Moderate Temperature Dense Phase Hydrogen Storage Materials within the US Department of Energy (DOE) H2 Storage Program: Trends toward Future Development

Abstract
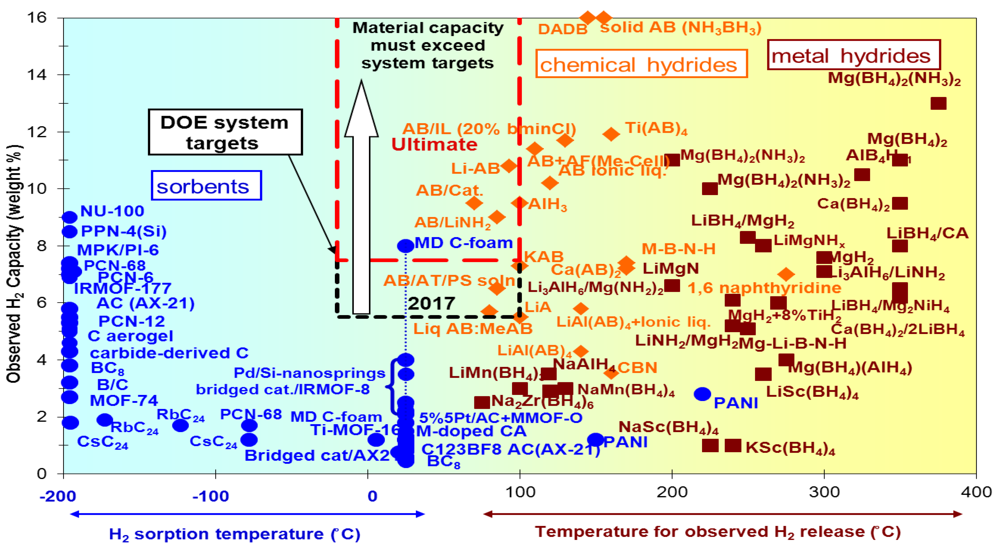
:1. Introduction
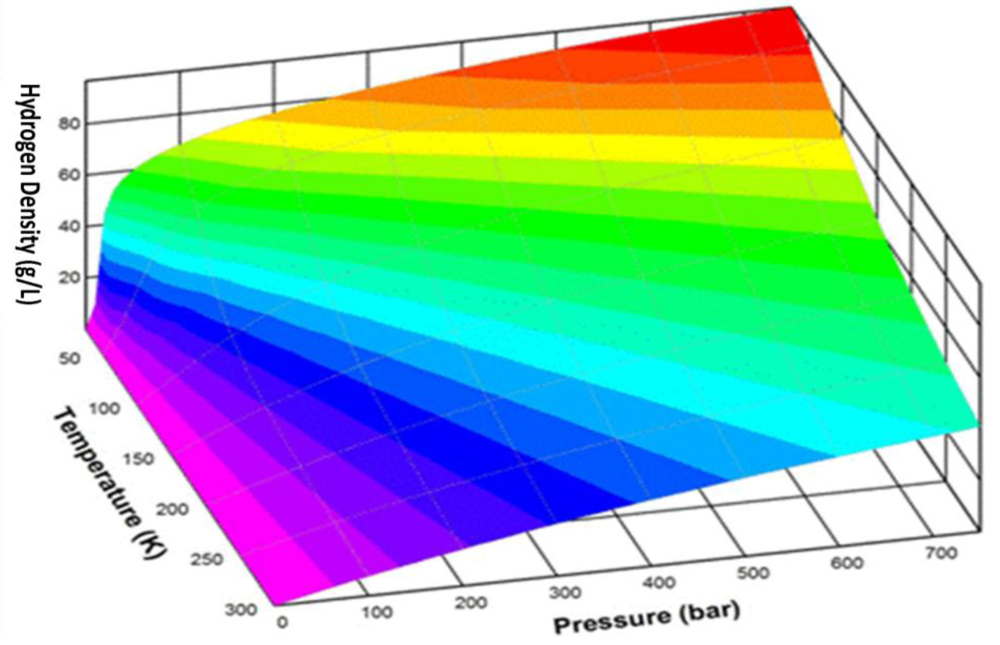
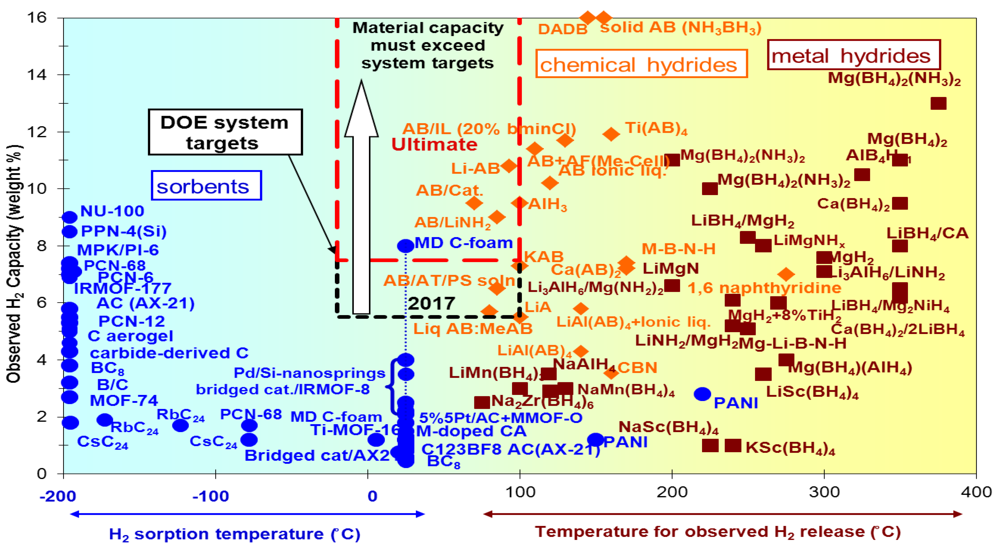
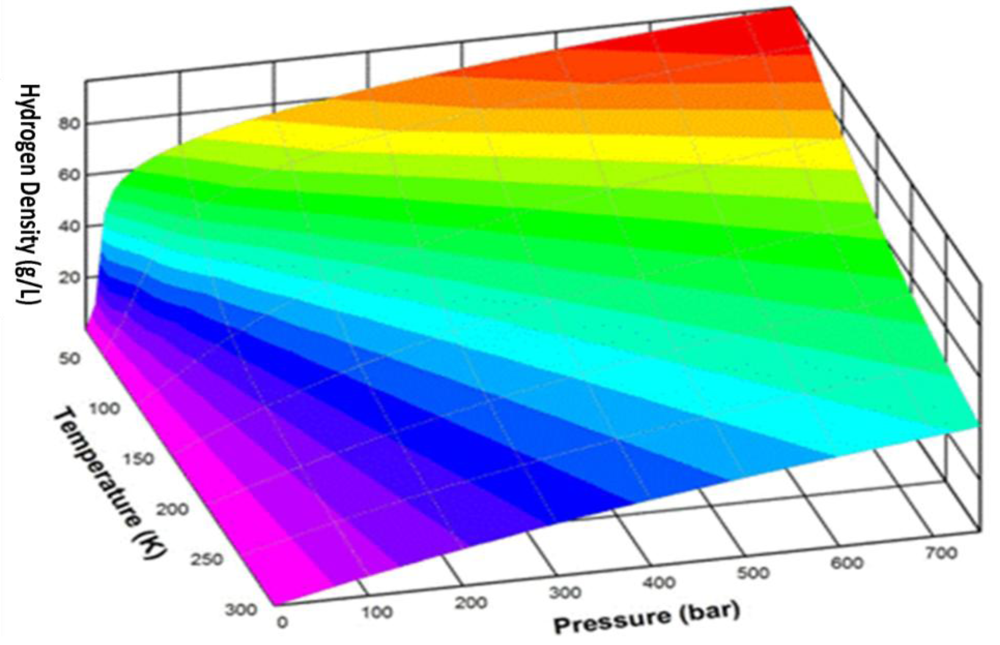
2. Moderate Temperature Condensed Phase Hydrogen Storage Materials
2.1. Metal Hydrides

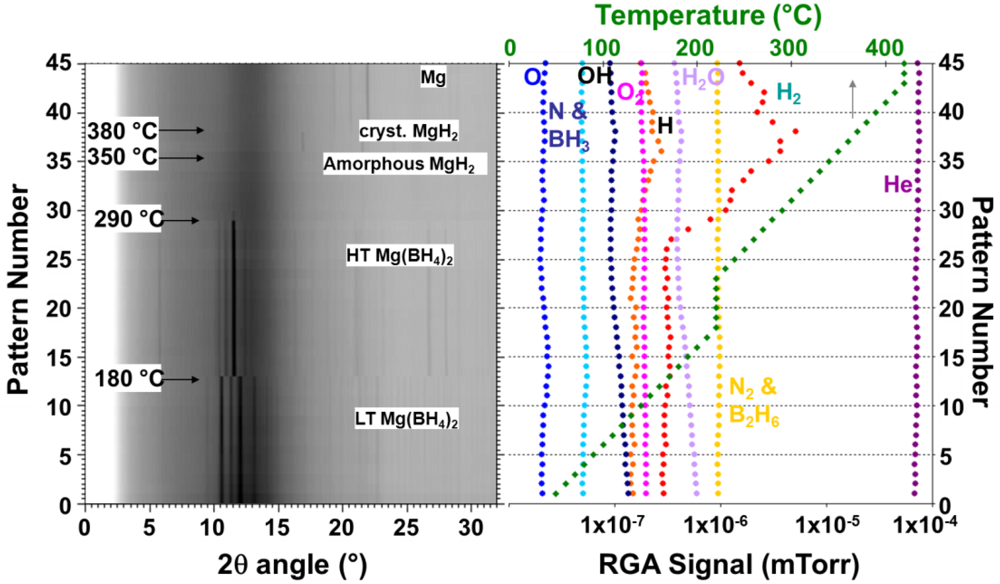
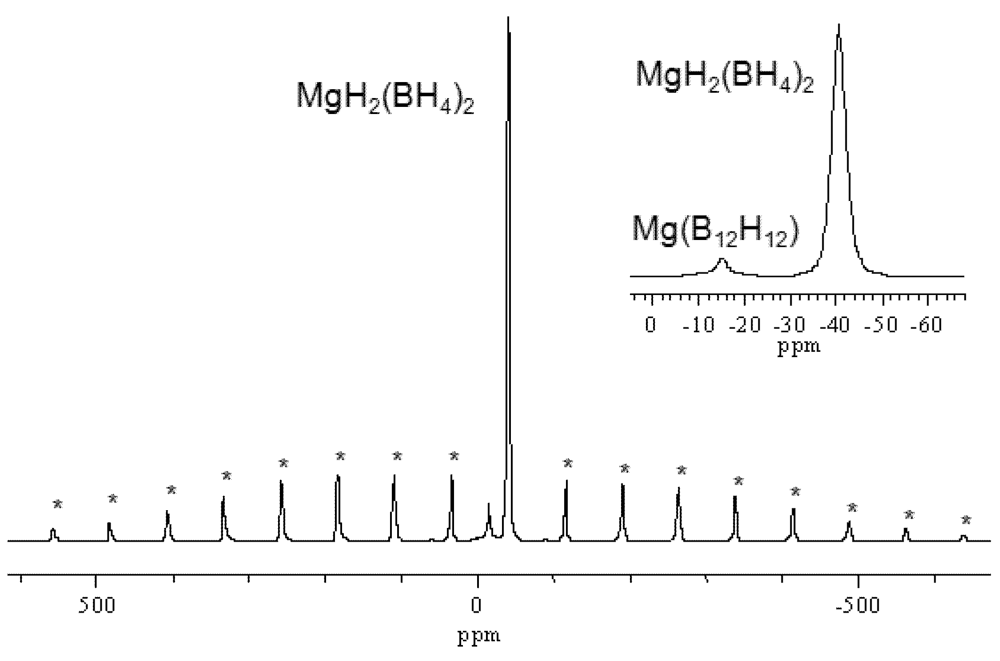
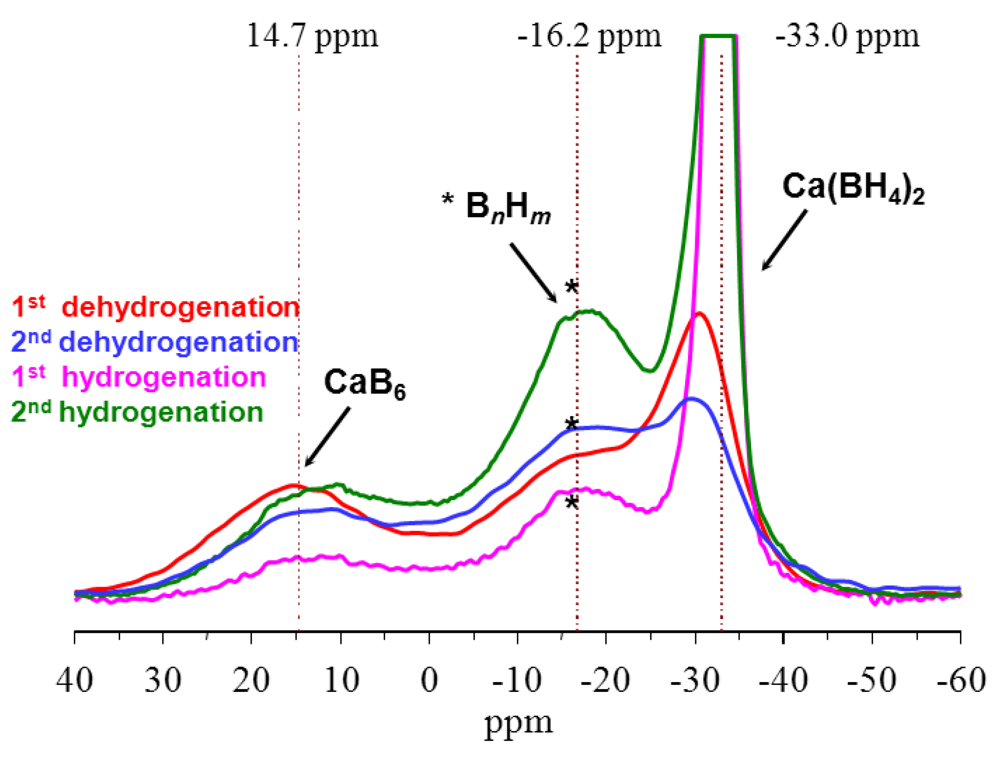
2.1.1. Complex Anionic Hydrides
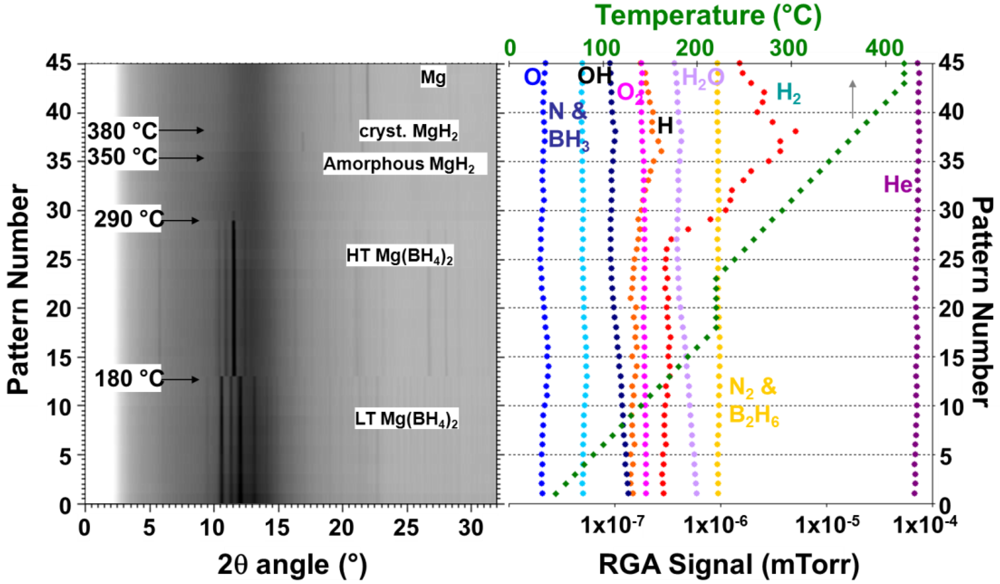

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rxn. No. | Hydrogen release reaction | wt.% H2 | g- H2/L | Reversible | Tdes | ∆ Hdes | Cycling | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | 2Al(BH4)3 + B2H6 ↔ 2AlB4H11 + 4H2 | 13.5 | Y | 493 | [14] | |||
