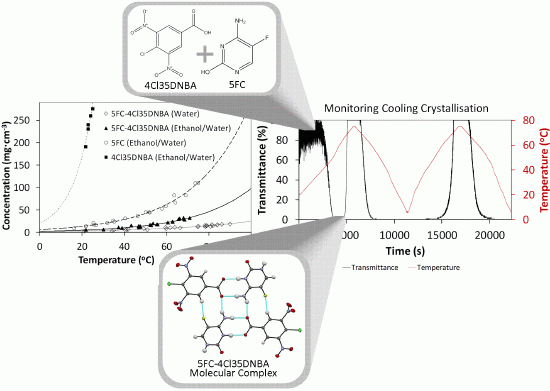
2.1. Molecular Complexes of Cytosine and 5-Fluorocytosine with 4-chloro-3,5-Dinitrobenzoic Acid
Two new multi-component molecular complexes were synthesised through evaporative crystallisation studies involving cytosine, 5-fluorocytosine (5FC) and 4-chloro-3,5-dinitrobenzoic acid (4Cl35DNBA). Sample preparation and crystallographic data collection details are given in the experimental section.
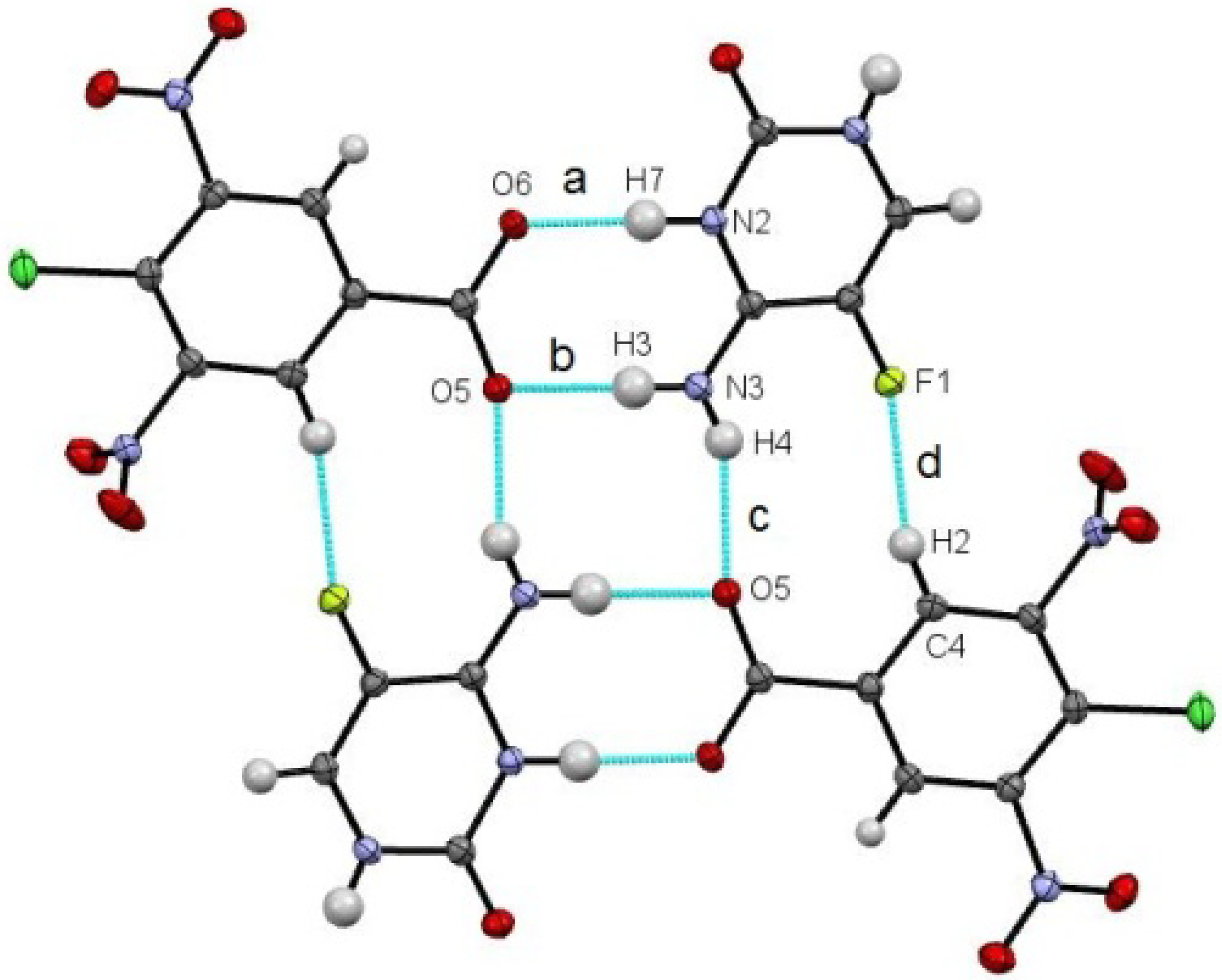
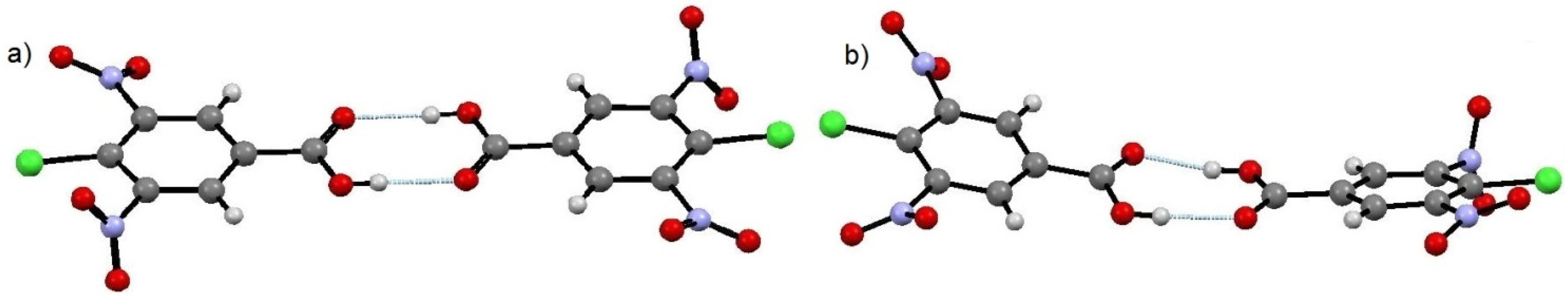
The 1:1 molecular complex of 5-fluorocytosine and 4-chloro-3,5-dinitrobenzoic acid (5FC-4Cl35DNBA) is a salt with proton transfer indicated from the carboxylic acid group of 4Cl35DNBA to the lone pair of the free N2 of 5FC. Heterodimers are formed through two charge-assisted N–H···O hydrogen bonds (N···O distances 2.722(1) Å and 2.803(1) Å) (
Figure 1a,b). Two heterodimers related by a centre of inversion combine to form a tetramer through an additional N–H···O hydrogen bond (N···O 2.871(1) Å) as well as a C–H···F weak hydrogen bond (C···F 3.091(1) Å). Each tetramer unit forms two further hydrogen bonds to neighbouring tetramers through atoms O6 (H-bond acceptor) and N1 (H-bond donor). These connected chains pack in a zig-zag, layered arrangement along the −1 0 1 plane.
Figure 1.
A tetramer unit of the 5FC-4Cl35DNBA molecular complex. (a) and (b) are the N–H···O hydrogen bonds forming the dimer; (c) and (d) are the N–H···O and C–H···F hydrogen bonds, respectively, that connect the two dimers together into the tetramer.
Figure 1.
A tetramer unit of the 5FC-4Cl35DNBA molecular complex. (a) and (b) are the N–H···O hydrogen bonds forming the dimer; (c) and (d) are the N–H···O and C–H···F hydrogen bonds, respectively, that connect the two dimers together into the tetramer.
