
Halogen and Hydrogen Bonding in Multicomponent Crystals of Tetrabromo-1H-Benzotriazole


Abstract

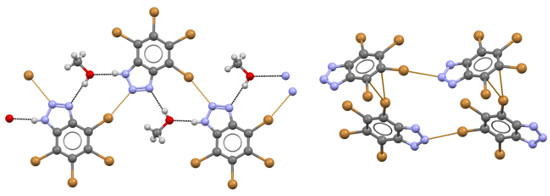{kind=link}
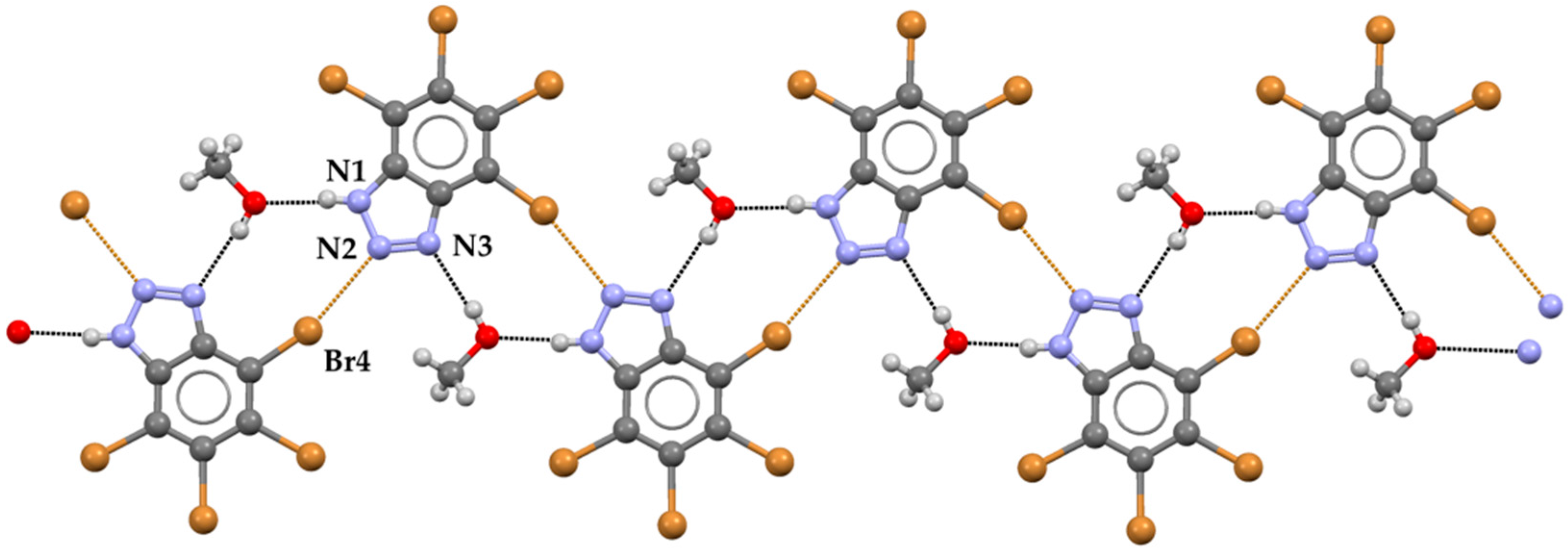{kind=link}
{kind=link}
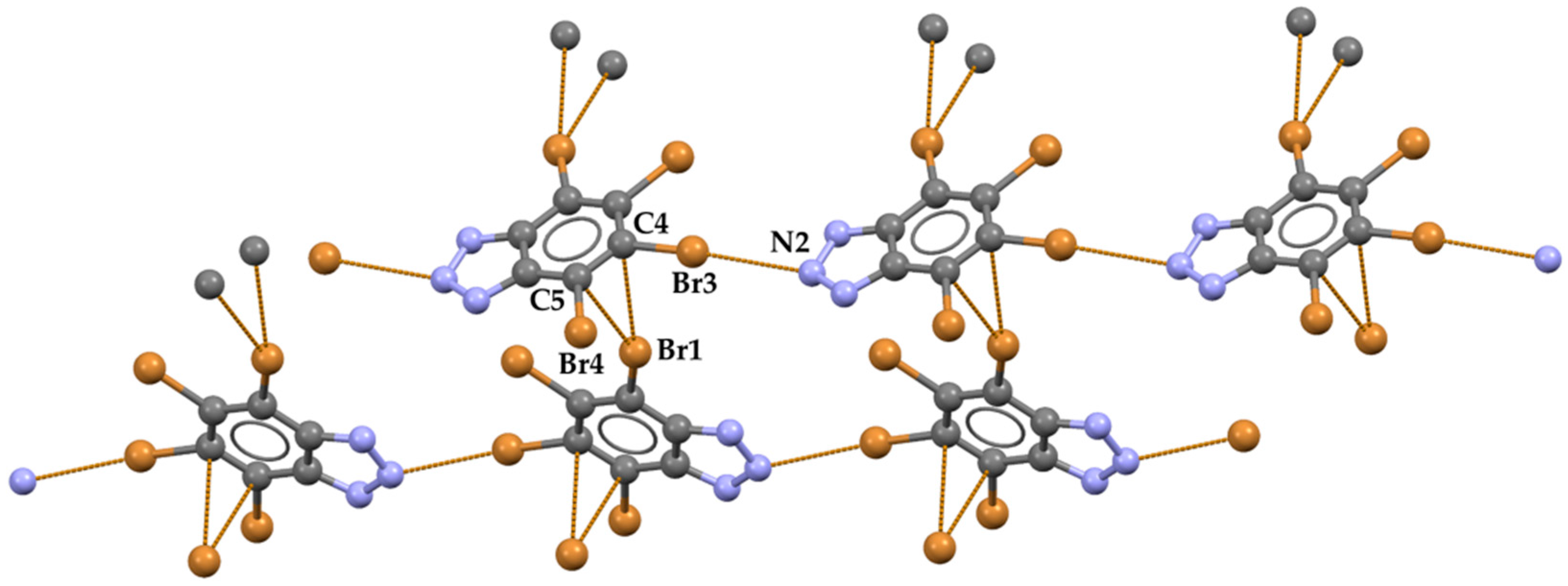{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Battistutta, R.; De Moliner, E.; Sarno, S.; Zanotti, G.; Pinna, L.A. Structural features underlying selective inhibition of protein kinase CK2 by ATP site-directed tetrabromo-2-benzotriazole. Protein Sci. 2001, 10, 2200–2206. [Google Scholar] [CrossRef] [PubMed]
- Sarno, S.; Reddy, H.; Meggio, F.; Ruzzene, M.; Davies, S.P.; Donella-Deana, A.; Shugar, D.; Pinna, L.A. Selectivity of 4,5,6,7-tetrabromobenzotriazole, an ATP site-directed inhibitor of protein kinase CK2 (’casein kinase-2’). FEBS Lett. 2001, 496, 44–48. [Google Scholar] [CrossRef]
- Zień, P.; Bretner, M.; Zasta̧piło, K.; Szyszka, R.; Shugar, D. Selectivity of 4,5,6,7-tetrabromobenzimidazole as an ATP-competitive potent inhibitor of protein kinase CK2 from various sources. Biochem. Biophys. Res. Commun. 2003, 306, 129–133. [Google Scholar] [CrossRef]
- Sarno, S.; Moro, S.; Meggio, F.; Zagotto, G.; Dal Ben, D.; Ghisellini, P.; Battistutta, R.; Zanotti, G.; Pinna, L.A. Toward the rational design of protein kinase casein kinase-2 inhibitors. Pharmacol. Ther. 2002, 93, 159–168. [Google Scholar] [CrossRef]
- Zien, P.; Duncan, J.S.; Skierski, J.; Bretner, M.; Litchfield, D.W.; Shugar, D. Tetrabromobenzotriazole (TBBt) and tetrabromobenzimidazole (TBBz) as selective inhibitors of protein kinase CK2: Evaluation of their effects on cells and different molecular forms of human CK2. Biochim. Biophys. Acta 2005, 1754, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Bretner, M.; Najda-Bernatowicz, A.; Łebska, M.; Muszyńska, G.; Kilanowicz, A.; Sapota, A. New inhibitors of protein kinase CK2, analogues of benzimidazole and benzotriazole. Mol. Cell. Biochem. 2008, 316, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.A.; Bain, J.; Kazimierczuk, Z.; Sarno, S.; Ruzzene, M.; Di Maira, G.; Elliott, M.; Orzeszko, A.; Cozza, G.; Meggio, F.; et al. The selectivity of inhibitors of protein kinase CK2: An update. Biochem. J. 2008, 415, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Unger, G.M.; Davis, A.T.; Slaton, J.W.; Ahmed, K. Protein kinase CK2 as regulator of cell survival: Implications for cancer therapy. Curr. Cancer Drug Targets 2004, 4, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [PubMed]