| 2 | 4LiBH4 + 5Mg2NiH4 ↔ 2MgNi2.5B2 + 4LiH + 8MgH2 + 8H2 | 6.5 | Y | 693 | 15 | 10 | [16] | |
| 3 a | 3LiAlH4 ↔ Li3AlH6 + 2Al + 3H2 | 5.3 | 74 | Y | 353–453 | −10 | 5 | [17,18] |
| 3 b | Li3AlH6 ↔ 3LiH + Al +
 H2 H2 | 2.6 | Y | 25 | ||||
| 4 | Ti-doped LiAlH4 | ~7 | 353–453 | 3 | [17] | |||
| 5 | 2AlH3 → 2AlH2 + H2 | 10 | 147 | N | 398–448 | 7.6 | [19,20] | |
| 6 a | NaAl4 ↔  Na3AlH6 + Al + H2 Na3AlH6 + Al + H2 | 4 | 80 | Y | 393 | 37 | 100 | [18,21,22] |
| 6 b | Na3Al6 ↔ 3NaH + Al +
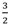 H2 H2 | 1.9 | 43 | Y | 453 | 47 | ||
| 7 | LiH + 2LiNH3 + KBH4 → Li3BN2 + KH + 4H2 | 7.48 b | 508 b | 43.61 b | [23] | |||
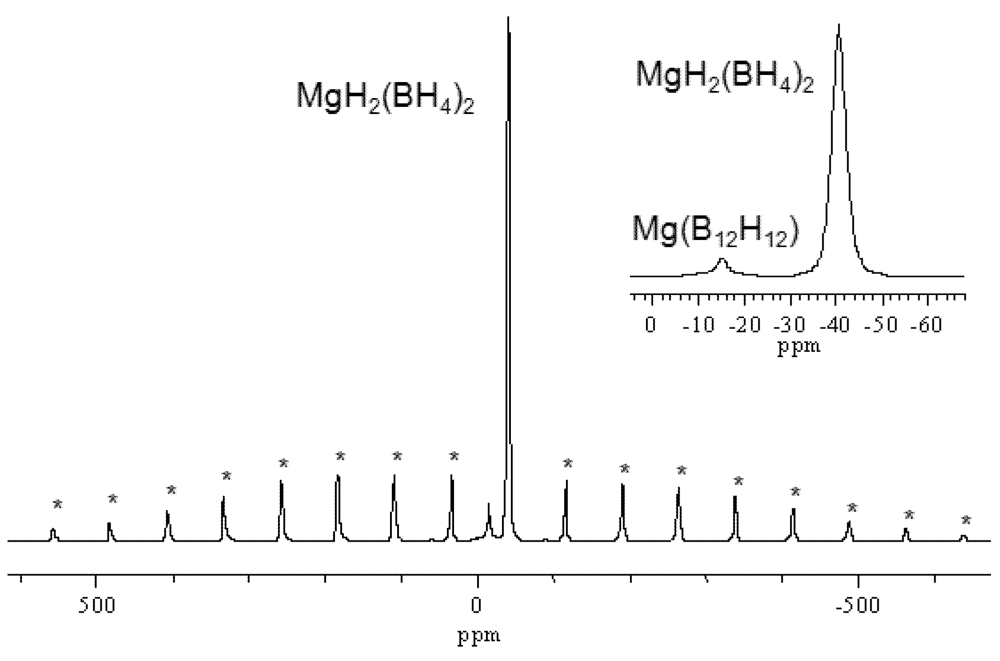