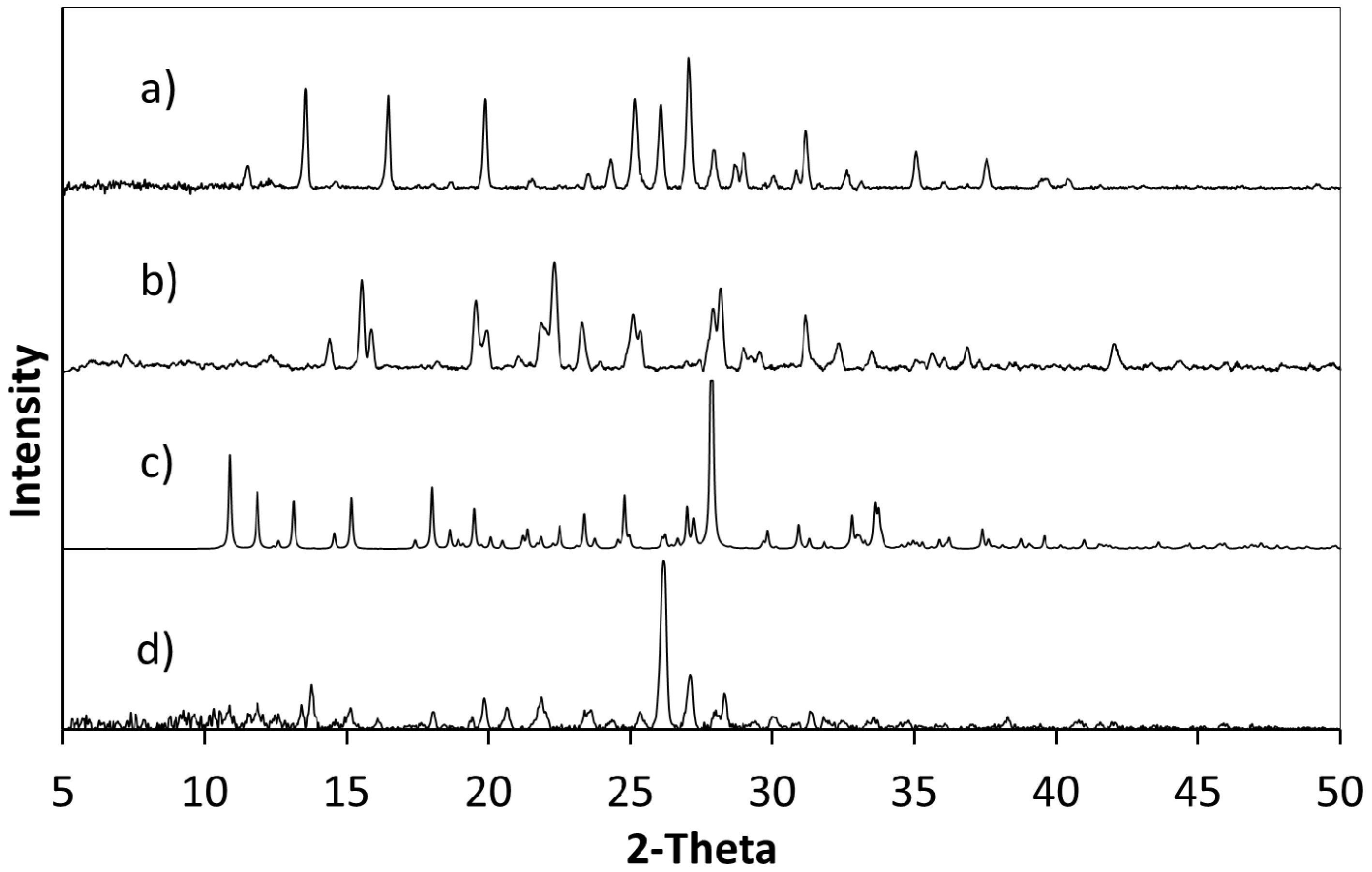
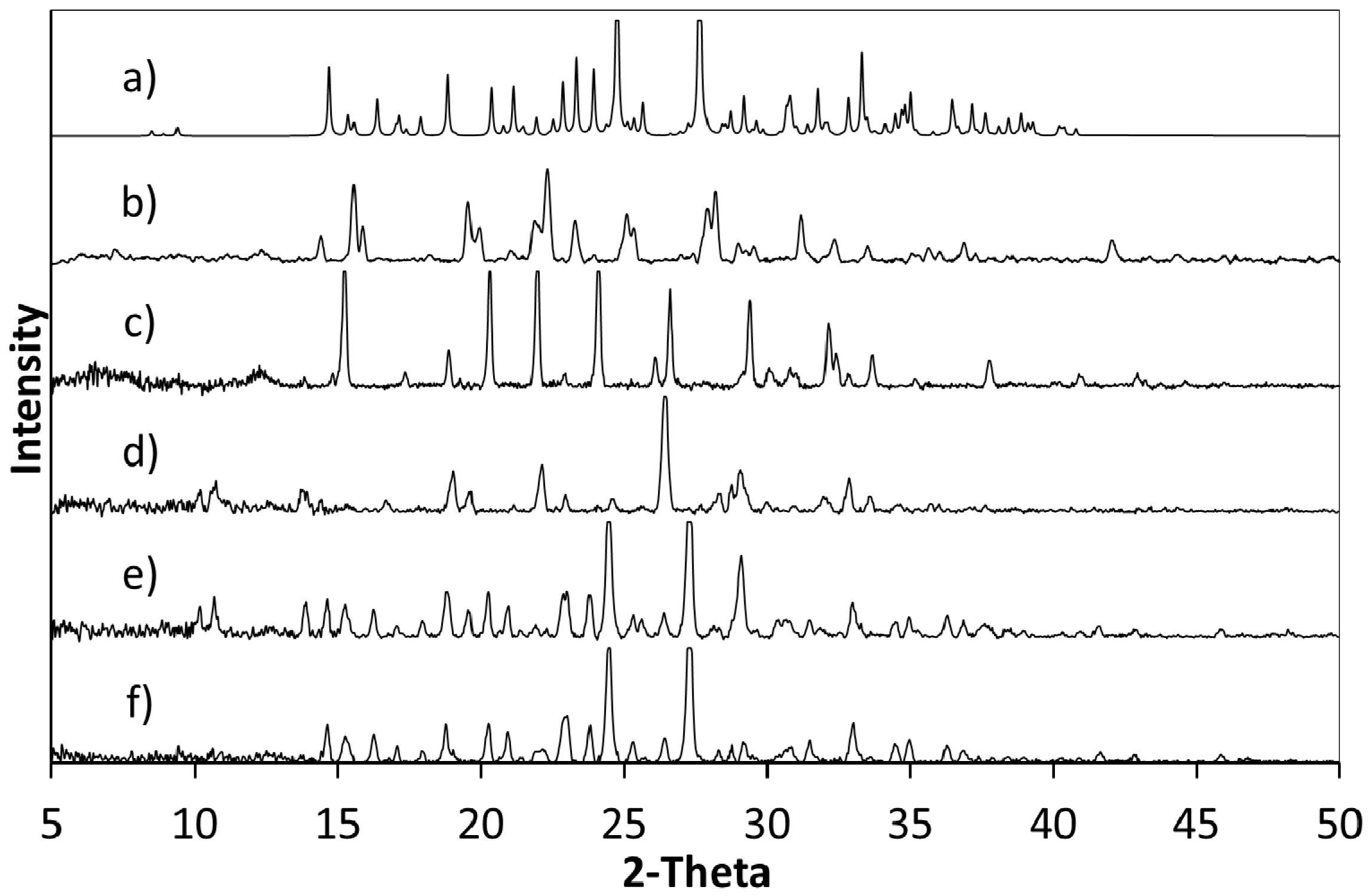
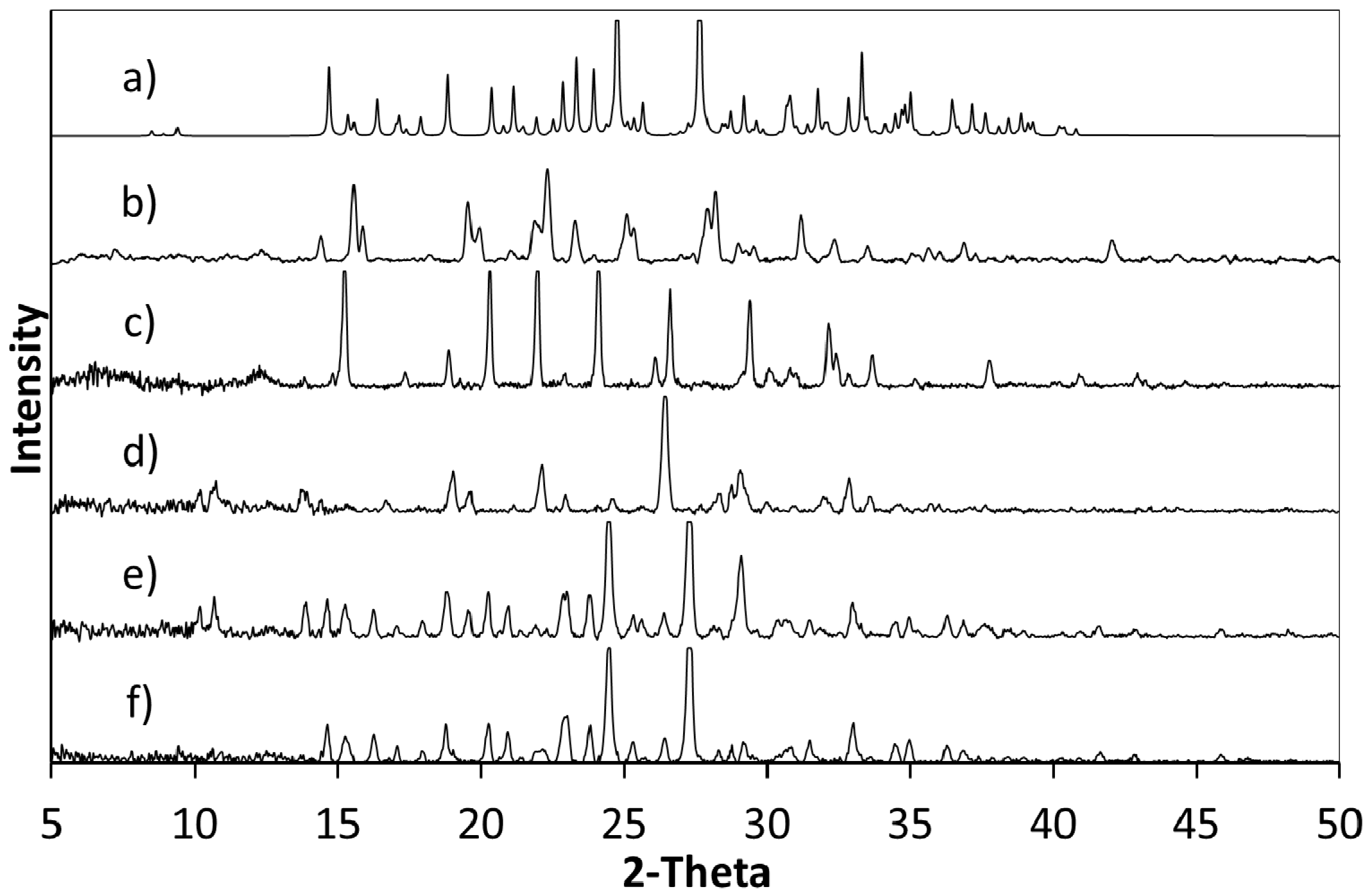
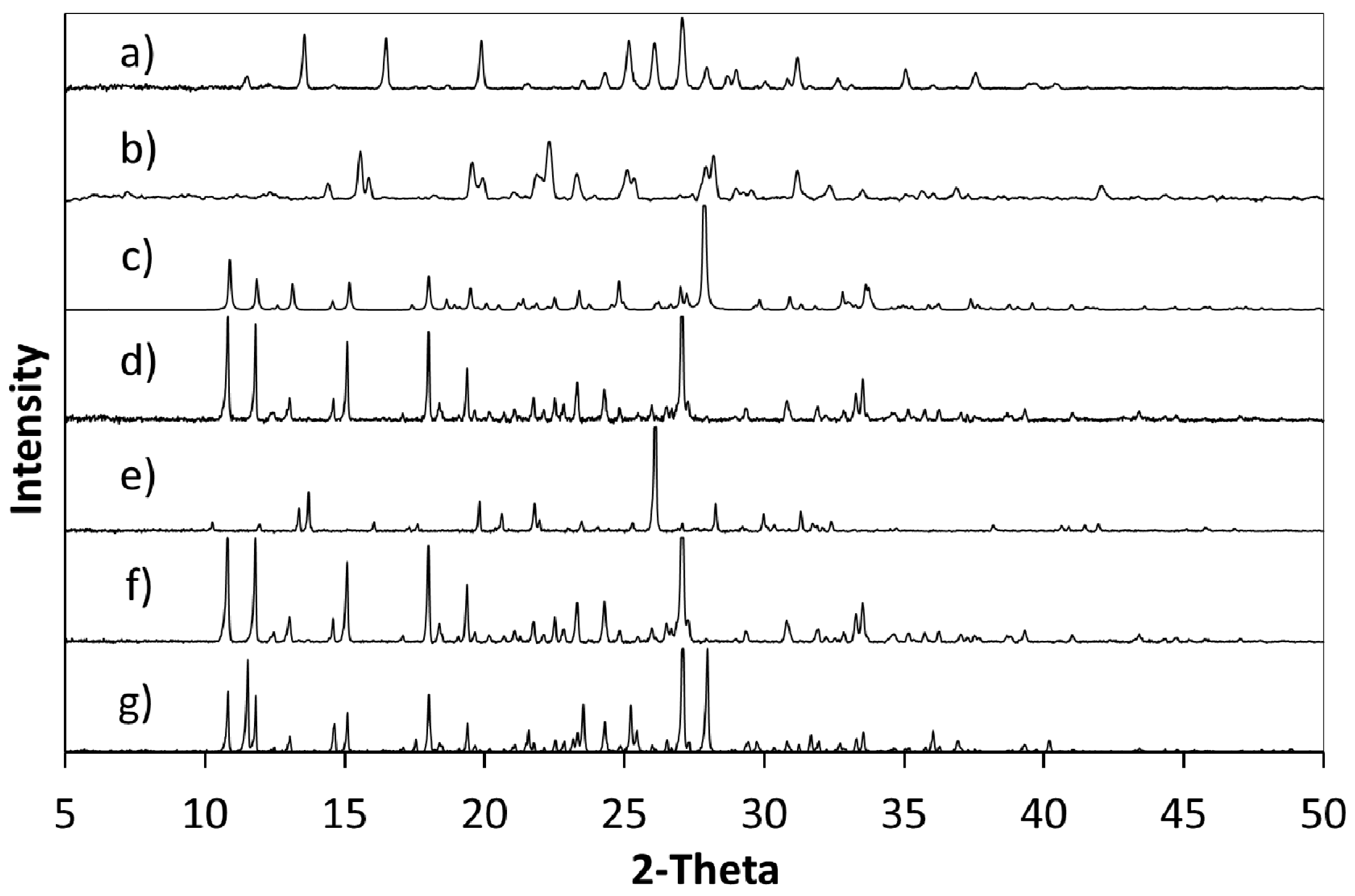
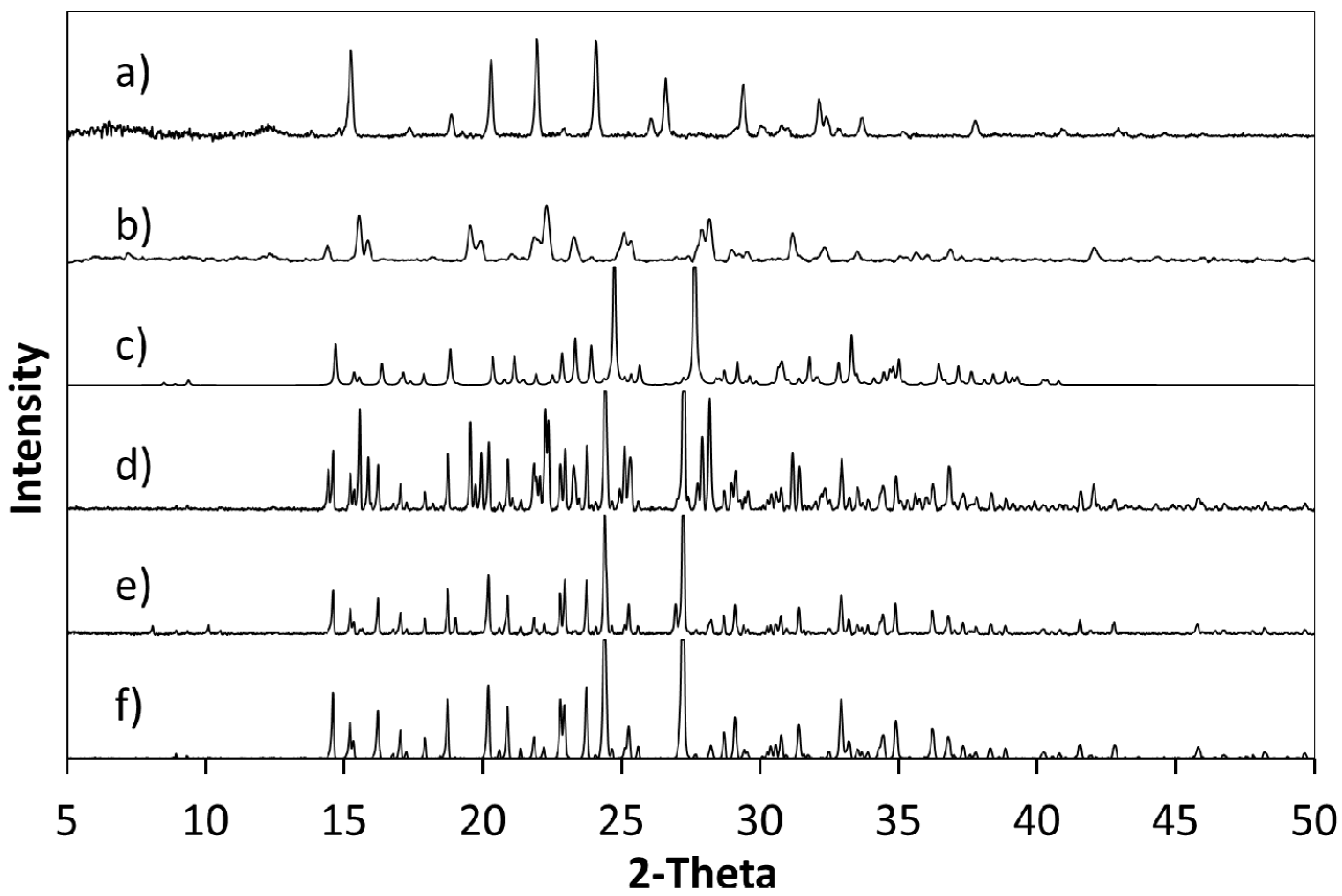
Powder X-ray diffraction (PXRD) shows that when crystallisation is initiated from a solution of a 1:1 ratio or a 2:1 ratio of starting materials (5FC:4Cl35DNBA), the same 1:1 5FC-4Cl35DNBA complex is formed, which is indicated by the intense peaks at 24.4° 2θ and 27.3° 2θ in the PXRD (
Figure 2). However, when crystallised using a 1:2 starting ratio of 5FC to 4Cl35DNBA a unique and intense PXRD peak can be seen at 26.4° 2θ. This does not correspond to the 5FC-4Cl35DNBA complex, nor is it representative of either 5FC or 4Cl35DNBA (including any known hydrates