai, T.; Amann, U.; Hohlwein, D.; Ihringer, J.; Fiebig, M. Magnetic phase control by an electric field. Nature 2004, 430, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Kimura, T.; Lawes, G.; Ramirez, A.P.; Tokura, Y. Ferroelectricity and Giant Magnetocapacitance in Perovskite Rare-Earth Manganites. Phys. Rev. Lett. 2004, 92, 257201. [Google Scholar] [CrossRef] [PubMed]
- Prokhnenko, O.; Feyerherm, R.; Dudzik, E.; Landsgesell, S.; Aliouane, N.; Chapon, L.C.; Argyriou, D.N. Enhanced Ferroelectric Polarization by Induced Dy Spin Order in Multiferroic DyMnO3. Phys. Rev. Lett. 2007, 98, 057206. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Murugavel, P.; Lee, D.; Noh, T.W.; Jo, Y.; Jung, M.H.; Jang, K.H.; Park, J.G. Multiferroic properties of epitaxially stabilized hexagonal DyMnO3 thin films. Appl. Phys. Lett. 2007, 90, 012903. [Google Scholar] [CrossRef]
- Tishin, A.M.; Spichkin, Y.L. The Magnetocaloric Effect and Its Applications; IOP Publishing Ltd.: London, UK, 2003. [Google Scholar]
- Balli, M.; Mahmed, C.; Bonhote, P.; Sari, O. On the magnetic forces in magnetic cooling machines: Numerical calculations and experimental investigations. IEEE Trans. Magn. 2011, 47, 3383–3386. [Google Scholar] [CrossRef]
- Balli, M.; Fruchart, D.; Zach, R. Negative and conventional magnetocaloric effects of a MnRhAs single crystal. J. Appl. Phys. 2014, 115, 203909. [Google Scholar] [CrossRef]
- Balli, M.; Fruchart, D.; Gignoux, D.; Zach, R. The “colossal” magnetocaloric effect in Mn1-xFexAs: What are we really measuring? Appl. Phys. Lett. 2009, 95, 072509. [Google Scholar] [CrossRef]
- Balli, M.; Sari, O.; Fruchart, D.; Forchelet, J. Influence of the materials magnetic state on the accurate determination of the magnetocaloric effect. Eur. Phys. J. Web Conf. 2012, 29, 00005. [Google Scholar] [CrossRef]
- Gschneidner, K.A., Jr.; Pecharsky, V.K. Magnetocaloric Materials. Annu. Rev. Mater. Sci. 2000, 30, 387–429. [Google Scholar] [CrossRef]
- Niknia, I.; Trevizoli, P.V.; Govindappa, P.; Campbell, O.; Christiaanse, T.V.; Teyber, R.; Rowe, A. A material screening technique for optimum performance of an AMR. In Proceedings of the Seventh IIF-IIR International Conference on Magnetic Refrigeration at Room Temperature, Thermag VII, Torino, Italy, 11–14 September 2016.
- Lee, S.; Pirogov, A.; Han, J.H.; Park, J.G.; Hoshikawa, A.; Kamiyama, T. Direct observation of a coupling between spin, lattice and electric dipole moment in multiferroic YMnO3. Phys. Rev. B 2005, 71, 180413. [Google Scholar] [CrossRef]
- Bary’achtar, V.G.; L’vov, V.A.; Jablonskii, D.A. Theory of inhomogeneous magnetoelectric effect. Sov. JETP Lett. 1983, 37, 565–567. [Google Scholar]
- Stefanovskii, E.P.; Jablonskii, D.A. Theory of electrical polarization of multisublattice orthorhombic antiferromagnets with a double-exchange superlattice. Sov. J. Low Temp. Phys. 1986, 12, 478–480. [Google Scholar]
- Mostovoy, M. Ferroelectricity in Spiral Magnets. Phys. Rev. Lett. 2006, 96, 067601. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, G. Study of (La1-xCax)MnO3. I. Magnetic Structure of LaMnO3. J. Phys. Soc. Jpn. 1970, 29, 606–615. [Google Scholar] [CrossRef]
- Wollan, E.O.; Koehler, W.C. Neutron Diffraction Study of the Magnetic Properties of the Series of Perovskite-Type Compounds [(1-x)La, xCa]MnO3. Phys. Rev. 1955, 100, 545. [Google Scholar] [CrossRef]
- Katsura, H.; Nagaosa, N.; Balatsky, V. Spin current and magnetoelectric effect in noncollinear magnets. Phys. Rev. Lett. 2005, 95, 057205. [Google Scholar] [CrossRef] [PubMed]
- Sergienko, I.A.; Dagotto, E. Role of the Dzyaloshinskii-Moriya interaction in multiferroic perovskites. Phys. Rev. B 2006, 73, 094434. [Google Scholar] [CrossRef]