| 8 a | 6Mg(BH4)2 → 5MgH2 + Mg(B12H12) + 13H2↑ | |||||||
| 8 b | 5MgH2 + Mg(B12H12) → 5Mg + 5H2↑ + Mg(B12H12) | 12 | 147 | Y | 673, 803 | 41 | 2 | [17,24] |
| 8 c | 5Mg + Mg(B12H12) → 6MgB2 + 6H2↑ | |||||||
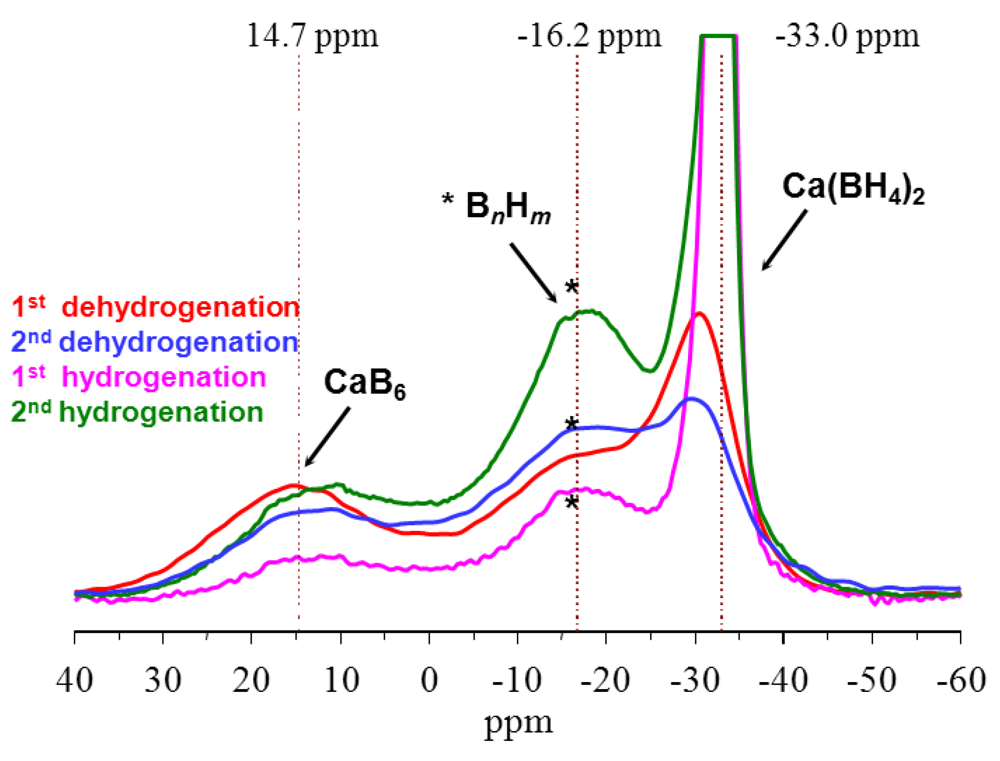
| 9 a | 3Ca(BH4)2 → CaB6 + 2CaH2 + 10H2 | 9.5 | 108 | Y | 573–623 | 41.4 b | [25,26] | |
| 9 b | CaB6 + 2CaH2 → 3Ca(BH4)2 | |||||||
| 10 | Li3N + 2H2 ↔ Li2NH + LiH + H2 ↔ LiNH2 + 2LiH | 11.5 | Y | 528 | [27] | |||
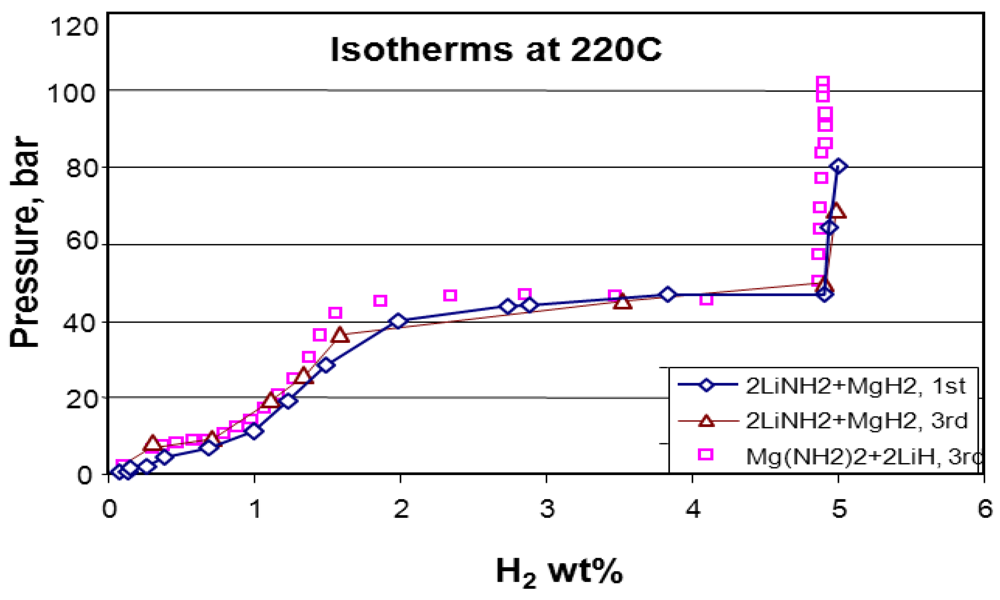
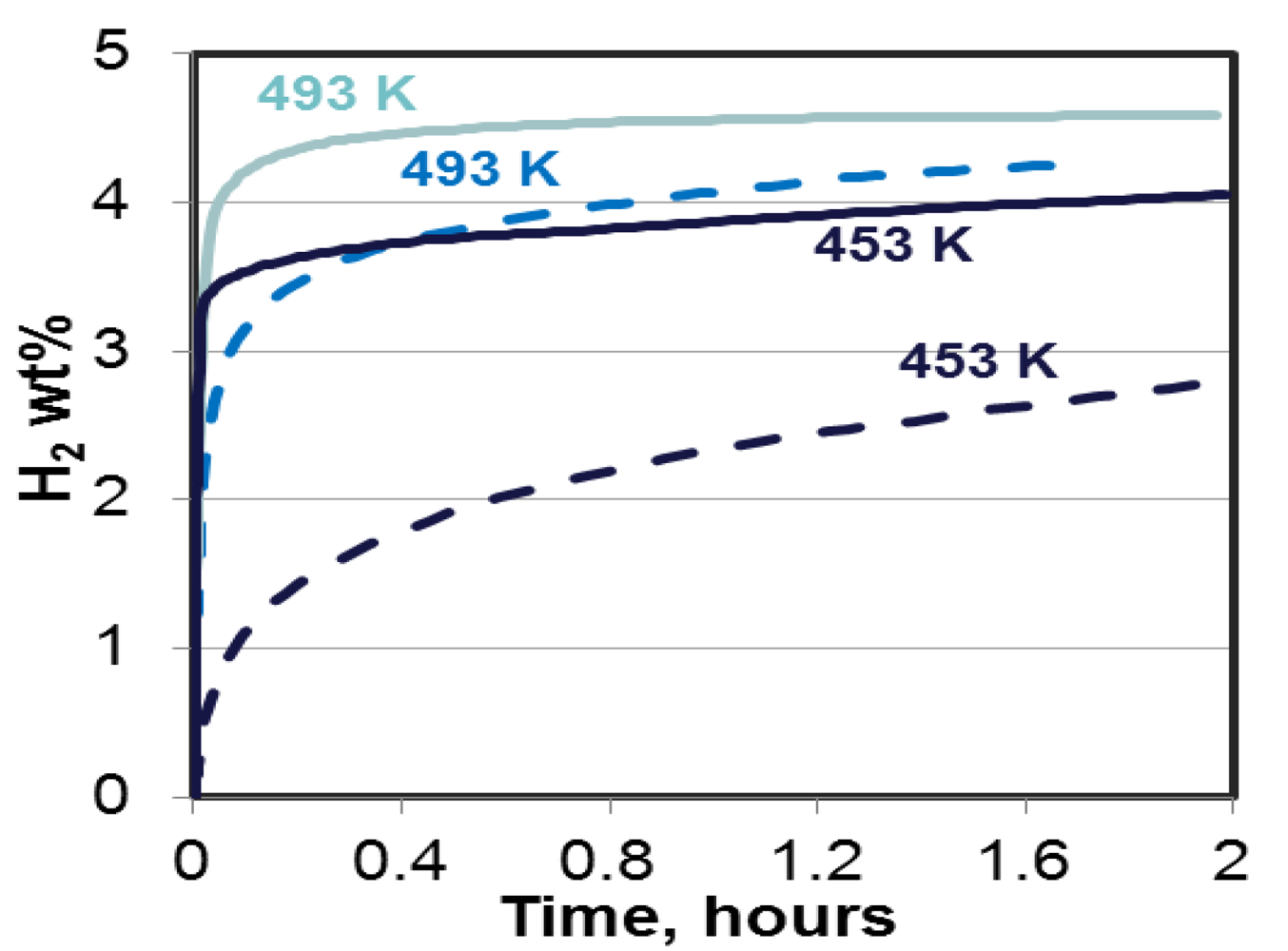
| 11 | 2LiNH2 + MgH2 → Mg(NH2)2 + 2LiH ↔ MgLi(NH2) + 2H2↑ | 5 | 70 | Potential | 41.8 | 264 cycles; 23% loss in capacity | [18] | |
| 12 | MgH2 + LiNH2 → LiMgN + 2H2↑ | 8.2 | Y | 525,543 | 33–38 | [28] | ||