or solvates). This would infer that there is new material present, which could be a new polymorph, a new multi-component molecular complex or another solvate of the starting materials; further analysis is required to elucidate this phase.
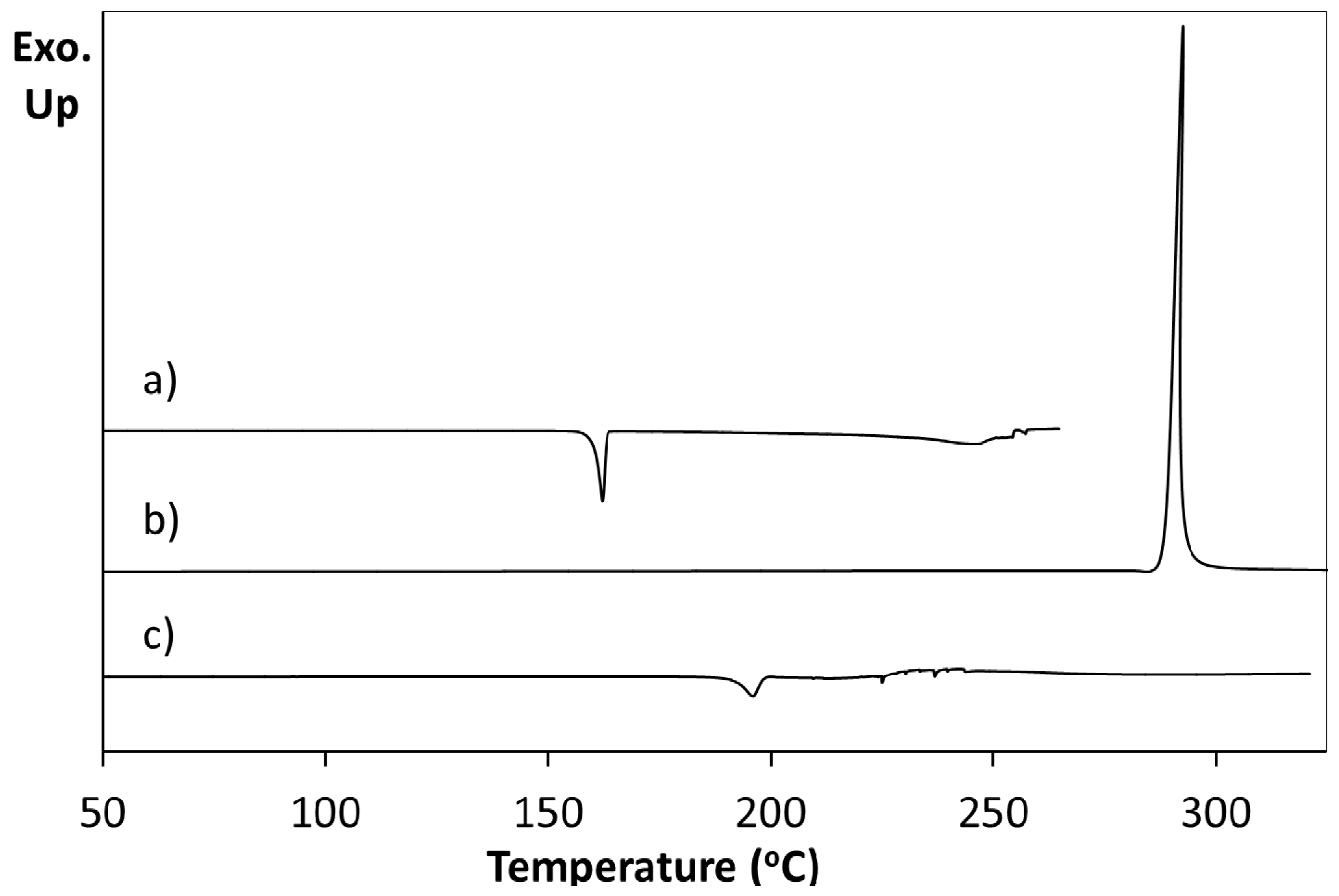
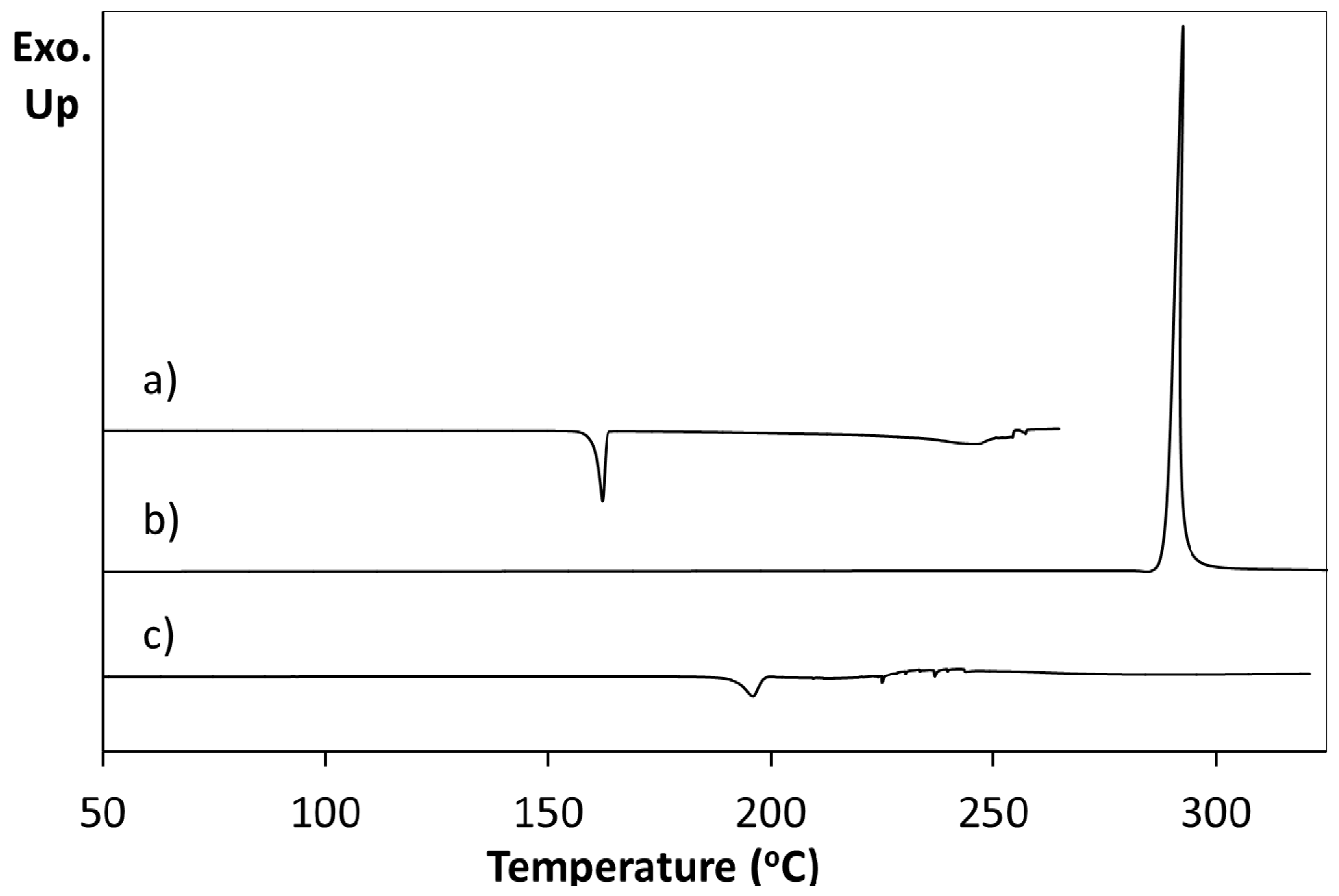
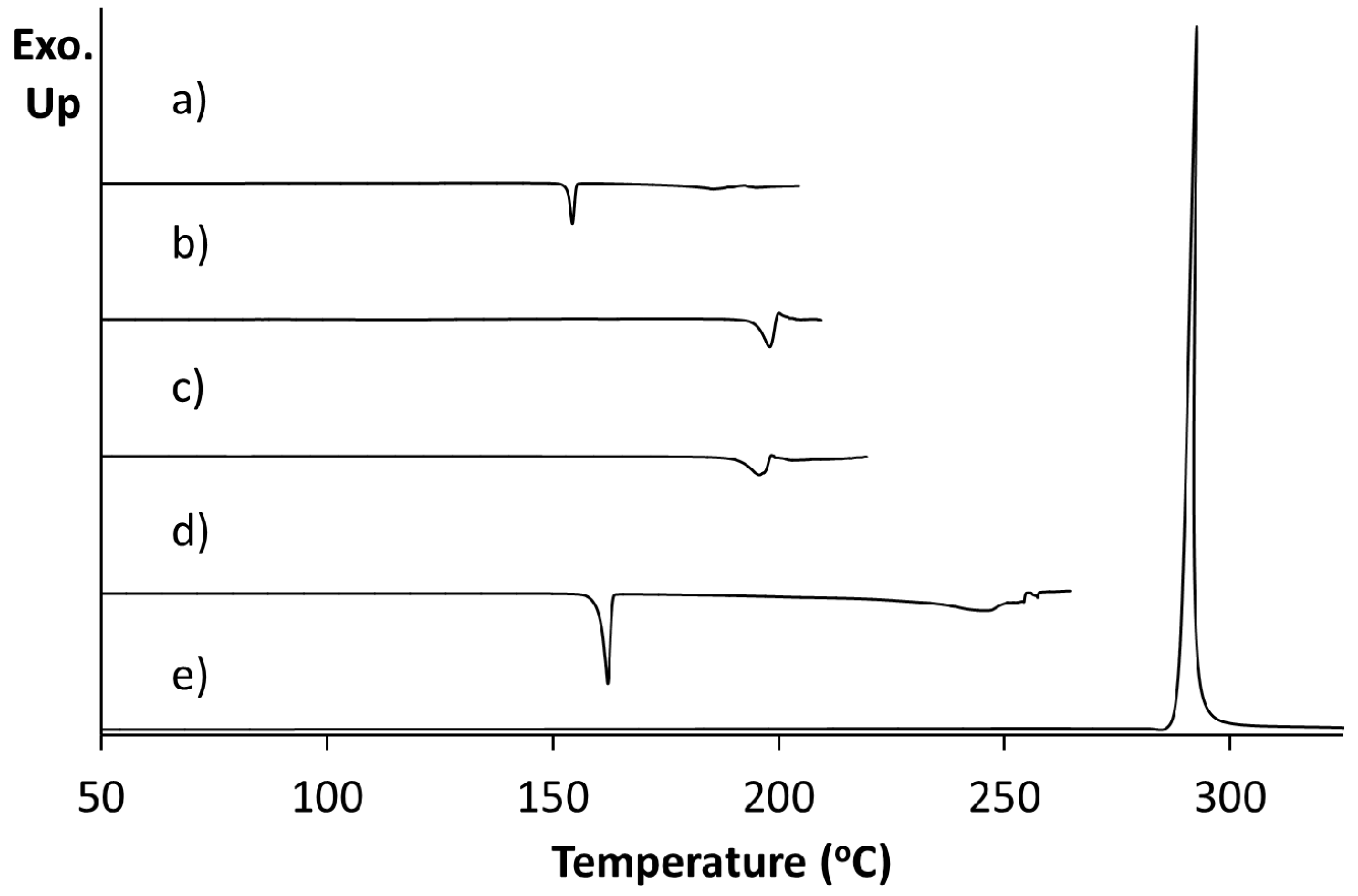
Differential scanning calorimetry (DSC) of a pure sample of 5FC-4Cl35DNBA arising from 1:1 evaporative crystallisation (as characterised through PXRD) shows a single endothermic event at 196.1 °C prior to decomposition (
Figure 3a). This does not correspond to the DSC traces of the 4Cl35DNBA and 5FC starting materials. 4Cl35DNBA displays an endothermic event at 162.2 °C (