- Martins, L.R.; Lúcio, P.; Melão, A.; Antunes, I.; Cardoso, B.A.; Stansfield, R.; Bertilaccio, M.T.S.; Ghia, P.; Drygin, D.; Silva, M.G.; et al. Activity of the clinical-stage CK2-specific inhibitor CX-4945 against chronic lymphocytic leukemia. Leukemia 2014, 28, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Wa̧sik, R.; Wińska, P.; Poznański, J.; Shugar, D. Synthesis and physico-chemical properties in aqueous medium of all possible isomeric bromo analogues of benzo-1H-triazole, potential inhibitors of protein kinases. J. Phys. Chem. B 2012, 116, 7259–7268. [Google Scholar] [CrossRef] [PubMed]
- Gianoncelli, A.; Cozza, G.; Orzeszko, A.; Meggio, F.; Kazimierczuk, Z.; Pinna, L.A. Tetraiodobenzimidazoles are potent inhibitors of protein kinase CK2. Bioorg. Med. Chem. 2009, 17, 7281–7289. [Google Scholar] [CrossRef] [PubMed]
- Shan, N.; Zaworotko, M.J. The role of cocrystals in pharmaceutical science. Drug Discov. Today 2008, 13, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K. Modification of physicochemical characteristics of active pharmaceutical ingredients and application of supersaturatable dosage forms for improving bioavailability of poorly absorbed drugs. Adv. Drug Deliv. Rev. 2012, 64, 480–495. [Google Scholar] [CrossRef] [PubMed]
- Elder, D.P.; Holm, R.; De Diego, H.L. Use of pharmaceutical salts and cocrystals to address the issue of poor solubility. Int. J. Pharm. 2013, 453, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Babu, N.J.; Nangia, A. Solubility advantage of amorphous drugs and pharmaceutical cocrystals. Cryst. Growth Des. 2011, 11, 2662–2679. [Google Scholar] [CrossRef]
- Anderson, K.M.; Goeta, A.E.; Steed, J.W. Supramolecular Synthon Frustration Leads to Crystal Structures with Z′ > 1. Cryst. Growth Des. 2008, 8, 2517–2524. [Google Scholar] [CrossRef]
- Babu, N.J.; Cherukuvada, S.; Thakuria, R.; Nangia, A. Conformational and Synthon Polymorphism in Furosemide (Lasix). Cryst. Growth Des. 2010, 10, 1979–1989. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Wijethunga, T.K.; Haj, M.A.; Desper, J.; Moore, C. The structural landscape of heteroaryl-2-imidazoles: Competing halogen- and hydrogen-bond interactions. CrystEngComm 2014, 16, 7218. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Panikkattu, S.; Chopade, P.D.; Desper, J. Competing hydrogen-bond and halogen-bond donors in crystal engineering. CrystEngComm 2013, 15, 3125–3136. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Metrangolo, P.; Neukirch, H.; Pilati, T.; Resnati, G. Halogen bonding based recognition processes: A world parallel to hydrogen bonding. Acc. Chem. Res. 2005, 38, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Beale, T.M.; Chudzinski, M.G.; Sarwar, M.G.; Taylor, M.S. Halogen bonding in solution: Thermodynamics and applications. Chem. Soc. Rev. 2013, 42, 1667–1680. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed]