| 13 a | LiNH2 + MgH2 → 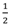 Mg(NH2)2 + MgH2 + LiH ↔ Li2Mg(NH2)2 + MgH2 ↔ LiMgN Mg(NH2)2 + MgH2 + LiH ↔ Li2Mg(NH2)2 + MgH2 ↔ LiMgN | 6.5 | 107 | Y | 10 | [18] | ||
| 13 b | Mg(NH2)2 + MgH2 + LiH ↔ Li2Mg(NH2)2 + MgH2 ↔ LiMgN | |||||||
| 14 | Nano-confined MgH2\carbon aerogel | 9–16 | 42 | 4 | [17] | |||
| 15 | 2MgH2 + Mg(NH2)2 → Mg3N2 + 4H2 | 7.4 b | 453 b | 26 b | [23] | |||
| 16 a | xNH3BH3 → [NH2BH2]x + (x-1)H2 | 6.5 | 373 | −21.7 | [29,30,31] | |||
| 16 b | [NH2BH2]x→ [NHBH]x + H2 | 6.9 | N | 423 | −(23.9–15.4) | |||
| 16 c | [NHBH]x → BNx + H2 | 96 a | >823 | |||||
| 17 | Ca(NH2BH3)2 ↔ CaBN + H2 | 7.5 | 50 c | N | 443 | 3.5 | [32,33] | |
| 18 | LiNH2BH3 ↔ LiBNHx + H2 | 10.9 | 52 | N | ~403 | ~(-2) | [34,35] | |
| 19 | NaNH2BH3 ↔ NaBNH2 + H2 | 7.6 | 43 | N | ~353 | -(2-5) | [34,35] | |