Figure 3b), which corresponds to its literature melting point of 159–162 °C and 5FC displays an exothermic event at 292.6 °C, which corresponds to 5FC thermal decomposition (296 °C) (
Figure 3c).
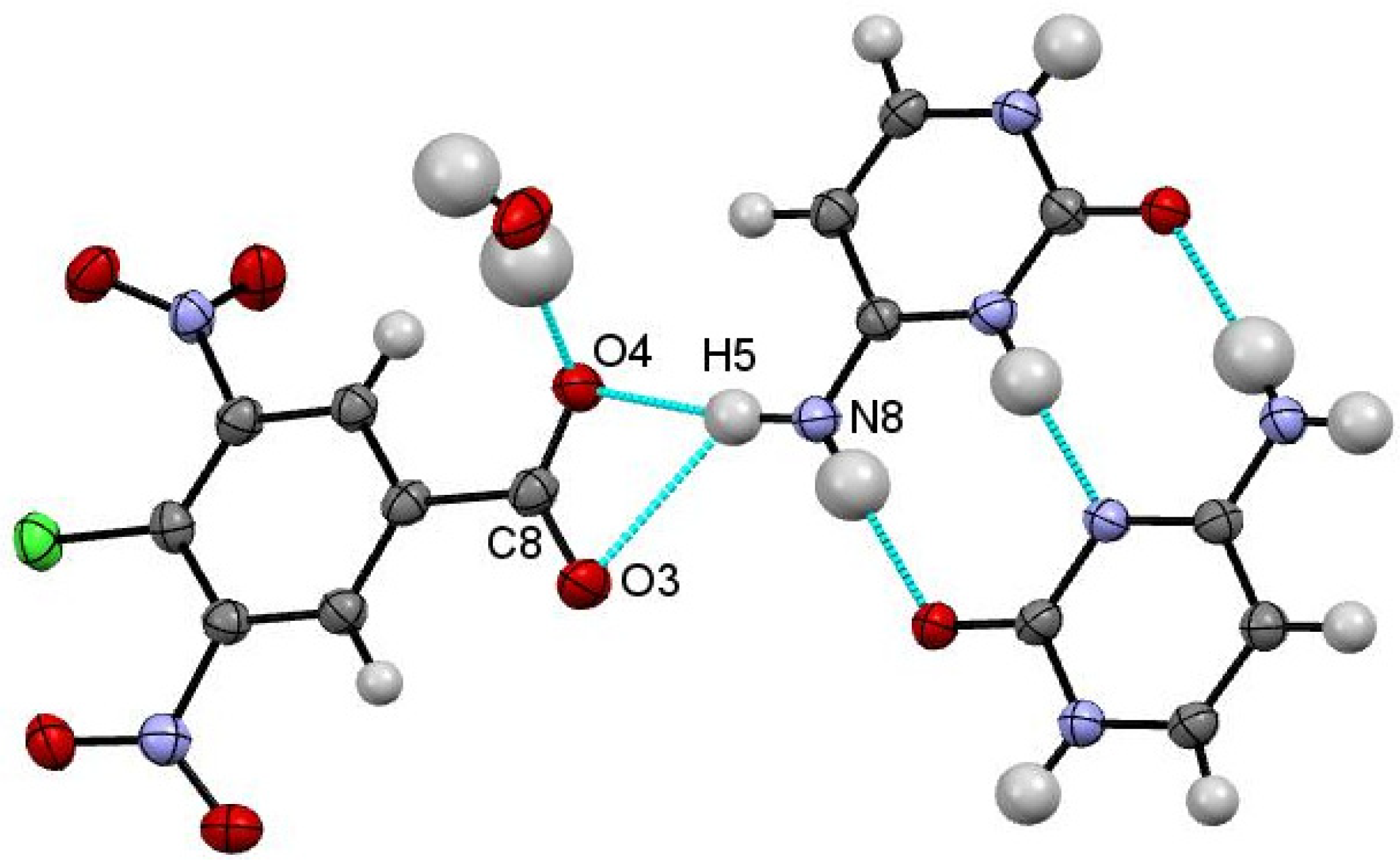
In contrast to the 5FC-4Cl35DNBA molecular complex, cytosine (CYT) forms a monohydrated 2:1 complex with 4Cl35DNBA (CYT-4Cl35DNBA). The asymmetric unit contains a water molecule, a single 4Cl35DNBA molecule and two cytosine molecules (
Figure 4). Interestingly, only one cytosine molecule remains neutral while the other accepts a proton from the carboxylic acid group of the 4Cl35DNBA molecule. De-protonation of the carboxylic acid group is indicated by the similarities in the C8–O3 and C8–O4 bond distances, at 1.230(3) Å and 1.258(3) Å, respectively.