- Parisini, E.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen bonding in halocarbon-protein complexes: A structural survey. Chem. Soc. Rev. 2011, 40, 2267–2278. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, P.; Hays, F.A.; Westhof, E.; Ho, P.S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 16789–16794. [Google Scholar] [CrossRef] [PubMed]
- Voth, A.R.; Hays, F.A.; Ho, P.S. Directing macromolecular conformation through halogen bonds. Proc. Natl. Acad. Sci. USA 2007, 104, 6188–6193. [Google Scholar] [CrossRef] [PubMed]
- Baldrighi, M.; Bartesaghi, D.; Cavallo, G.; Chierotti, M.R.; Gobetto, R.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Polymorphs and co-crystals of haloprogin: an antifungal agent. CrystEngComm 2014, 16, 5897–5904. [Google Scholar] [CrossRef]
- Baldrighi, M.; Cavallo, G.; Chierotti, M.R.; Gobetto, R.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen bonding and pharmaceutical cocrystals: The case of a widely used preservative. Mol. Pharm. 2013, 10, 1760–1772. [Google Scholar] [CrossRef] [PubMed]
- Aakeröy, C.B.; Welideniya, D.; Desper, J.; Moore, C. Halogen-bond driven co-crystallization of potential anti-cancer compounds: A structural study. CrystEngComm 2014, 16, 10203–10209. [Google Scholar] [CrossRef]
- Miroslaw, B.; Plech, T.; Wujec, M. Halogen bonding in the antibacterial 1,2,4-triazole-3-thione derivative—Spectroscopic properties, crystal structure and conformational analysis. J. Mol. Struct. 2015, 1083, 187–193. [Google Scholar] [CrossRef]
- Fucke, K.; McIntyre, G.J.; Lemée-Cailleau, M.H.; Wilkinson, C.; Edwards, A.J.; Howard, J.A.K.; Steed, J.W. Insights into the Crystallisation Process from Anhydrous, Hydrated and Solvated Crystal Forms of Diatrizoic Acid. Chem. A Eur. J. 2015, 21, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Valkonen, A.; Chukhlieb, M.; Moilanen, J.; Tuononen, H.M.; Rissanen, K. Halogen and hydrogen bonded complexes of 5-iodouracil. Cryst. Growth Des. 2013, 13, 4769–4775. [Google Scholar] [CrossRef]
- Gerhardt, V.; Egert, E. Cocrystals of 6-chlorouracil and 6-chloro-3-methyluracil: Exploring their hydrogen-bond-based synthon motifs with several triazine and pyrimidine derivatives. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2015, 71, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Golob, S.; Perry, M.; Lusi, M.; Chierotti, M.R.; Grabnar, I.; Lassiani, L.; Voinovich, D.; Zaworotko, M.J. Improving Biopharmaceutical Properties of Vinpocetine Through Cocrystallization. J. Pharm. Sci. 2016, 105, 3626–3633. [Google Scholar] [CrossRef] [PubMed]