| 20 | cycle CBN → NH-BH + 3H2 | 5–7 | Potential | 27.9 | [36,37] |

2.1.2. Complex Amide/Imide Hydrides
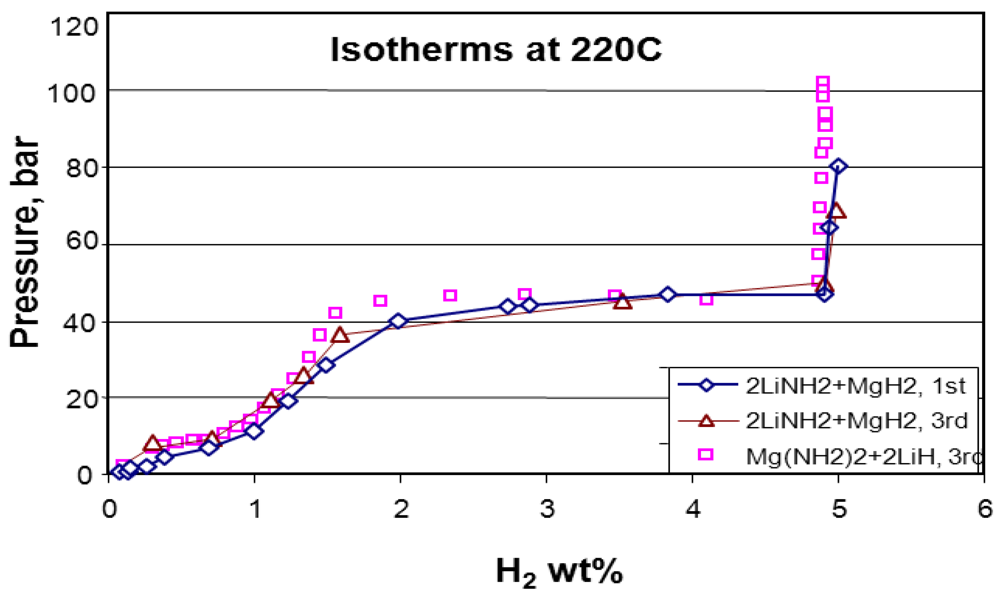

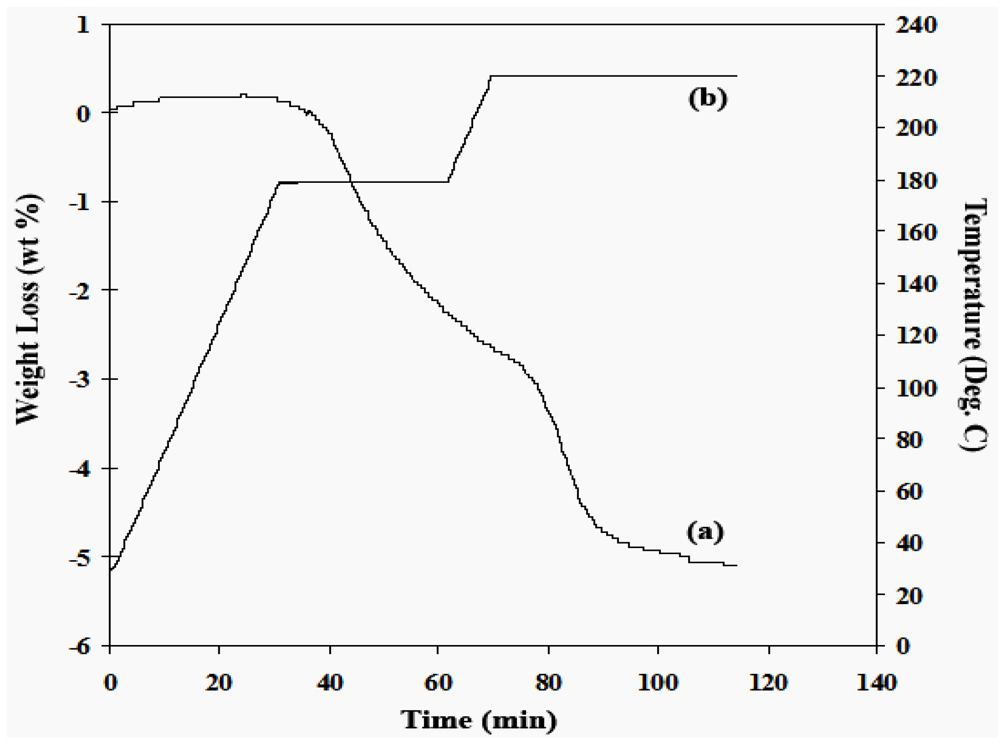
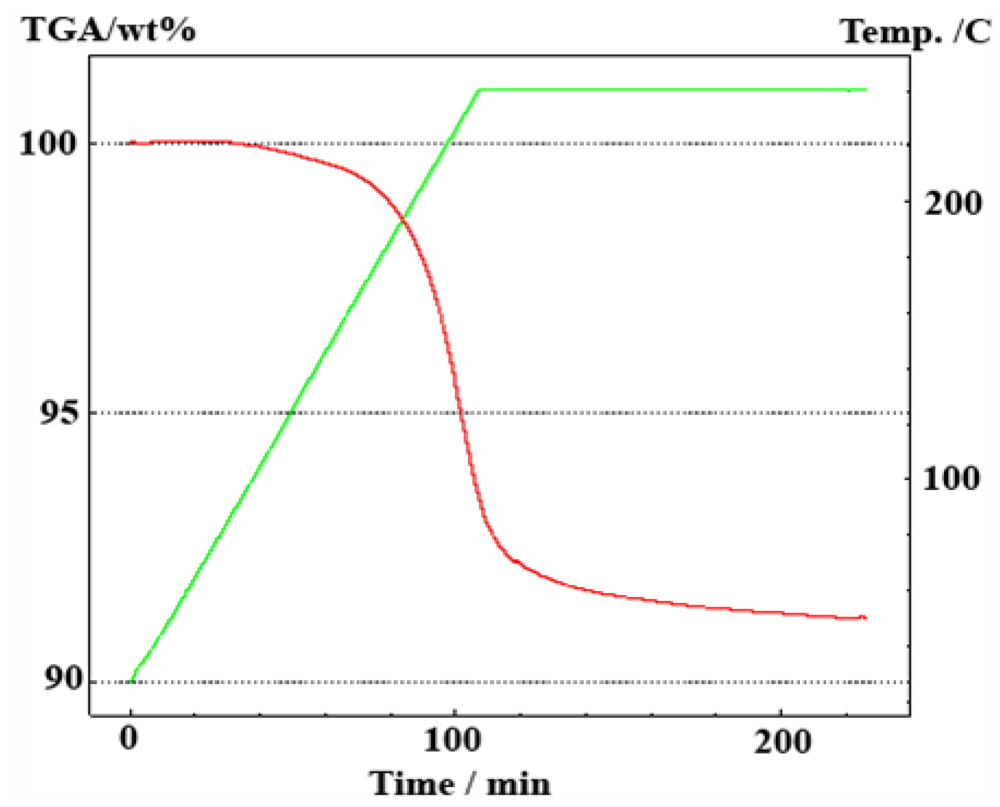
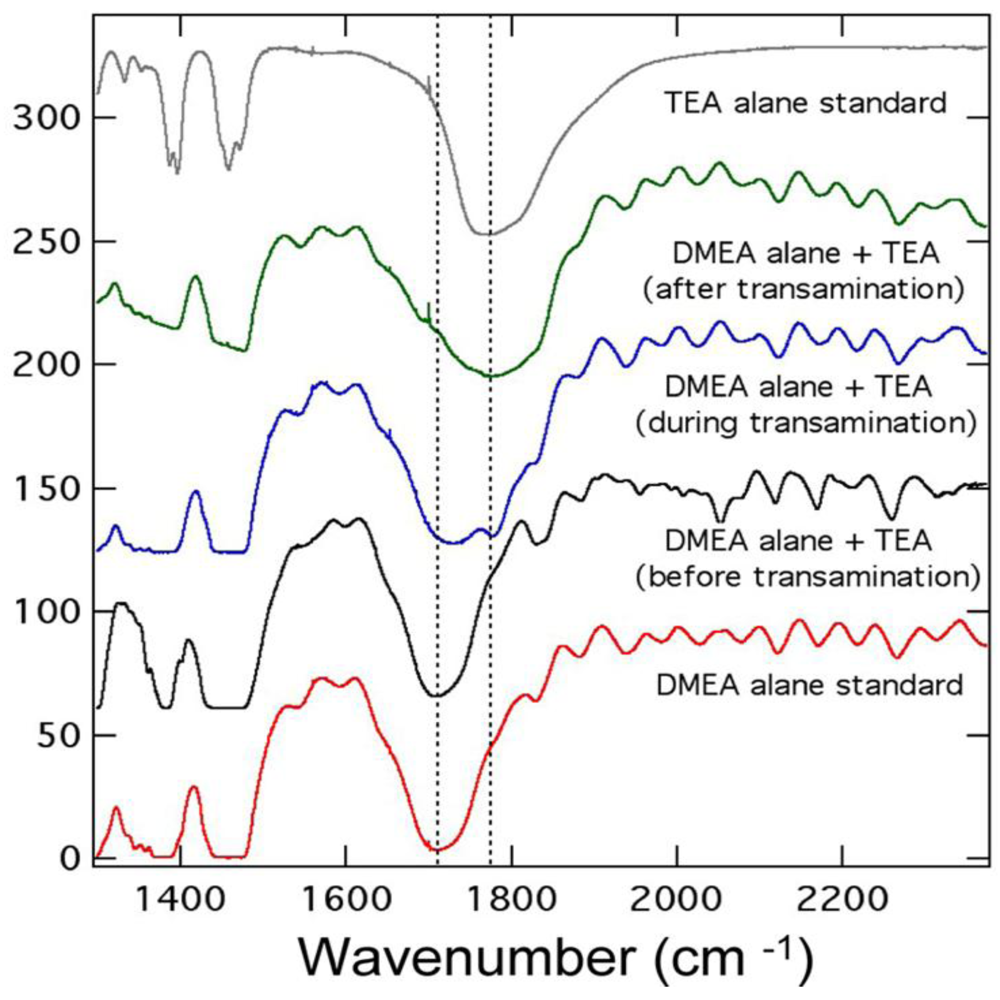
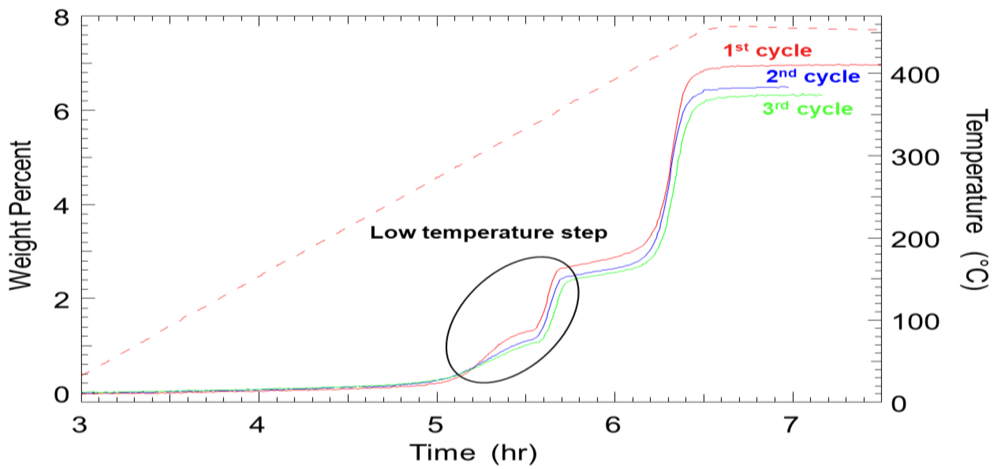
2.1.3. Aluminum Hydride (Alane)



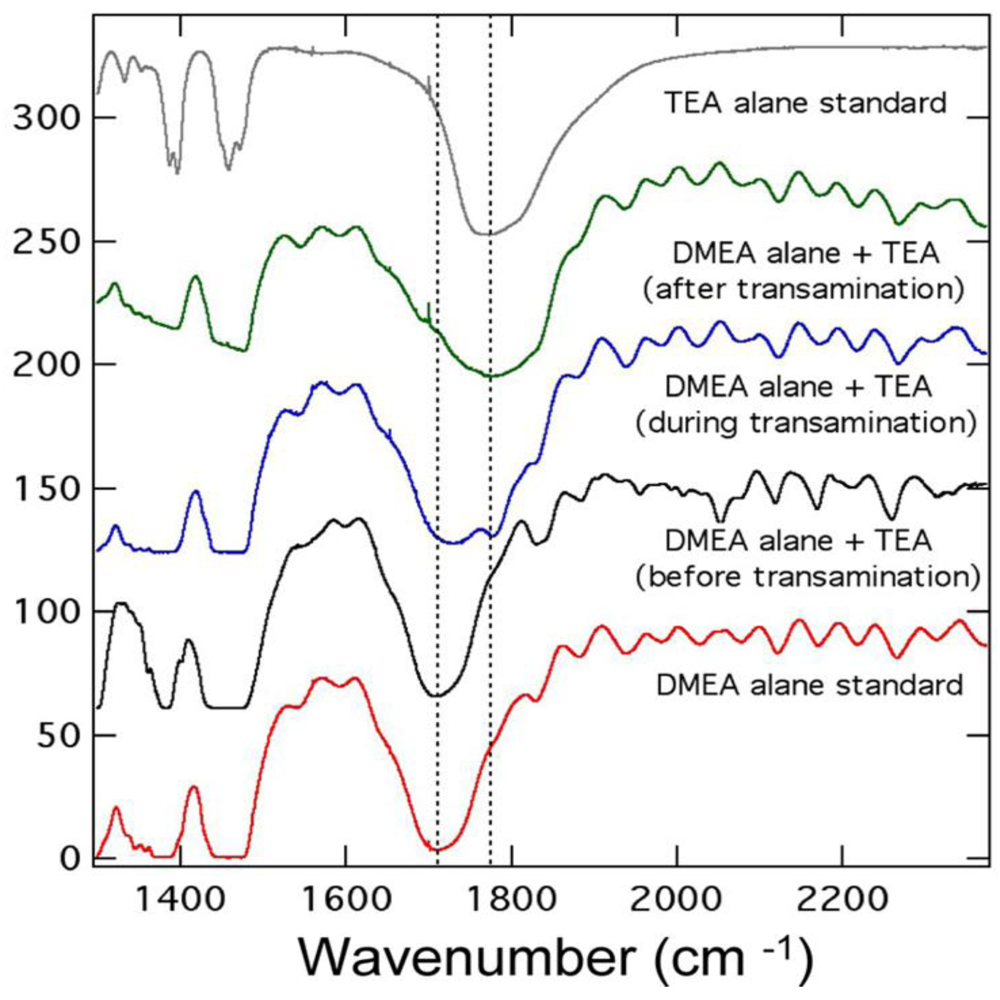

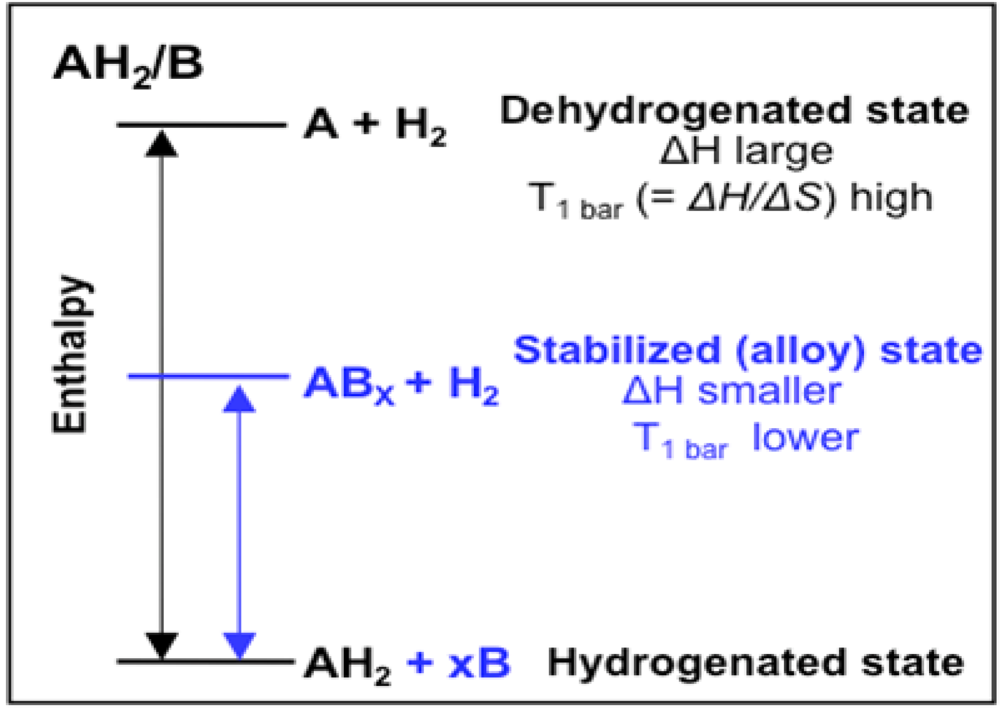
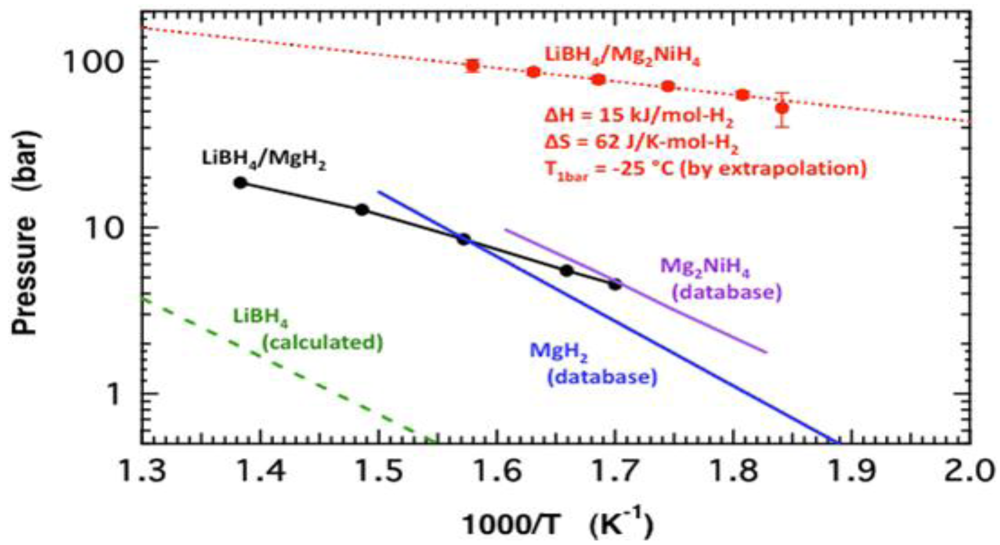
2.1.4. Destabilized Hydrides
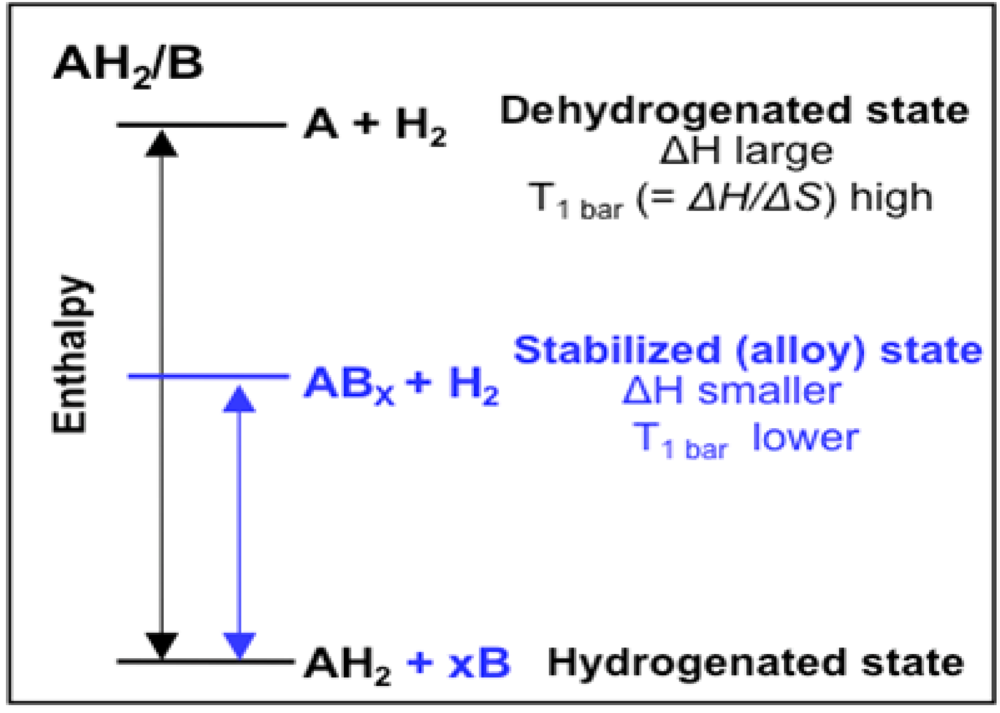
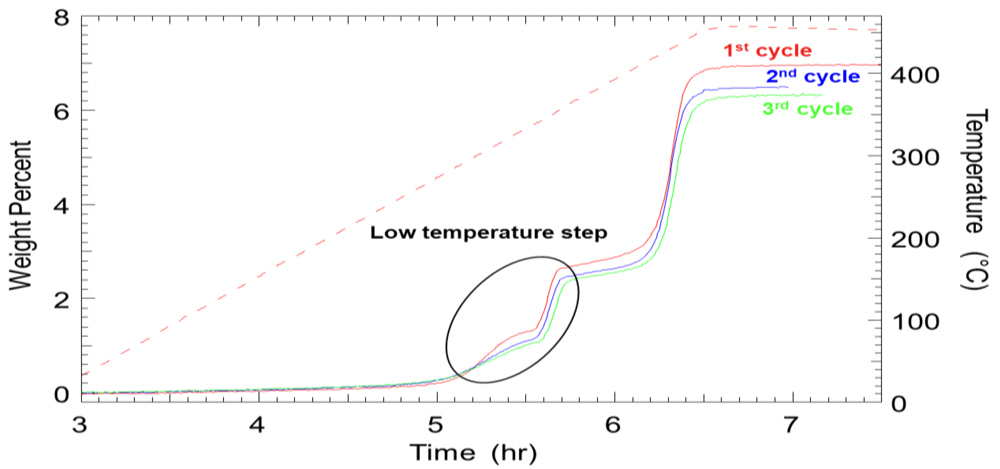
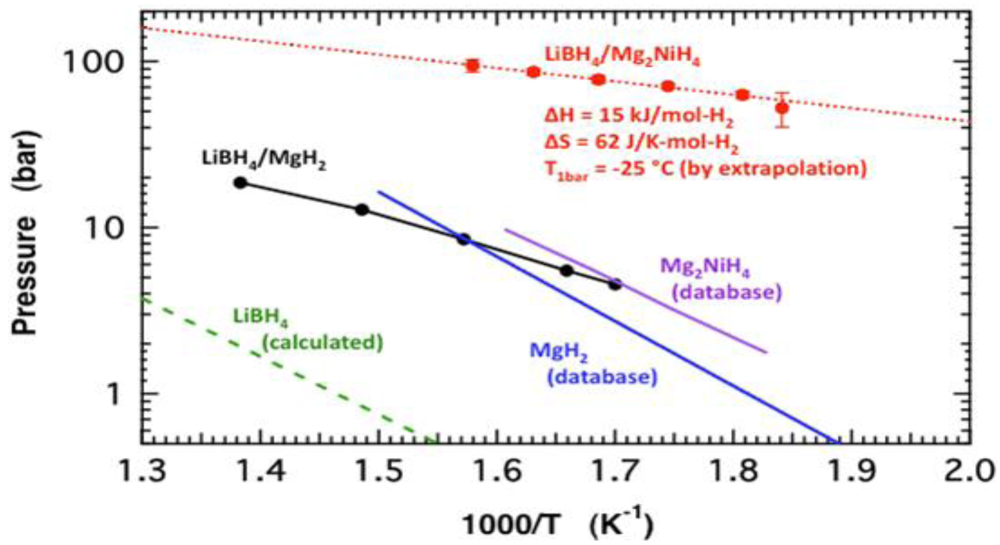
2.1.5. Computational Guidance of Metal Hydride Development
2.1.6. Summary and Research Trends
2.2. Regenerable Chemical Hydrogen Storage Materials
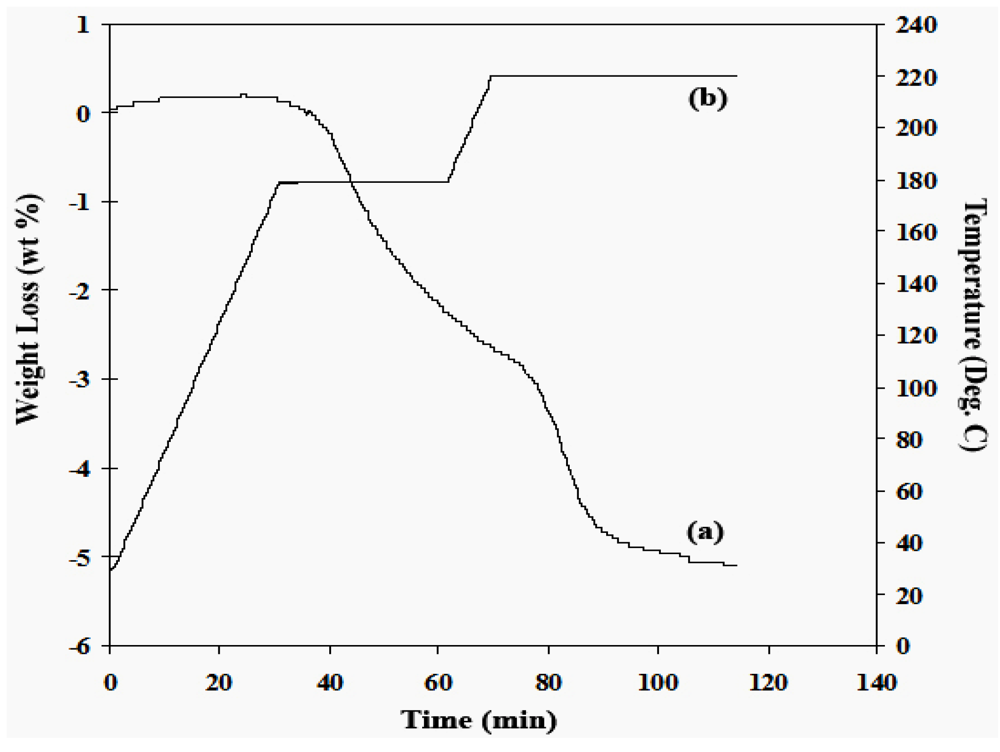