Figure 2.
(a) PXRD pattern generated from the single crystal determined structure of 5FC-4Cl35DNBA for comparison with PXRD patterns of (b) 4Cl35DNBA; (c) 5FC; and products of evaporative crystallisation of 5FC: 4Cl35DNBA in various ratios: (d) 1:2; (e) 2:1; and (f) 1:1 (all crystallisations in ethanol and water 1:1 by volume).
Figure 2.
(a) PXRD pattern generated from the single crystal determined structure of 5FC-4Cl35DNBA for comparison with PXRD patterns of (b) 4Cl35DNBA; (c) 5FC; and products of evaporative crystallisation of 5FC: 4Cl35DNBA in various ratios: (d) 1:2; (e) 2:1; and (f) 1:1 (all crystallisations in ethanol and water 1:1 by volume).
Figure 3.
Differential scanning calorimetry of (a) 5FC-4Cl35DNBA; (b) 4Cl35DNBA; (c) 5FC.
Figure 3.
Differential scanning calorimetry of (a) 5FC-4Cl35DNBA; (b) 4Cl35DNBA; (c) 5FC.
Figure 4.
Asymmetric unit of CYT-4Cl35DNBA displaying the hydrogen bonding interactions of the deprotonated 4Cl35DNBA carboxylic acid group with the cytosine homodimer.
Figure 4.
Asymmetric unit of CYT-4Cl35DNBA displaying the hydrogen bonding interactions of the deprotonated 4Cl35DNBA carboxylic acid group with the cytosine homodimer.
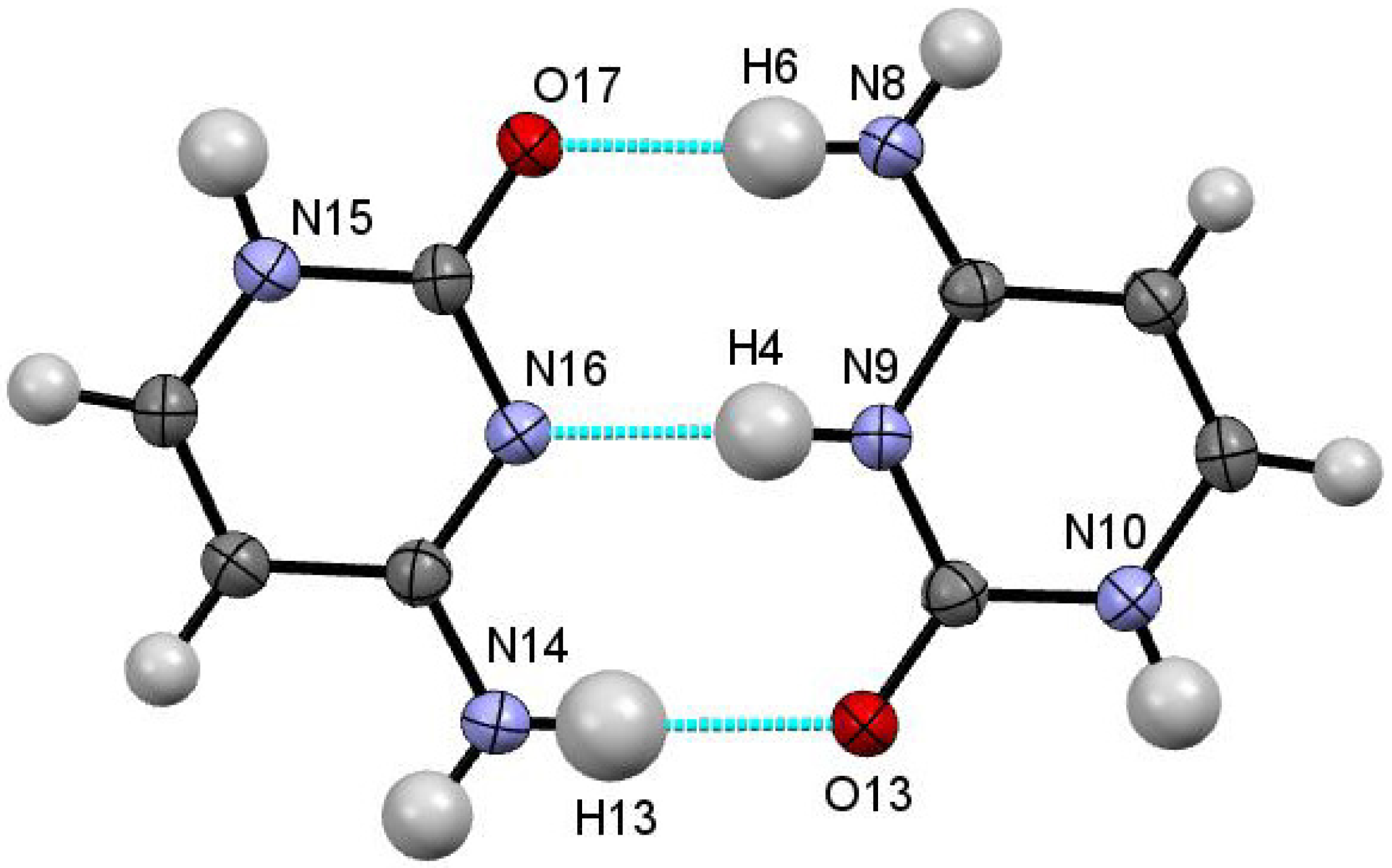
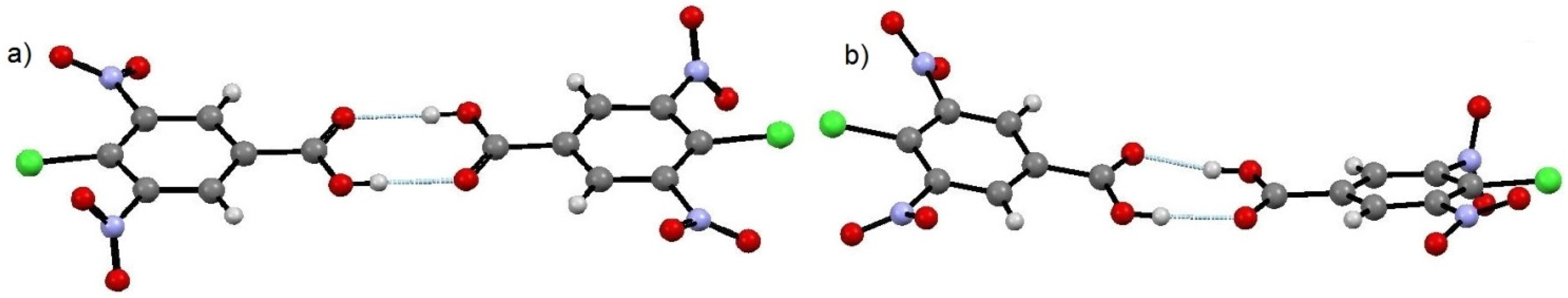
Variation in the protonation states of the two cytosine species facilitates formation of a cytosine homodimer bound through a pseudo Watson-Crick hydrogen bonding interaction consisting of three moderately strong hydrogen bonds N8–H6···O17, N9–H4···N16 and N14–H13···O13 with D···A distances of 2.809 (3) Å, 2.820 (3) Å and 2.865 (3) Å, respectively (
Figure 5). Three further prominent hydrogen bonds link the three components of the molecular complex, and are also shown in
Figure 4. Atom H5 acts as a bifurcated hydrogen bond donor to the carboxylate ion of 4Cl35DNBA and the water molecule forms a moderate strength hydrogen bond with atom O4 of the carboxylate group. The cytosine and 4Cl35DNBA molecules lie parallel to the 1 0 −1 plane, with each of the inversion related layers hydrogen bonding to another through a water molecule.
Figure 5.
A cytosine dimer with pseudo Watson-Crick hydrogen bonding arrangement within CYT-4Cl35DNBA.
Figure 5.
A cytosine dimer with pseudo Watson-Crick hydrogen bonding arrangement within CYT-4Cl35DNBA.