- Dzyaloshinskii, I. Theory of Helicoidal Structures in Antiferromagnets. I. Nonmetals. Sov. Phys. JETP 1964, 19, 960–971. [Google Scholar]
- Moriya, T. Anisotropic Superexchange Interaction and Weak Ferromagnetism. Phys. Rev. 1960, 120, 91–98. [Google Scholar] [CrossRef]
- Kenzelmann, M.; Harris, A.B.; Jonas, S.; Broholm, C.; Schefer, J.; Kim, S.B.; Zhang, C.L.; Cheong, S.-W.; Vajk, O.P.; Lynn, J.W. Magnetic Inversion Symmetry Breaking and Ferroelectricity in TbMnO3. Phys. Rev. Lett. 2005, 95, 087206. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Ishihara, S.; Shintani, H.; Arima, T.; Takahashi, K.T.; Ishizaka, K.; Tokura, Y. Distorted perovskite with e1g configuration as a frustrated spin system. Phys. Rev. B 2003, 68, 060403(R). [Google Scholar] [CrossRef]
- Fukunaga, M.; Noda, Y. Classification and interpretation of the polarization of multiferroic RMn2O5. J. Phys. Soc. Jpn. 2010, 79, 054705. [Google Scholar] [CrossRef]
- Xiang, H.J.; Wei, S.H.; Whangbo, M.H.; Da Silva, J.L. Spin-Orbit Coupling and Ion Displacements in Multiferroic TbMnO3. Phys. Rev. Lett. 2008, 101, 037209. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Jang, H.M. Modulated spin structure responsible for the magnetic-field-induced polarization switching in multiferroic TbMn2O5. Phys. Rev. B 2005, 91, 014403. [Google Scholar] [CrossRef]
- Oh, Y.S.; Jeon, B.G.; Haam, S.Y.; Park, S.; Correa, V.F.; Lacerda, A.H.; Cheong, S.-W.; Jeon, G.S.; Kim, K.H. Strong magnetoelastic effect on the magnetoelectric phenomena of TbMn2O5. Phys. Rev. B 2011, 83, 060405(R). [Google Scholar] [CrossRef]
- Harikrishnan, S.; Rößler, S.; Kumar, C.N.; Bhat, H.L.; Rößler, U.K.; Wirth, S.; Steglich, F.; Elizabeth, S. Phase transitions and rare-earth magnetism in hexagonal and orthorhombic DyMnO3 single crystals. J. Phys. Condens. Matter 2009, 21, 096002. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, V.Y.; Mukhin, A.A.; Prokhorov, A.S.; Balbashov, A.M.; Iskhakova, L.D. Magnetic properties and phase transitions in hexagonal DyMnO3 single crystals. Phys. Solid State 2006, 48, 1726–1729. [Google Scholar] [CrossRef]
- Kimura, T.; Lawes, G.; Goto, T.; Tokura, Y.; Ramirez, A.P. Magnetoelectric phase diagrams of orthorhombic RMnO3 (R = Gd, Tb, and Dy). Phys. Rev. B 2005, 71, 224425. [Google Scholar] [CrossRef]
- Feyerherm, R.; Dudzik, E.; Aliouane, N.; Argyriou, D.N. Commensurate Dy magnetic ordering associated with incommensurate lattice distortion in multiferroic DyMnO3. Phys. Rev. B 2006, 73, 180401(R). [Google Scholar] [CrossRef]
- Nandi, S.; Kreyssig, A.; Yan, J.Q.; Vannette, M.D.; Lang, J.C.; Tan, L.; Kim, J.W.; Prozorov, R.; Lagrasso, T.A.; McQueeney, R.J.; et al. Magnetic structure of Dy3+ in hexagonal multiferroic DyMnO3. Phys. Rev. B 2008, 78, 075118. [Google Scholar] [CrossRef]
- Kimura, T.; Tokura, Y. Magnetoelectric phase control in a magnetic system showing cycloidal/conical spin order. J. Phys. Condens. Matter 2008, 20, 434204. [Google Scholar] [CrossRef]
- Wehrenfennig, C.; Meier, D.; Lottermoser, T.; Lonkai, T.; Hoffmann, J.U.; Aliouane, N.; Argyriou, D.N.; Fiebig, M. Incompatible magnetic order in multiferroic hexagonal DyMnO3. Phys. Rev. B 2010, 82, 100414 (R). [Google Scholar] [CrossRef]
- Pękała, M.; Wolff-Fabris, F.; Fagnard, J.F.; Vanderbemden, P.; Mucha, J.; Gospodinov, M.M.; Lovchinov, V.; Ausloos, M. Magnetic properties and anisotropy of orthorhombic DyMnO3 single crystal. J. Magn. Magn. Mater. 2013, 335, 46–52. [Google Scholar] [CrossRef]
- Sari, O.; Balli, M.; Trottet, G.; Bonhote, P.; Egolf, P.W.; Muller, C.; Heitzler, J.C.; Bour, S. Initial results of a test-bed magnetic refrigeration machine with practical running conditions. In Proceedings of the 3rd International Conference on Magnetic Refrigeration at Room Temperature, Des Moines, IA, USA, 2009; pp. 371–380.