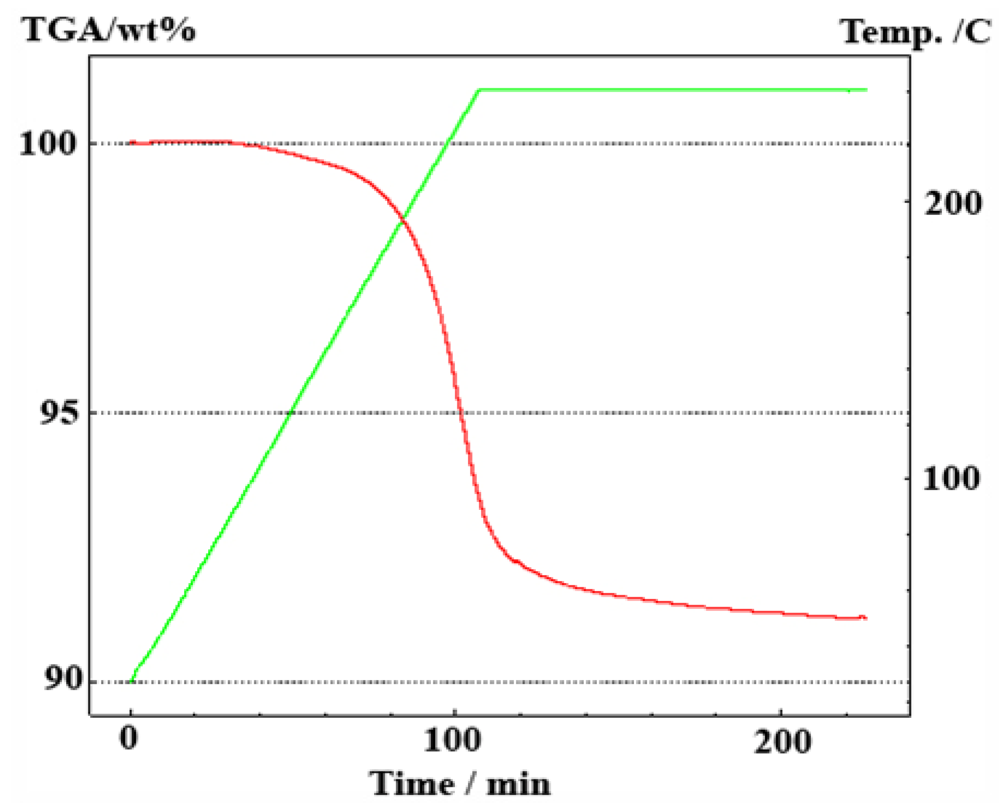
2.2.1. Ammonia Borane
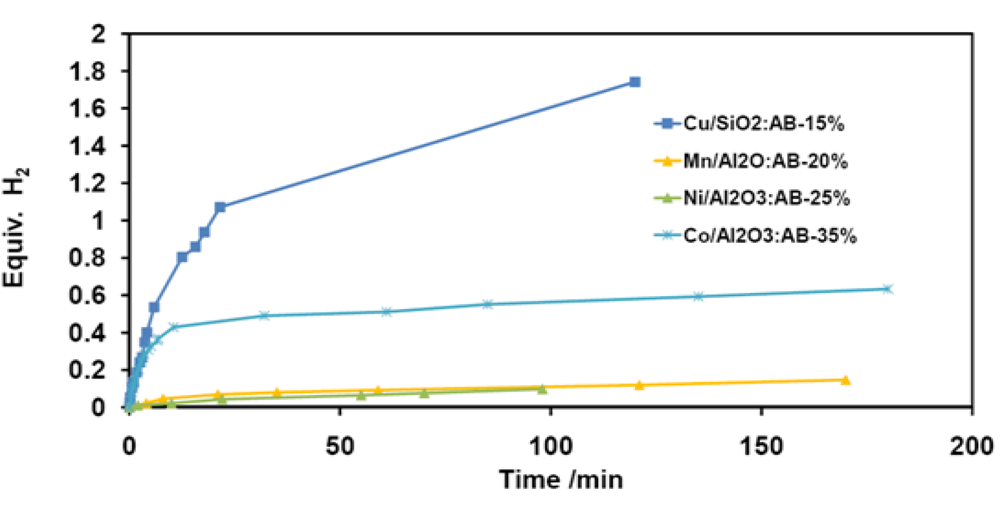
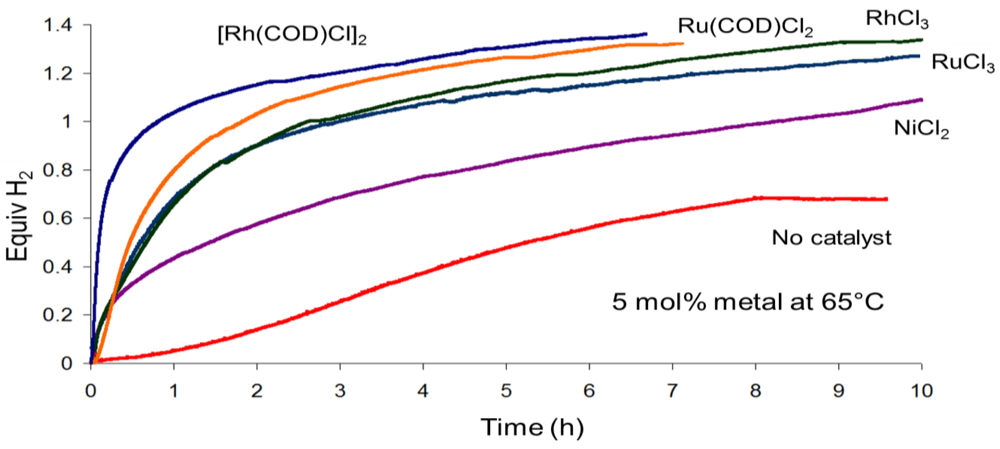
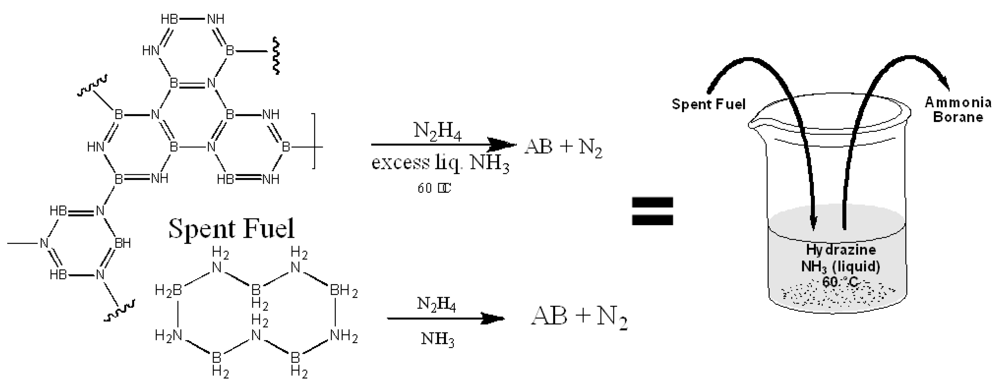
2.2.2. Regeneration of Spent AB
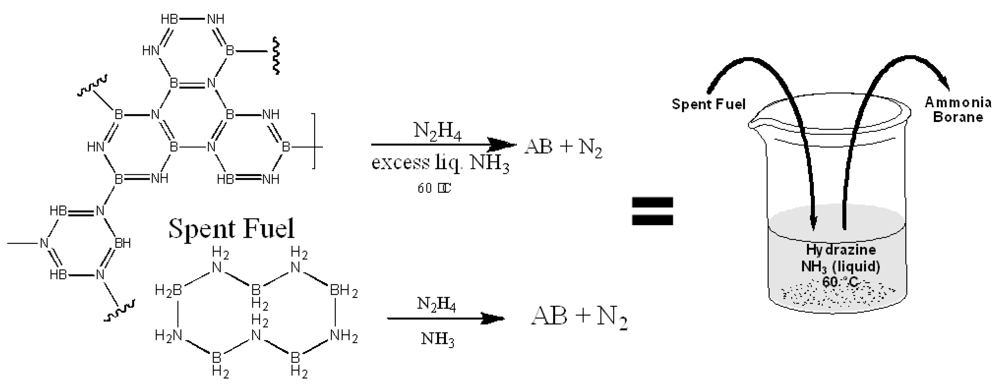
2.2.3. Metal Amidoboranes
2.2.4. Cyclo-CBN Compounds
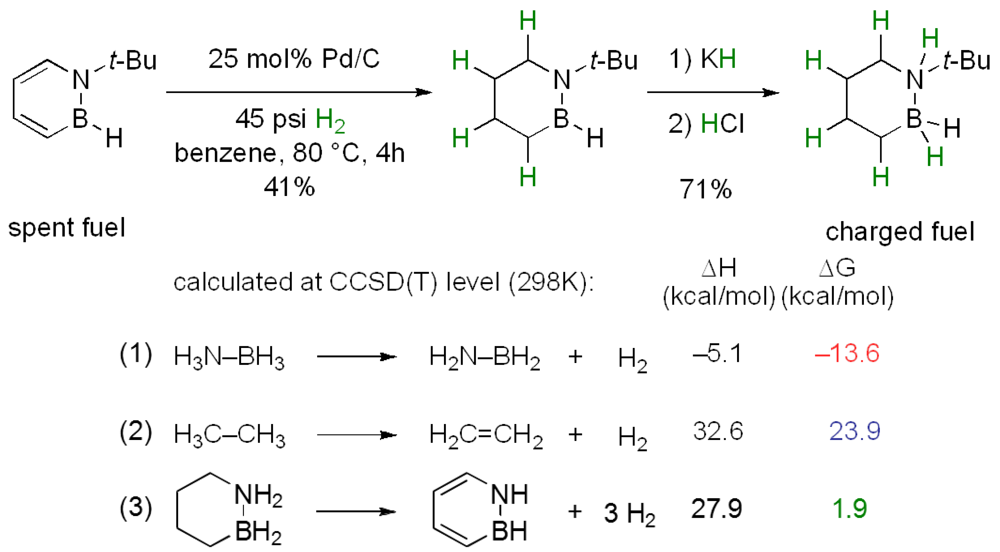
2.2.5. Computational Guidance of Chemical Hydrogen Storage Systems
2.2.6. Summary and Research Trends
3. Conclusions
Conflict of Interest
References
- DOE. Office of Energy Efficiency and Renewable Energy Hydrogen, Fuel Cell Technologies Program Multi-year Research, Development and Demonstration Plan: Planned Program Activities for 2005–2015. Available online: http://www1.eere.energy.gov/hydrogenandfuelcells/ mypp/index.html (accessed on 4 May 2012).