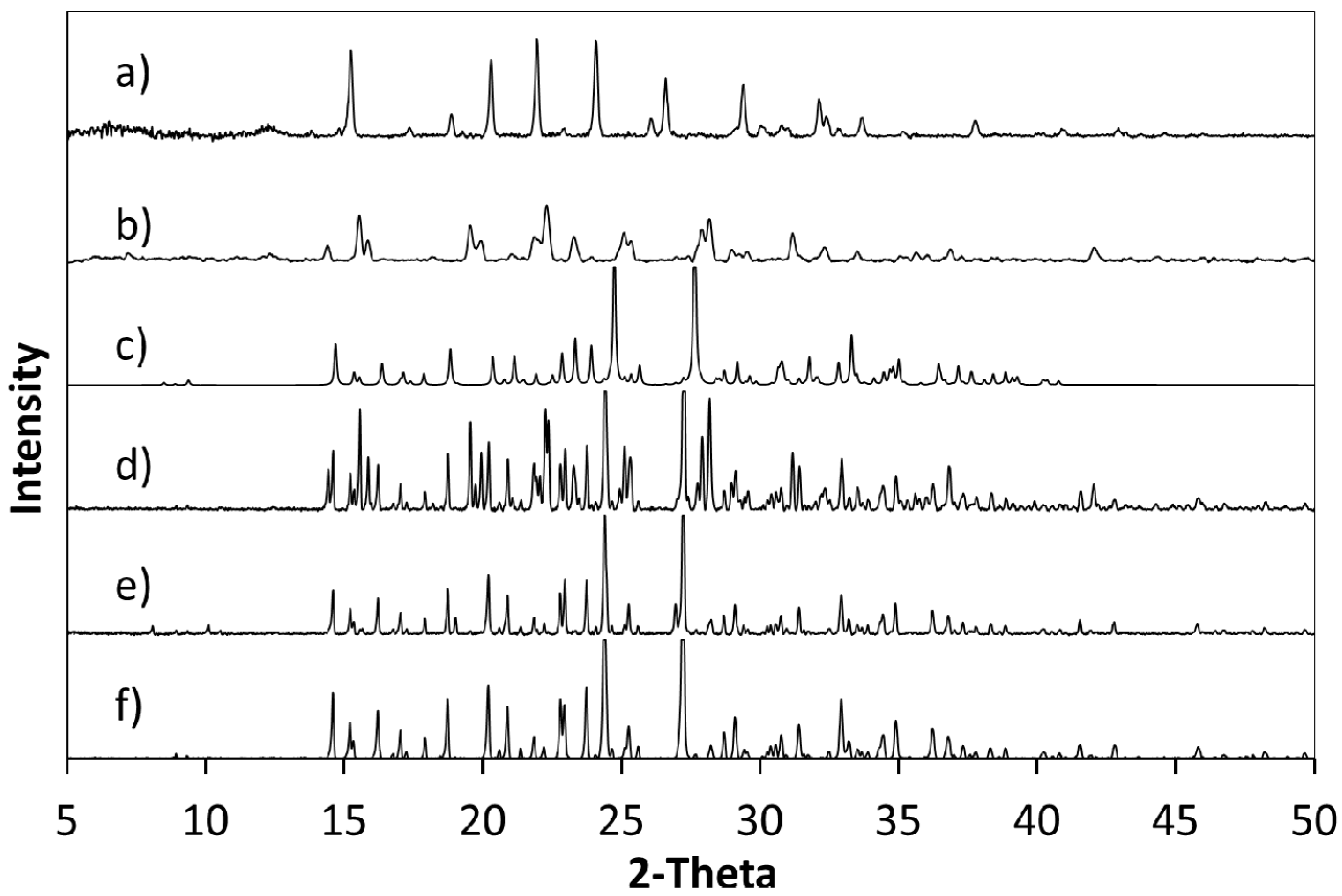
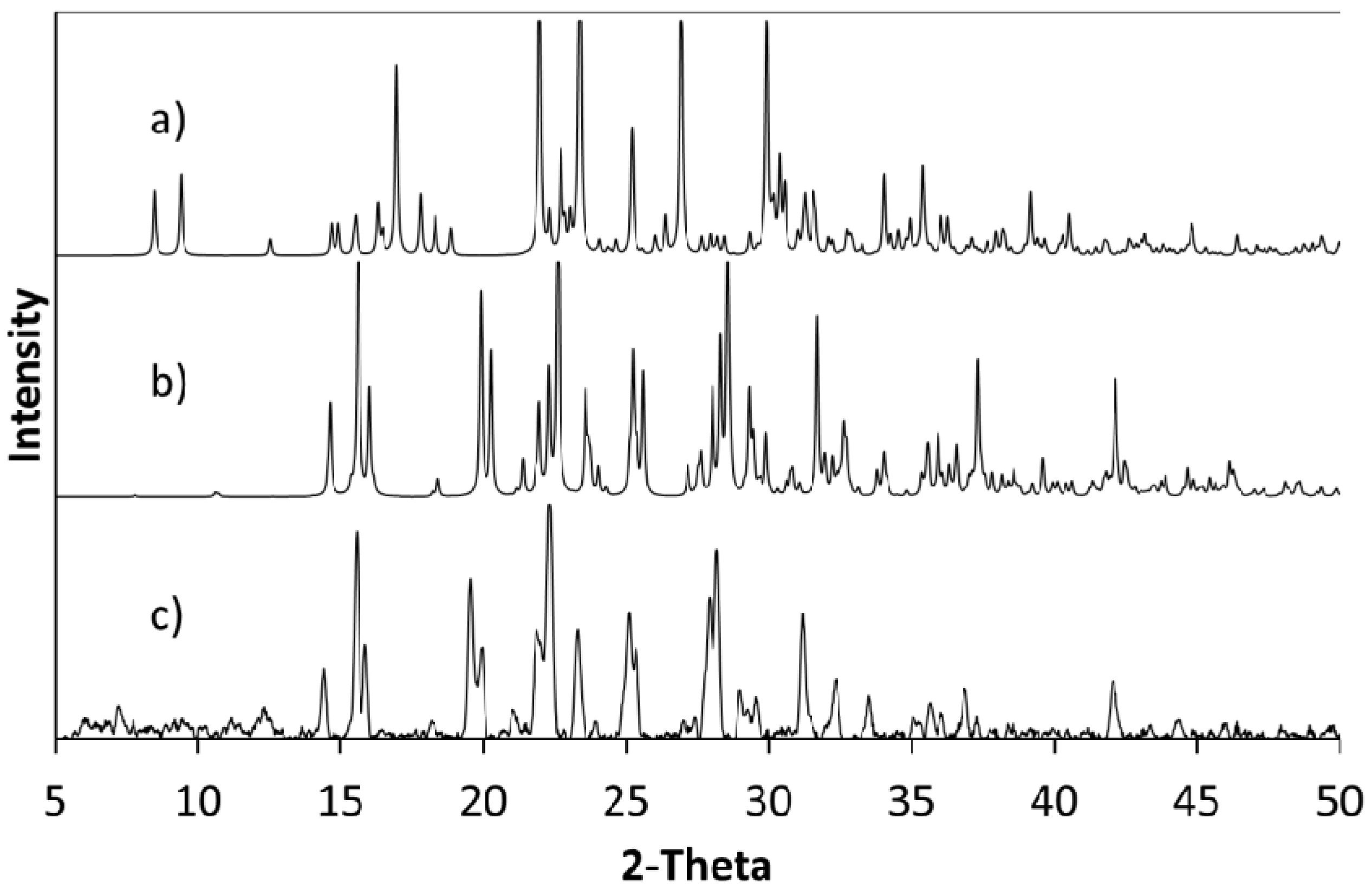
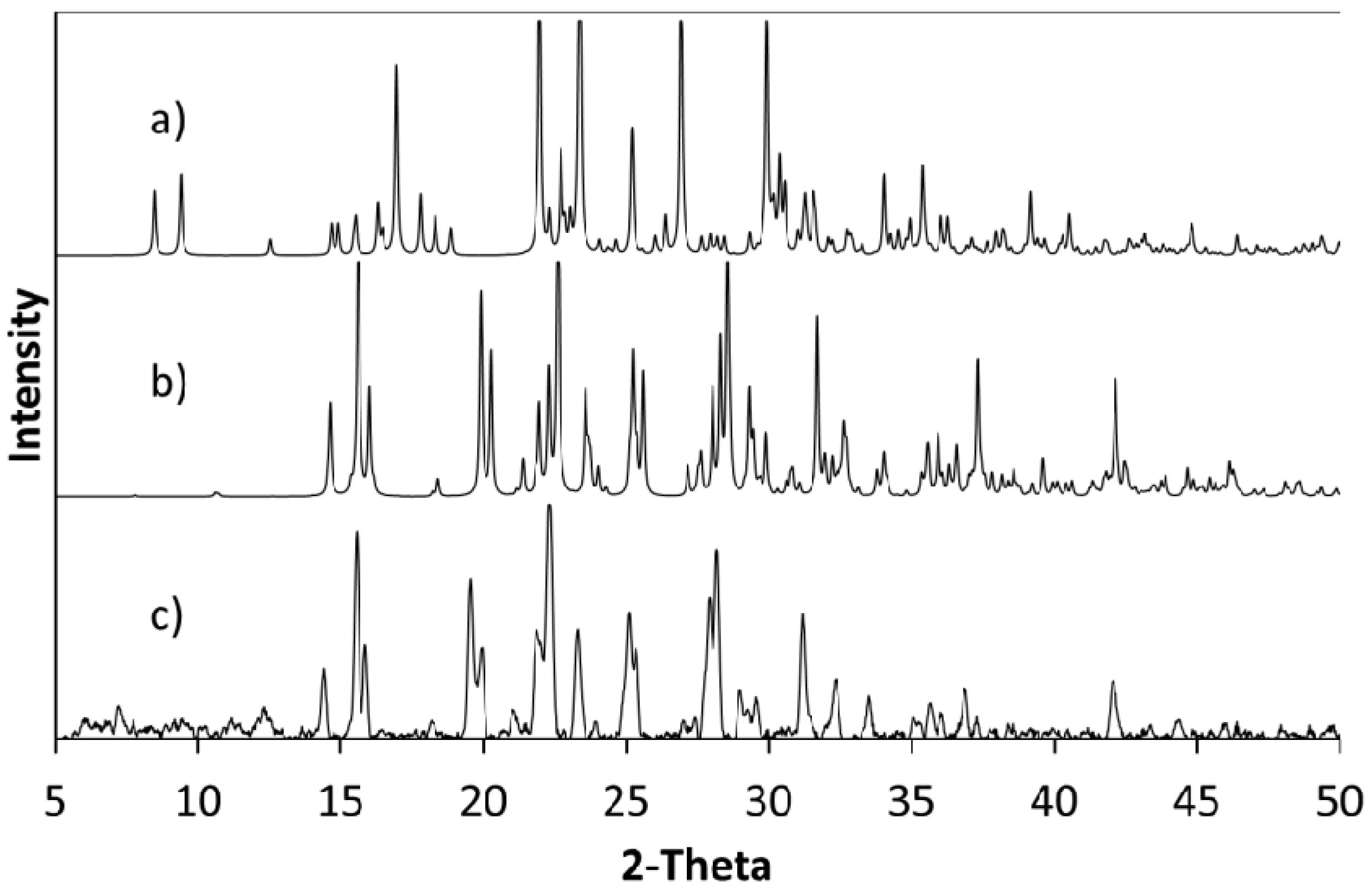
The products of further evaporative crystallisations of cytosine with 4Cl35DNBA were analysed using a combination of single crystal X-ray diffraction (SXRD) and PXRD (
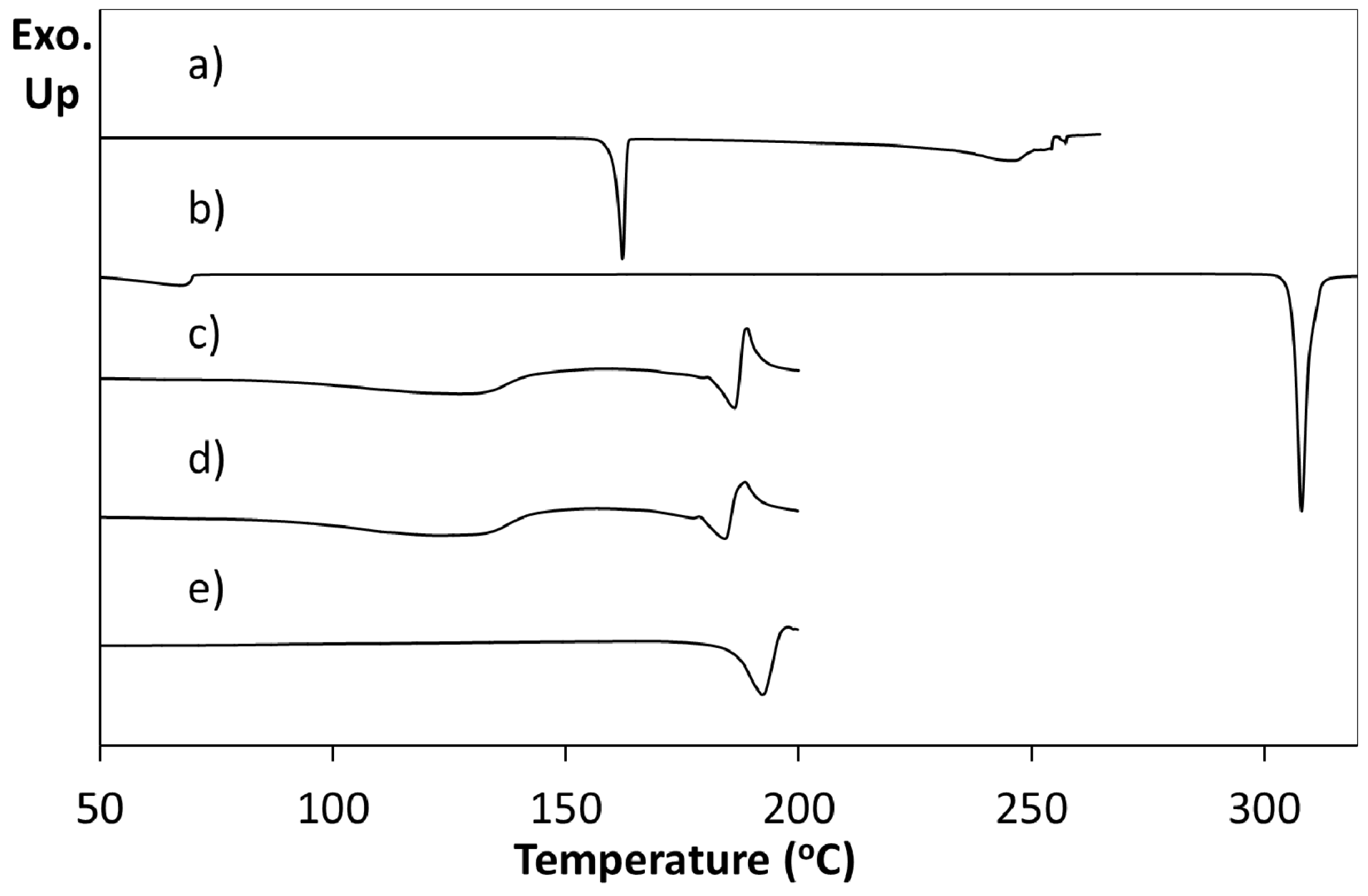
Figure 6). This confirmed that when using a 2:1 ratio of cytosine to 4Cl35DNBA the initial stoichiometry was retained and the 2:1 molecular complex CYT-4Cl35DNBA was produced. However, when a 1:1 stoichiometry of starting materials was used a crystalline powder was formed and the resulting PXRD pattern does not correspond to any of the starting materials (including polymorphs or solvates) or to the 2:1 molecular complex; it may, therefore, represent an additional unknown complex which could be a 1:1 molecular complex of cytosine and 4Cl35DNBA or a non-hydrated molecular complex of cytosine and 4Cl35DNBA. The DSC trace for this form is simpler than that observed for the 2:1 CYT-4Cl35DNBA a single endothermic event at a similar onset temperature to the second event in the 2:1 molecular complex (1:1, 192.3 °C; 2:1 184.2 °C) (
Figure 7). The DSC trace for the 2:1 complex has an initial broad endothermic event at 95 °C which may reflect a desolvation process or a transformation into a different polymorphic or stoichiometric form.
Figure 6.
PXRD patterns of (a) cytosine; (b) 4Cl35DNBA and (c) a pattern calculated from the single crystal structure of CYT-4Cl35DNBA. These were compared with PXRD patterns of bulk products of evaporative crystallisation of cytosine and 4Cl35DNBA from different crystallisation conditions using (d) a 2:1 starting material ratio from ethanol and water (1:1 by volume); (e) a 1:1 starting material ratio from acetone and water (1:1 by volume); (f) a 2:1 starting material ratio in acetone and water (1:1 by volume) and (g) supersaturated preparation from which the single crystal used for full SXRD data was obtained.
Figure 6.
PXRD patterns of (a) cytosine; (b) 4Cl35DNBA and (c) a pattern calculated from the single crystal structure of CYT-4Cl35DNBA. These were compared with PXRD patterns of bulk products of evaporative crystallisation of cytosine and 4Cl35DNBA from different crystallisation conditions using (d) a 2:1 starting material ratio from ethanol and water (1:1 by volume); (e) a 1:1 starting material ratio from acetone and water (1:1 by volume); (f) a 2:1 starting material ratio in acetone and water (1:1 by volume) and (g) supersaturated preparation from which the single crystal used for full SXRD data was obtained.