- De Moliner, E.; Brown, N.R.; Johnson, L.N. Alternative binding modes of an inhibitor to two different kinases. Eur. J. Biochem. 2003, 270, 3174–3181. [Google Scholar] [CrossRef] [PubMed]
- A normalized contact (Nc) is the ratio between the observed separation of interacting atoms and the sum of their respective van der Waals radii; it allows the different interactions lengths to be more meaningfully compared than when absolute separations are used.
- Rather long type-I bromine-bromine interactions (Br1⋅⋅⋅Br3 distance is 360.66(8) Å, corresponding to Nc = 0.97; C−Br⋅⋅⋅Br angles are 126.1(1)° and 126.5(1)°) connect adjacent and coplanar ribbons and form planes which develop along the b crystallographic axis. No contacts below van der Waals radii exist between adjacent planes.
- Childs, S.L.; Stahly, G.P.; Park, A. The salt-cocrystal continuum: The influence of crystal structure on ionization state. Mol. Pharm. 2007, 4, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SADABS, Empirical Absorption Correction Program; University of Gottingen: Gottingen, Germany, 1997. [Google Scholar]
- Blessing, R.H. An empirical correction for absorption anisotropy. Acta Crystallogr. Sect. A Found. Crystallogr. 1995, 51, 33–38. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Wiley, R.H.; Hussung, K.F. Halogenated Benzotriazoles. J. Am. Chem. Soc. 1957, 79, 4395–4400. [Google Scholar] [CrossRef]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baldrighi, M.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen and Hydrogen Bonding in Multicomponent Crystals of Tetrabromo-1H-Benzotriazole. Crystals 2017, 7, 332. https://doi.org/10.3390/cryst7110332
Baldrighi M, Metrangolo P, Pilati T, Resnati G, Terraneo G. Halogen and Hydrogen Bonding in Multicomponent Crystals of Tetrabromo-1H-Benzotriazole. Crystals. 2017; 7(11):332. https://doi.org/10.3390/cryst7110332
Chicago/Turabian StyleBaldrighi, Michele, Pierangelo Metrangolo, Tullio Pilati, Giuseppe Resnati, and Giancarlo Terraneo. 2017. "Halogen and Hydrogen Bonding in Multicomponent Crystals of Tetrabromo-1H-Benzotriazole" Crystals 7, no. 11: 332. https://doi.org/10.3390/cryst7110332
APA StyleBaldrighi, M., Metrangolo, P., Pilati, T., Resnati, G., & Terraneo, G. (2017). Halogen and Hydrogen Bonding in Multicomponent Crystals of Tetrabromo-1H-Benzotriazole. Crystals, 7(11), 332. https://doi.org/10.3390/cryst7110332