- Ahluwalia, R.K.; Hua, T.Q.; Peng, J.-K. Fuel cycle efficiencies of different automotive on-board hydrogen storage options. Int. J. Hydrog. Energy 2007, 32, 3592–3602. [Google Scholar] [CrossRef]
- Ahluwalia, R.; Hua, T.; Peng, J.-K.; Kumar, R. System Level Analysis of Hydrogen Storage Options. Proceedings of the 2008 U.S. DOE Hydrogen Program Annual Merit Review, Arlington, VA, USA, 9–13 June 2008; Available online: http://www.hydrogen.energy.gov/ pdfs/review08/st_2_ahluwalia.pdf (accessed on 4 May 2012).
- Ahluwalia, R.K.; Hua, T.Q.; Peng, J.-K. On-board and off-board performance of hydrogen storage options for light-duty vehicles. Int. J. Hydrog. Energy 2011, 37, 2891–2910. [Google Scholar]
- US DOE Targets for Onboard Hydrogen Storage Systems for Light-Duty Vehicles. Available online: http://www1.eere.energy.gov/hydrogenandfuelcells/storage/pdfs/targets_onboard_hydro_storage.pdf (accessed on 4 May 2012).
- Bourlinos, A.B.; Steriotis, T.A.; Karakassides, M.; Sanakis, Y.; Tzitzios, V.; Trapalis, C.; Kouvelos, E.; Stubos, A. Synthesis, characterization and gas sorption properties of a molecularly-derived graphite oxide-like foam. Carbon 2007, 45, 852–857. [Google Scholar]
- Miller, M.A.; Page, R. National Testing Laboratory for Solid-State Hydrogen Storage Technologies. Proceedings of the 2008 U.S. DOE Hydrogen Program Annual Merit Review, Washington, DC, USA, 9–13 June 2008; Available online: http://www.hydrogen.energy.gov/ pdfs/review08/st_28_miller.pdf (accessed 4 May 2012).
- Miller, M.A.; Page, R. National Testing Laboratory for Solid-State Hydrogen Storage Technologies. Proceedings of the 2009 U.S. DOE Hydrogen Program Annual Merit Review, Washington, DC, USA, 18–22 May 2009; Available online: http://www.hydrogen.energy.gov/ pdfs/review09/stp_45_miller.pdf (accessed 4 May 2012).
- Van Vancht, J.H.N.; Kuijpers, F.A.; Bruning, H.C.A.M. Reversible room-temperature absorption of large quantities of hydrogen by intermetallic compounds. Philips Res. Rep. 1970, 25, 133–146. [Google Scholar]
- Bogdanovic, B.; Schwickardi, M. Ti-doped alkali metal aluminum hydrides as potential novel reversible hydrogen storage materials. J. Alloys Compd. 1997, 253–254, 1–9. [Google Scholar]
- Satyapal, S.; Petrovic, J.; Read, C.; Thomas, G.; Ordaz, G. The U.S. department of energy’s national hydrogen storage project: Progress towards meeting hydrogen-powered vehicle requirements. Catal. Today 2007, 120, 246–256. [Google Scholar]
- Konoplev, V.N. Synthesis of magnesium tetrahydroborate. Zhurnal Neorganicheskoi Khimii 1980, 25, 1737–1740. [Google Scholar]
- Soloveichik, G.L.; Gao, Y.; Rijssenbeek, J.; Andrus, M.; Kniajanski, S.; Bowman, J., R.C.; Hwang, S.-J.; Zhao, J.-C. Magnesium borohydride as a hydrogen storage material: Properties and dehydrogenation pathway of unsolvated Mg(BH4)2. Int. J. Hydrog. Energy 2009, 34, 916–928. [Google Scholar]
- Zhao, J.-C.; Andrus, M.; Cui, J.; Gao, Y.; Kniajanski, S.; Lemmon, J.; Raber, T.; Rijssenbeek, J.; Rubinsztajn, G.; Soloveichik, G.L. Lightweight Intermetallics for Hydrogen Storage. Proceedings of the 2007 U.S. DOE Hydrogen Program Annual Merit Review, Washington, DC, USA, 14–18 May 2007; Available online: http://www.hydrogen.energy.gov/pdfs/review07/ st_16_zhao.pdf (accessed on 4 May 2012).
- Zhao, J.-C.; Cui, J.; Gao, Y.; Kniajanski, S.; Lemmon, J.; Raber, T.; Rijssenbeek, J.; Rubinsztajn, G.; Soloveichik, G.L. Lightweight Intermetallics for Hydrogen Storage. Proceedings of the 2008 U.S. DOE Hydrogen Program Annual Merit Review, Washington, DC, USA, 9–13 June 2008; Available online: http://www.hydrogen.energy.gov/pdfs/review08/stp_22_zhao.pdf (accessed on 4 May 2012).
- Liu, P.; Vajo, J. Thermodynamically Tuned Nanophase Materials for Reversible Hydrogen Storage. Proceedings of the 2010 U.S. DOE Hydrogen Program Annual Merit Review, Washington, DC, USA, 7–11 June 2010; Available online: http://www.hydrogen.energy.gov/ pdfs/review10/st065_liu_2010_p_web.pdf (accessed on 4 May 2012).
- Jensen, C.; McGrady, S. Fundamental Studies of Advanced, High-Capacity Reversible Metal Hydrides. Proceedings of the 2009 U.S. DOE Hydrogen Program Annual Merit Review, Washington, DC, USA, 18–22 May 2009; Available online: http://www.hydrogen.energy.gov/ pdfs/review09/st_07_jensen.pdf (accessed on 4 May 20112).
- Klebanoff, L.; Keller, J. 5-Year Review of Metal Hydride Center of Excellence. Proceedings of the 2010 U.S. DOE Hydrogen Program Annual Merit Review, Washington, DC, USA, 7–11 June 2010; Available online: http://www.hydrogen.energy.gov/pdfs/review10/st029_klebanoff_ 2010_o_web.pdf (accessed on 4 May 2012).
- Graetz, J.; Wegrzyn, J.; Johnson, J.K.; Celebi, Y.; Zhou, W.M.; Reilly, J. Aluminum Hydride Regeneration. Proceedings of the 2010 U.S. DOE Hydrogen Program Annual Merit Review, Washington, DC, USA, 7–11 June 2010; Available online: http://www.hydrogen.energy.gov/ pdfs/review10/st034_graetz_2010_o_web.pdf (accessed on 4 May 2012).
- Graetz, J.; Reilly, J.J. Kinetically stabilized hydrogen storage materials. Scr. Mater. 2007, 56, 835–839. [Google Scholar] [CrossRef]
- Bogdanovic, B.; Brand, R.A.; Marjanovic, A.; Schwickardi, M.; Tolle, J. Metal doped sodium aluminum hydrides as potential new hydrogen storage materials. J. Alloys Comp. 2000, 302, 36–58. [Google Scholar] [CrossRef]
- Bogdanovic, B.; Eberle, U.; Felderhoff, M.; Schuth, F. Complex aluminum hydrides. Scr. Mater. 2007, 56, 813–816. [Google Scholar]
- Johnson, J.K.; Sholl, D.S. First-Principles Modeling of Hydrogen Storage in Metal Hydride Systems. Proceedings of the 2010 U.S. DOE Hydrogen Program Annual Merit Review, Washington, DC, USA, 7–11 June 2010; Available online: http://www.hydrogen.energy.gov/ pdfs/review10/st064_johnson_2010_p_web.pdf (accessed on 4 May 2012).
- Severa, G.; Ronnebro, E.; Jensen, C.M. Direct hydrogenation of magnesium boride to magnesium borohydride: Demonstration of >11 weight percent reversible hydrogen storage. Chem. Commun. 2010, 46, 421–423. [Google Scholar]
- Ozolins, V.; Majzoub, E.H.; Wolverton, C. First-principles prediction of thermodynamically reversible hydrogen storage reactions in the Li-Mg-Ca-B-H system. J. Am. Chem. Soc. 2009, 131, 230–237. [Google Scholar]
- Ronnebro, E.; Majzoub, E.H. Calcium borohydride for hydrogen storage: Catalysis and reversibility. J. Phys. Chem. B 2007, 111, 12045–12047. [Google Scholar] [CrossRef]
- Chen, P.; Xiong, Z.T.; Luo, J.Z.; Lin, J.Y.; Tan, K.L. Interaction of hydrogen with metal nitrides and imides. Nature 2002, 420, 302–303. [Google Scholar]
- Fang, Z.Z.; Sohn, H.Y. Discovery of H2 Storage Materials: LiMgN and Ng-Ti-H. Proceedings of the 2010 U.S. DOE Hydrogen Program Annual Merit Review, Washington, DC, USA, June 7–11 2010; Available online: http://www.hydrogen.energy.gov/pdfs/review10/st062_fang _2010 _p_web.pdf (accessed on 4 May 2012).
- Ott, K. 2010 Overview and Wrapup: DOE Chemical Hydrogen Storage Center of Excellence. Proceedings of the 2010 U.S. DOE Hydrogen Program Annual Merit Review, Washington, DC, USA, 7–11 June 2010; Available online: http://www.hydrogen.energy.gov/pdfs/review10/ st036_ott_2010_o_web.pdf (accessed on 4 May 2012).
- Nakagawa, T.; Shrestha, R.; Davis, B.; Diyabalanage, H.; Burrell, A.; Henson, N.; Semelsberger, T.; Stephens, F.; Gordon, J.; Ott, K.; et al. Chemical Hydrogen Storage R&D at Los Alamos National Laboratory. Proceedings of the 2010 U.S. DOE Hydrogen Program Annual Merit Review, Washington, DC, USA, 7–11 June 2010; Available online: http://www.hydrogen.energy.gov/pdfs/review10/st040_burrell_2010_o_web.pdf (accessed on 4 May 2012).