- Lorenz, B.; Yen, F.; Gospodinov, M.M.; Chu, C.W. Field-induced phases in HoMnO3 at low temperatures. Phys. Rev. B 2005, 71, 014438. [Google Scholar] [CrossRef]
- Midya, A.; Mandal, P.; Das, S.; Banerjee, S.; Chandra, L.S.; Ganesan, V.; Barman, S.R. Magnetocaloric effect in HoMnO3 crystal. Appl. Phys. Lett. 2010, 96, 142514. [Google Scholar] [CrossRef]
- Munoz, A.; Alonso, J.A.; Martínez-Lope, M.J.; Casáis, M.T.; Martínez, J.L.; Fernandez-Diaz, M.T. Evolution of the magnetic structure of hexagonal HoMnO3 from neutron powder diffraction data. Chem. Mater. 2001, 13, 1497–1505. [Google Scholar] [CrossRef]
- Fiebig, M.; Lottermoser, T.; Pisarev, R.V. Spin-rotation phenomena and magnetic phase diagrams of hexagonal RMnO3. J. Appl. Phys. 2003, 93, 8194–8196. [Google Scholar] [CrossRef]
- Lonkai, T.; Hohlwein, D.; Ihringer, J.; Prandl, W. The magnetic structures of YMnO3-δ and HoMnO3. Appl. Phys. A Mater. Sci. Process 2002, 74, s843–s845. [Google Scholar] [CrossRef]
- Hur, N.; Jeong, I.K.; Hundley, M.F.; Kim, S.B.; Cheong, S.W. Giant magnetoelectric effect in multiferroic HoMnO3 with a high ferroelectric transition temperature. Phys. Rev. B 2009, 79, 134120. [Google Scholar] [CrossRef]
- De Lacheisserie, E.D.T. Magnétisme: Fondements; EDP Sciences: Les Ulis, France, 2000. [Google Scholar]
- Kim, J.W.; Nenkov, K.; Schultz, L.; Dörr, K. Magnetic properties of thick multiferroic hexagonal HoMnO3 films. J. Magn. Magn. Mater. 2009, 321, 1727–1730. [Google Scholar] [CrossRef]
- Wilkins, S.B.; Forrest, T.R.; Beale, T.A.W.; Bland, S.R.; Walker, H.C.; Mannix, D.; Yakhou, F.; Prabhakaran, D.; Boothroyd, A.T.; Hill, J.P.; et al. Nature of the Magnetic Order and Origin of Induced Ferroelectricity in TbMnO3. Phys. Rev. Lett. 2009, 103, 207602. [Google Scholar] [CrossRef] [PubMed]
- Kajimoto, R.; Yoshizawa, H.; Shintani, H.; Kimura, T.; Tokura, Y. Magnetic structure of TbMnO3 by neutron diffraction. Phys. Rev. B 2004, 70, 012401. [Google Scholar] [CrossRef]
- Salama, H.A.; Stewart, G.A. Exchange-induced Tm magnetism in multiferroic h-TmMnO3. J. Phys. Condens. Matter 2009, 21, 386001. [Google Scholar] [CrossRef] [PubMed]
- Yen, F.; Dela Cruz, C.; Lorenz, B.; Galstyan, E.; Sun, Y.Y.; Gospodinov, M.; Chu, C.W. Magnetic phase diagrams of multiferroic hexagonal RMnO3 (R = Er, Yb, Tm, and Ho). J. Mater. Res. 2007, 22, 2163–2173. [Google Scholar] [CrossRef]
- Fabreges, X.; Mirebeau, I.; Bonville, P.; Petit, S.; Lebras-Jasmin, G.; Forget, A.; André, G.; Pailhes, S. Magnetic order in YbMnO3 studied by neutron diffraction and Mössbauer spectroscopy. Phys. Rev. B 2008, 78, 214422. [Google Scholar] [CrossRef]
- Wagh, A.A.; Suresh, K.G.; Kumar, P.A.; Elizabeth, S. Low temperature giant magnetocaloric effect in multiferroic GdMnO3 single crystals. J. Phys. D 2015, 48, 135001. [Google Scholar] [CrossRef]
- Hemberger, J.; Lobina, S.; Von Nidda, H.A.K.; Tristan, N.; Ivanov, V.Y.; Mukhin, A.A.; Balbashov, A.M.; Loidl, A. Complex interplay of 3d and 4f magnetism in La1-xGdxMnO3. Phys. Rev. B 2004, 70, 024414. [Google Scholar] [CrossRef]
- Chandra, S.; Biswas, A.; Phan, M.H.; Srikanth, H. Impacts of nanostructuring and magnetic ordering of Nd3+ on themagnetic and magnetocaloric response in NdMnO3. J. Magn. Magn. Mater. 2015, 384, 138–143. [Google Scholar] [CrossRef]