Figure 7.
Differential scanning calorimetry of (a) 4Cl35DNBA and (b) cytosine, together with those for products of the evaporative crystallisation of cytosine and 4ClDNBA in: (c) a 2:1 starting material ratio in ethanol and water (1:1 by volume); (d) a 2:1 starting material ratio in acetone and water (1:1 by volume); (e) a 1:1 starting material ratio in acetone and water (1:1 by volume).
Figure 7.
Differential scanning calorimetry of (a) 4Cl35DNBA and (b) cytosine, together with those for products of the evaporative crystallisation of cytosine and 4ClDNBA in: (c) a 2:1 starting material ratio in ethanol and water (1:1 by volume); (d) a 2:1 starting material ratio in acetone and water (1:1 by volume); (e) a 1:1 starting material ratio in acetone and water (1:1 by volume).
2.4. Translation into Cooling Crystallisation
Four cooling crystallisations of 4Cl35DNBA with cytosine or 5FC with differing conditions (
Table 1); all produced very fine, yellow needle crystals. SXRD analysis of each sample showed that several crystals in each preparation corresponded to the targeted molecular complex previously discovered through evaporative routes as described above.
Table 1.
Conditions of cooling crystallisations using a ReactArray Reaction Block.
Table 1.
Conditions of cooling crystallisations using a ReactArray Reaction Block.
| Sample | Components | Ratio | Solvent | Solvent Ratio | Cooling profile |
|---|
| A | cytosine, 4Cl35DNBA | 1:1 | acetone and water | 1:1 | 1 |
| B | 5FC, 4Cl35DNBA | 1:1 | acetone and water | 1:1 | 1 |
| C | 5FC, 4Cl35DNBA | 1:1 | ethanol and water | 1:1 | 2 |
| D | 5FC, 4Cl35DNBA | 1:2 | ethanol and water | 1:1 | 2 |
PXRD analysis of sample A (
Figure 12) confirmed that CYT-4Cl35DNBA was produced, though it is important to note that the peak intensities do vary due to preferred orientation. The bulk of samples B, C and D were confirmed to be 5FC-4Cl35DNBA (
Figure 13).
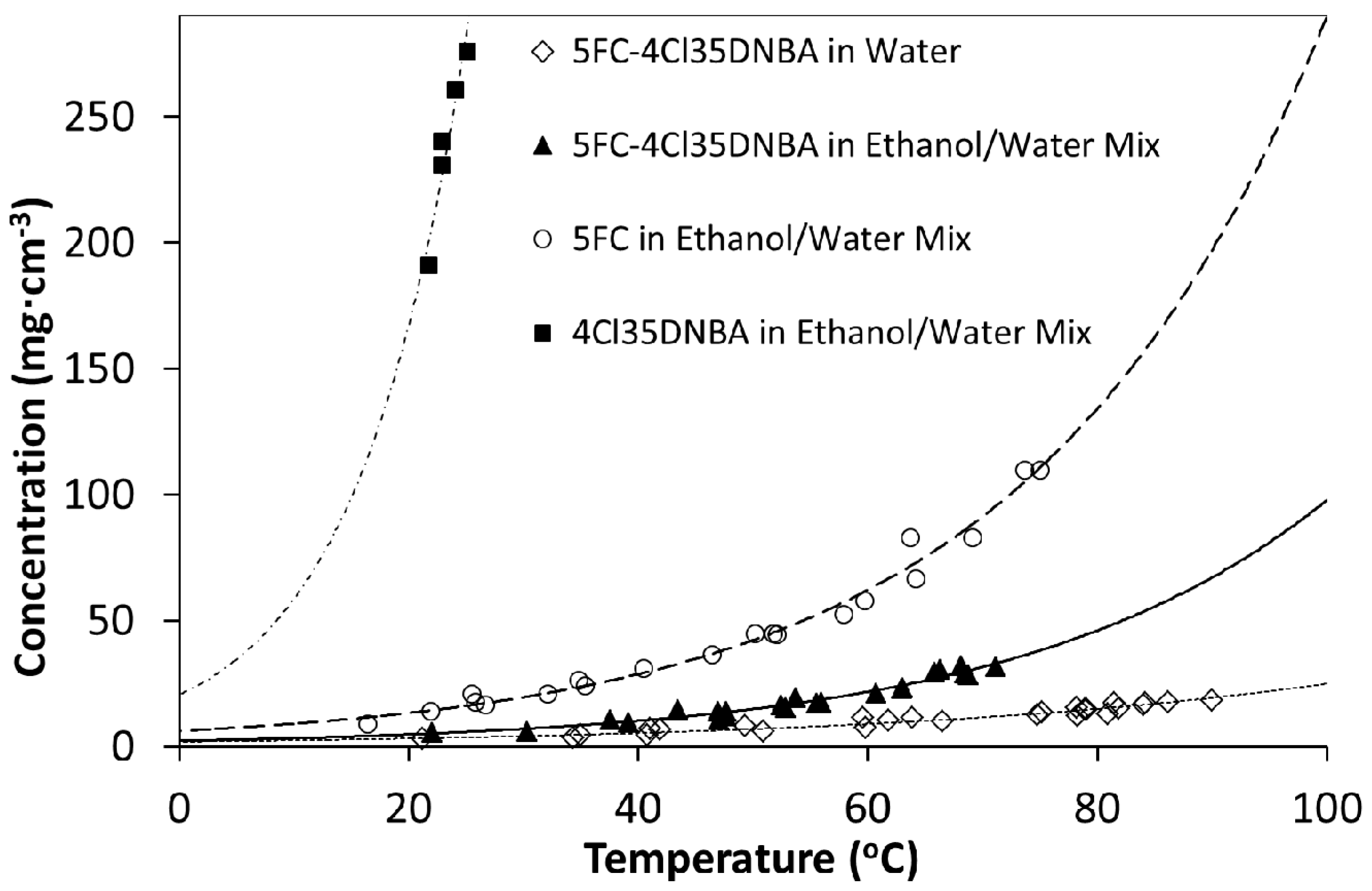
Solubility analysis showed 4Cl35DNBA to be approximately six times more soluble in ethanol than 5FC. 5FC was more soluble in ethanol than in water and 4Cl35DNBA did not dissolve in water alone. In light of this a mixture of ethanol and water was used to help to reduce the solubility of 4Cl35DNBA. Turbidity measurements showed a 7:3 mixture of ethanol to water (by volume) to be a suitable ratio of solvents for this system.