- Da Silva, C.A.; Silva, R.S.; Plaza, E.J.R.; Moreno, N.O. Magnetic State and Magnetocaloric Effect of SmMnO3. J. Supercond. Nov. Magn. 2013, 26, 2497–2499. [Google Scholar] [CrossRef]
- Sagar, E.; Pavan Kumar, N.; Esakkimuthuraju, M.; Reddy, P.V. Magnetocaloric effect in multiferroic EuMnO3. Phys. Express 2015, 5, 3. [Google Scholar]
- Samanta, T.; Das, I.; Banerjee, S. Giant magnetocaloric effect in antiferromagnetic ErRu2Si2 compound. Appl. Phys. Lett. 2007, 91, 152506. [Google Scholar] [CrossRef]
- Midya, A.; Mandal, P.; Rubi, K.; Chen, R.; Wang, J.S.; Mahendiran, R.; Lorusso, G.; Evangelisti, M. Large adiabatic temperature and magnetic entropy changes in EuTiO3. Phy. Rev. B 2016, 93, 094422. [Google Scholar] [CrossRef]
- Mo, Z.J.; Hao, Z.H.; Shen, J.; Li, L.; Wu, J.F.; Hu, F.X.; Sun, J.R.; Shen, B.G. Observation of giant magnetocaloric effect in EuTi1-xCrxO3. J. Alloys Compd. 2015, 649, 674–678. [Google Scholar] [CrossRef]
- Von Ranke, P.J.; Alho, B.P.; Nóbrega, E.P.; De Sousa, V.S.R.; Alvarenga, T.S.T.; Carvalho, A.M.G.; De Oliveira, N.A. The influence of magnetic and electric coupling properties on the magnetocaloric effect in quantum paraelectric EuTiO3. J. Magn. Magn. Mater. 2012, 324, 1290–1295. [Google Scholar] [CrossRef]
- Ke, Y.J.; Zhang, X.Q.; Ma, Y.; Cheng, Z.H. Anisotropic magnetic entropy change in RFeO3 single crystals (R = Tb, Tm, or Y). Sci. Rep. 2016, 6, 19775. [Google Scholar] [CrossRef] [PubMed]
- Arrott, A.; Noakes, J.E. Approximate equation of state for nickel near its critical temperature. Phys. Rev. Lett. 1967, 19, 786. [Google Scholar] [CrossRef]
- Hu, W.J.; Du, J.; Li, B.; Zhang, Q.; Zhang, Z.D. Giant magnetocaloric effect in the Ising antiferromagnet DySb. Appl. Phys. Lett. 2008, 92, 192505. [Google Scholar] [CrossRef]
- Li, L.; Nishimura, K.; Hutchison, W.D.; Qian, Z.; Huo, D.; NamiKi, T. Giant reversible magnetocaloric effect in ErMn2Si2 compound with a second order magnetic phase transition. Appl. Phys. Lett. 2012, 100, 152403. [Google Scholar] [CrossRef]
- Balli, M.; Jandl, S.; Fournier, P.; Dimitrov, D.Z. On the conventional and rotating magnetocaloric effects in multiferroic TbMn2O5 single crystals. arXiv 2016. [Google Scholar]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balli, M.; Roberge, B.; Fournier, P.; Jandl, S. Review of the Magnetocaloric Effect in RMnO3 and RMn2O5 Multiferroic Crystals. Crystals 2017, 7, 44. https://doi.org/10.3390/cryst7020044
Balli M, Roberge B, Fournier P, Jandl S. Review of the Magnetocaloric Effect in RMnO3 and RMn2O5 Multiferroic Crystals. Crystals. 2017; 7(2):44. https://doi.org/10.3390/cryst7020044
Chicago/Turabian StyleBalli, Mohamed, Benoit Roberge, Patrick Fournier, and Serge Jandl. 2017. "Review of the Magnetocaloric Effect in RMnO3 and RMn2O5 Multiferroic Crystals" Crystals 7, no. 2: 44. https://doi.org/10.3390/cryst7020044
APA StyleBalli, M., Roberge, B., Fournier, P., & Jandl, S. (2017). Review of the Magnetocaloric Effect in RMnO3 and RMn2O5 Multiferroic Crystals. Crystals, 7(2), 44. https://doi.org/10.3390/cryst7020044