Figure 12.
PXRD analysis of product of cooling crystallisation A. The plot compares the PXRD patterns for starting materials: (a) CYT; (b) 4Cl35DNBA, and the PXRD pattern for (c) CYT-4Cl35DNBA calculated from single crystal data with that of (d) Product A. The peak positions show that A contains the same CYT-4Cl35DNBA complex as found in the 2:1 evaporative experiment, though the intensities are heavily affected by preferred orientation.
Figure 12.
PXRD analysis of product of cooling crystallisation A. The plot compares the PXRD patterns for starting materials: (a) CYT; (b) 4Cl35DNBA, and the PXRD pattern for (c) CYT-4Cl35DNBA calculated from single crystal data with that of (d) Product A. The peak positions show that A contains the same CYT-4Cl35DNBA complex as found in the 2:1 evaporative experiment, though the intensities are heavily affected by preferred orientation.
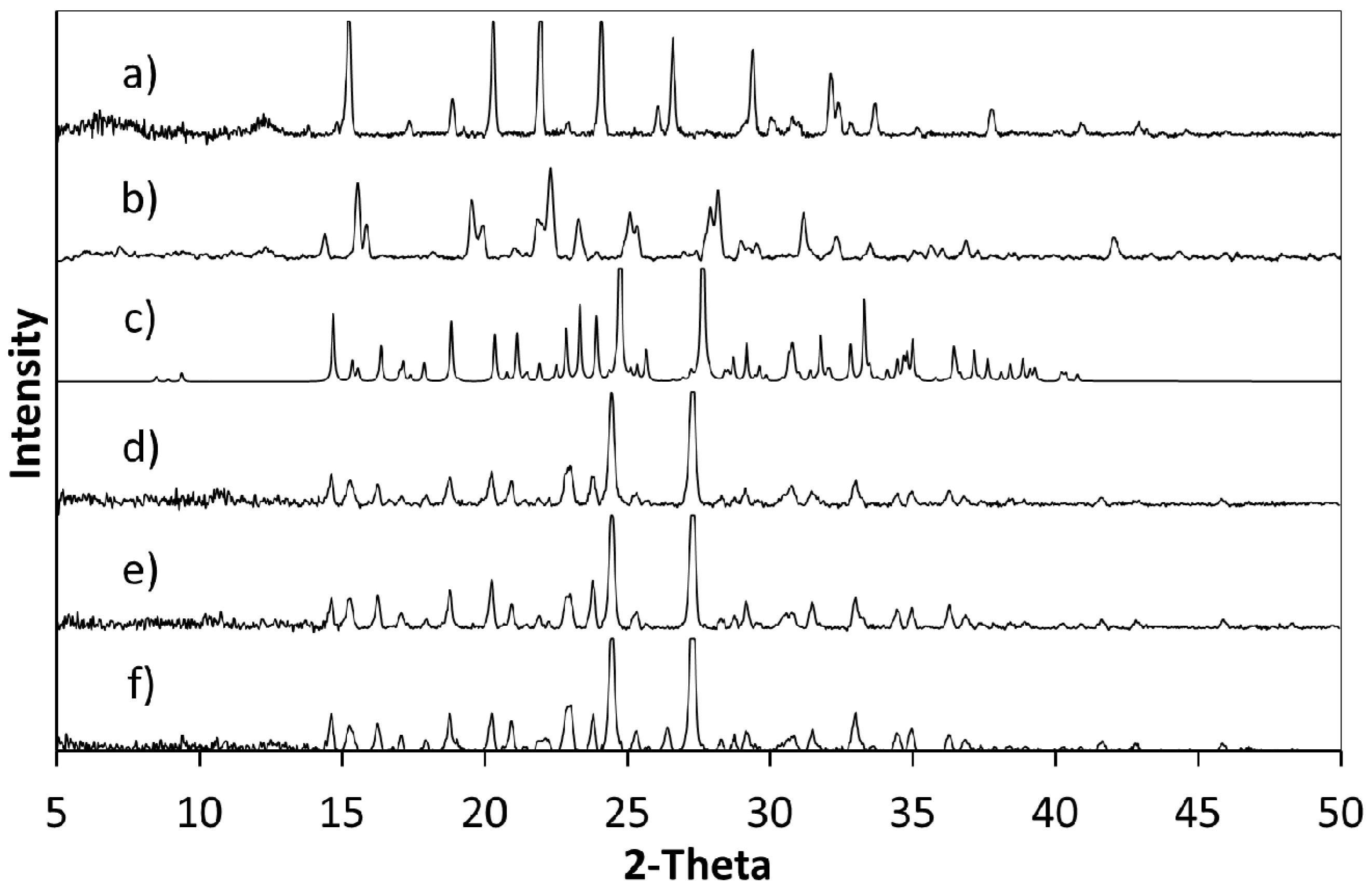
Figure 13.
PXRD analysis of cooling crystallisations B, C and D. (a) 5FC; (b) 4Cl35DNBA; (c) 5FC-4Cl35DNBA; (d) Product B; (e) Product C; (f) Product D.
Figure 13.
PXRD analysis of cooling crystallisations B, C and D. (a) 5FC; (b) 4Cl35DNBA; (c) 5FC-4Cl35DNBA; (d) Product B; (e) Product C; (f) Product D.
The formation of 5FC-4CL35DNBA could be followed during the cooling crystallisation using turbidimetric methods using a Crystal16 apparatus (
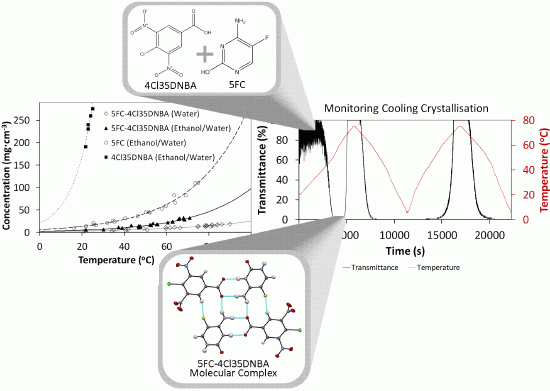
Figure 14). Initially the turbidity signal is noisy due to the particle size but ranges from 60% to 100% transmittance. As the temperature increases the solution becomes turbid and the transmittance drops to 0%. This increase in turbidity with an increase in temperature in the early stages of the process is perhaps counterintuitive and indicates the initial, rapid, crystallisation of the less soluble multi-component complex (as shown by previous solubility measurements). As the temperature continues to increase, 5FC-4Cl35DNBA dissolves and the solution develops 100% transmittance. 5FC-4Cl35DNBA then recrystallises upon cooling and the transmittance returns to 0%. Importantly, repetition of the heating and cooling temperature ramps then follows the expected trend, showing cloud points and clear points at the same temperatures as before. It should be noted that there are very few previous examples of the crystallisation of multi-component complexes by cooling at this small scale. This relatively simple methodology thus acts as an exemplar for the intended transfer of such processes into the continuous environment, by establishing the initial cooling stage.
It is also useful to note that these determinations illustrate that significant progress can be made in establishing such crystallisation processes using relatively simple analytical methods such as turbidity measurement. While acknowledging its inherent limitations, it is of value that this can be achieved even in the absence of full process analytical technologies (PAT), which will be implemented in future development of this work.
PXRD analysis of a range of samples from Crystal16 experiments confirm the formation of 5FC-4Cl35DNBA (
Figure 15) as indicated by the initial precipitation shown in the turbidity data. It should be noted, however, that when a slower cooling rate of 0.5 °C min
−1 was used, residual 4Cl35DNBA form II was present in the product as shown by the additional peaks in the PXRD pattern at 15.6° 2θ and 28.2° 2θ (
Figure 15d). While such a slow cooling rate is unlikely to be industrially relevant, for example, this element of the study was undertaken to explore the effect of cooling rate on the crystallisation products. The melt at 154.3 °C observed in DSC analysis (
Figure 16a) is much lower than those for the other samples, and is similar to the melting point of 4Cl35DNBA form II (162.2 °C). The other samples show endothermic events at 197.9 °C and 195.8 °C which are consistent with the 5FC-4Cl35DNBA sample produced from evaporative cooling as discussed above.
Figure 14.
Turbidity data from cooling crystallisation of 5FC with 4CL35DNBA (1:2) in ethanol and water (7:3 by volume). The temperature regimes used are shown as the red trace.
Figure 14.
Turbidity data from cooling crystallisation of 5FC with 4CL35DNBA (1:2) in ethanol and water (7:3 by volume). The temperature regimes used are shown as the red trace.
Figure 15.
PXRD analysis of cooling crystallisations carried out using the Crystal16 apparatus: (a) 5FC; (b) 4Cl35DNBA form II; (c) 5FC-4Cl35DNBA 5FC:4Cl35DNBA in ethanol:water (7:3 by volume) in (d) a 1:2 starting material ratio (0.5 °C min−1); (e) a 1:2 starting material ratio (2 °C min−1) and (f) a 1:1 starting material ratio (2 °C min−1).
Figure 15.
PXRD analysis of cooling crystallisations carried out using the Crystal16 apparatus: (a) 5FC; (b) 4Cl35DNBA form II; (c) 5FC-4Cl35DNBA 5FC:4Cl35DNBA in ethanol:water (7:3 by volume) in (d) a 1:2 starting material ratio (0.5 °C min−1); (e) a 1:2 starting material ratio (2 °C min−1) and (f) a 1:1 starting material ratio (2 °C min−1).
Figure 16.
Differential scanning calorimetry of products from cooling crystallisations using the Crystal16 involving 5FC-4Cl35DNBA 5FC:4Cl35DNBA in ethanol:water (7:3 by volume) in a: (a) a 1:2 ratio (0.5 °C min−1); (b) a 1:2 ratio (2 °C min−1); and (c) a 1:1 ratio (2 °C min−1); (d) 4Cl35DNBA; (e) 5FC.
Figure 16.
Differential scanning calorimetry of products from cooling crystallisations using the Crystal16 involving 5FC-4Cl35DNBA 5FC:4Cl35DNBA in ethanol:water (7:3 by volume) in a: (a) a 1:2 ratio (0.5 °C min−1); (b) a 1:2 ratio (2 °C min−1); and (c) a 1:1 ratio (2 °C min−1); (d) 4Cl35DNBA; (e) 5FC.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}