- Sneddon, L. Amineborane-Based Chemical Hydrogen Storage. Proceedings of the 2010 U.S. DOE Hydrogen Program Annual Merit Review, Washington, DC, USA, 7–11 June 2010; Available online: http://www.hydrogen.energy.gov/pdfs/review10/st039_sneddon_2010_o_ web.pdf (accessed on 4 May 2012).
- Burrell, A.K.; Shrestha, R.P.; Davis, B.; Diyabalanage, H.V.K.; Henson, N.; Inbody, M.; John, K.; Semelsberger, T.A.; Stephens, F.; Gordon, J.; et al. Chemical Hydrogen Storage R&D at Los Alamos National Laboratory. Proceedings of the 2009 U.S. DOE Hydrogen Program Annual Merit Review, Arlington, VA, USA, 18–22 May 2009; Available online: http://www.hydrogen.energygov/pdfs/review09/st_17_burrell.pdf (accessed on 4 May 2012).
- Diyabalanage, H.V.K.; Shrestha, R.P.; Semelsberger, T.A.; Scott, B.L.; Bowden, M.E.; Davis, B.L.; Burrell, A.K. Calcium amidotrihydroborate: A hydrogen storage material. Angew. Chem. Int. Ed. 2007, 46, 8995–8997. [Google Scholar]
- Autrey, T. PNNL Progress as Part of the Chemical Hydrogen Storage Center of Excellence. Proceedings of the 2009 U.S. DOE Hydrogen Program Annual Merit Review, Arlington, VA, USA, 18–22 May 2009; Available online: http://www.hydrogen.energy.gov/pdfs/review09/ st_18_autrey.pdf (accessed on 4 May 2012).
- Xiong, Z.; Yong, C.K.; Wu, G.; Chen, P.; Shaw, W.; Karkamkar, A.; Autrey, T.; Jones, M.O.; Johnson, S.R.; Edwards, P.P.; David, W.I.F. High-capacity hydrogen storage in lithium and sodium amidoboranes. Nat. Mater. 2008, 7, 138–141. [Google Scholar] [CrossRef]
- Campbell, P.G.; Zakharov, L.N.; Grant, D.J.; Dixon, D.A.; Liu, S.-Y. Hydrogen storage by boron-nitrogen heterocycles: A simple route for spent fuel regeneration. J. Am. Chem. Soc. 2010, 132, 3289–3291. [Google Scholar]
- Liu, S.-Y. Hydrogen Storage by Novel CBN Heterocycle Materials. Proceedings of the 2010 U.S. DOE Hydrogen Program Annual Merit Review, Washington, DC, USA, 7–11 June 2010; Available online: http://www.hydrogen.energy.gov/pdfs/review10/st038_liu_2010_o_web.pdf (accessed on 4 May 2012).
- Jensen, C.; McGrady, S. Advanced, High-Capacity Reversible Metal Hydrides. Proceedings of the 2010 U.S. DOE Hydrogen Program Annual Merit Review, Washington, DC, USA, 7–11 June 2010; Available online: http://www.hydrogen.energy.gov/pdfs/review10/st031_jensen_2010 _o_web.pdf (accessed on 4 May 2012).
- Allendorf, M.; Majzoub, E.H.; Stavila, V. Discovery and Development of Metal Hydrides for Reversible On-board Hydrogen Storage. Proceedings of the 2009 U.S. DOE Annual Merit Review, Washington, DC, USA, 18–22 May 2009; Available online: http://www.hydrogen. energy.gov/pdfs/review09/st_03_allendorf.pdf (accessed on 4 May 2012).
- Discovery and Development of Metal Hydrides for Reversible On-board Hydrogen Storage. Proceedings of the 2010 U.S. DOE Annual Merit Review; Washington, DC, USA: 7–11 June 2010. Available online: http://www.hydrogen.energy.gov/pdfs/review10/st033_allendorf_2010_o_web.pdf (accessed on 4 May 2012).
- Stavila, V.; Her, J.-H.; Zhou, W.; Hwang, S.-J.; Kim, C.; Ottley, L.A.M.; Udovic, T.J. Probing the structure, stability and hydrogen storage properties of calcium dodecahydro-closo-dodecaborate. J. Solid State Chem. 2010, 183, 1133–1140. [Google Scholar]
- Luo, W.; Ronnebro, E. Towards a viable hydrogen storage system for transportation application. J Alloys Compd. 2005, 404–406, 392–395. [Google Scholar]
- Klebanoff, L.; Keller, J. Metal Hydride Center of Excellence Overview. Proceedings of the 2006 U.S. DOE Annual Merit Review, Washington, DC, USA, 16–19 May 2006; Available online: http://www.hydrogen.energy.gov/pdfs/review06/st_13_klebanoff.pdf (accessed on 4 May 2012).
- Wang, J.; Liu, T.; Wu, G.; Li, W.; Liu, Y.; Araujo, C.M.; Scheicher, R.H.; Blomqvist, A.; Ahuja, R.; Xiong, Z.; et al. Potassium-modified Mg(NH2)2/2LiH system for hydrogen storage. Angew.Chem. Int. Ed. 2009, 48, 5828–5832. [Google Scholar]
- Lu, J.; Fang, Z.Z.; Choi, Y.J.; Sohn, H.Y. Potential of binary lithium magnesium nitride for hydrogen storage applications. J. Phys. Chem. C 2007, 111, 12129–12134. [Google Scholar] [CrossRef]
- Alapati, S.V.; Johnson, J.K.; Sholl, D.S. Identification of destabilized metal hydrides for hydrogen storage using first principles calculations. J. Phys. Chem. B 2006, 110, 8769–8776. [Google Scholar] [CrossRef]
- Alapati, S.V.; Johnson, J.K.; Sholl, D.S. Using first principles calculations to identify new destabilized metal hydride reactions for reversible hydrogen storage. Phys. Chem. Chem. Phys. 2007, 9, 1438–1452. [Google Scholar]
- Luo, W.; Wang, J.; Stewart, K.; Clift, M.; Gross, K. Li-Mg-N-H: Recent investigations and development. J. Alloys Compd. 2007, 446–447, 336–341. [Google Scholar]
- Graetz, J.; Chaudhuri, S.; Lee, Y.; Vogt, T.; Reilly, J.J. Pressure-indued structural and electronic changes in alpha-AlH3. Phys. Rev. B 2006, 74, 214114–214120. [Google Scholar] [CrossRef]
- Graetz, J. New approaches to hydrogen storage. Chem. Soc. Rev. 2009, 38, 73–82. [Google Scholar] [CrossRef]
- Ahluwalia, R.; Hua, T.; Peng, J.-K.; Kumar, R. System Level Analysis of Hydrogen Storage Options. Proceedings of the 2011 U.S. DOE Hydrogen Program Annual Merit Review, Arlington, VA, USA, 9–13 May 2011; Available online: http://www.hydrogen.energy.gov/pdfs/ review11/st001_ahluwalia_2011_o.pdf (accessed on 4 May 2012).
- Zidan, R.; Garcia-Diaz, B.L.; Fewox, C.S.; Stowe, A.C.; Gray, J.R.; Harter, A.G. Aluminum hydride: A reversible material for hydrogen storage. Chem. Commun. 2009. [Google Scholar] [CrossRef]
- Zidan, R.; Garcia-Diaz, B.L.; Martinez-Rodriguez, M.; Teprovich, J. Electrochemical Reversible Formation of Alane. Proceedings of the 2010 U.S. DOE Hydrogen Program Annual Merit Review, Washington, DC, USA, 7–11 June 2010; Available online: http://hydrogen.energy.gov/ pdfs/review10/st063_zidan_2010_p_web.pdf (accessed on 4 May 2012).
- Jensen, C.M.; McGrady, S. Fundamental Studies of Advanced, High-Capacity Reversible Metal Hydrides. Proceedings of the 2009 U.S. DOE Annual Merit Review, Washington, DC, USA, 18–22 May 2009; Available online: http://www.hydrogen.energy.gov/pdfs/review09/st_ 07_jensen.pdf (accessed on 4 May 2012).
- Alapati, S.V.; Johnson, J.K.; Sholl, D.S. Firist princilples screening of destabilized metal hydrides for high capacity H2 storage using scandium. J. Alloys Compd. 2007, 446–447, 23–27. [Google Scholar]
- Alapati, S.V.; Johnson, J.K.; Sholl, D.S. Stability analysis of doped materials for reversible hydrogen storage in destabilized metal hydrides. Phys. Rev. B 2007, 76, 104108. [Google Scholar] [CrossRef]
- Wu, H.; Zhou, W.; Udovic, T.J.; Rush, J.J.; Hartman, M.R.; Bowman, J., R.C.; Vajo, J.J. Neutron vibrational spectroscopy and first-principles study of novel ternary hydrides: Li4Si2H(D) and Li4Ge2H(D): Electronic structure and lattice dynamics. Phys. Rev. B 2007, 76, 224301–224306. [Google Scholar]
- Zarkevich, N.A.; Johnson, D.D. Comment on Structural stability of complex hydrides: LiBH4 revisited. Phys. Rev. Lett. 2006, 97, 119601–119605. [Google Scholar] [CrossRef]
- Zarkevich, N.A.; Tan, T.L.; Johnson, D.D. First-priniciples prediction of phase-segregating alloy phase diagrams and a rapid design estimates of their transition temperatures. Phys. Rev. B 2007, 75, 104203–104212. [Google Scholar]
- Bowman, J.R.C.; Hwang, S.-J.; Ahn, C.C.; Vajo, J.J. NMR and X-ray diffraction studies of phases in the destabilized LiH-Si system. Mater. Res. Soc. Symp. Proc. 2005, 837, N3.6.1. [Google Scholar]
- Dai, B.; Rankin, R.B.; Johnson, J.K.; Allendorf, M.D.; Sholl, D.S.; Zarkevich, N.A.; Johnson, D.D. Influence of surface reactions on complex hydride reversibility. J. Phys. Chem. C 2008, 112, 18270–18279. [Google Scholar]
- Kelly, S.T.; Clemens, B.M.; Van Atta, S.L.; Vajo, J.J.; Olson, G.L. Kinetic limitations of the Mg2Si system for reversible hydrogen storage. Nanotechnology 2009, 20, 204011–204017. [Google Scholar] [CrossRef]
- Lu, J.; Choi, Y.J.; Fang, Z.Z.; Sohn, H.Y. Effect of milling intensity on the formation of LiMgN from the dehydrogenation of LiNH2-MgH2 (1:1) mixture. J. Power Sources 2010, 195, 1992–1997. [Google Scholar]
- Lu, J.; Fang, Z.Z.; Sohn, H.Y. A new Li-Al-N-H system for reversible hydrogen storage. J. Phys. Chem. B 2006, 110, 14236–14239. [Google Scholar]
- Lu, J.; Fang, Z.Z.; Sohn, H.Y.; Bowman, J.R.C.; Hwang, S.-J. Potential and reaction mechanism of Li-Mg-Al-N-H system for reversible hydrogen storage. J. Phys. Chem. C 2007, 111, 16686–16692. [Google Scholar] [CrossRef]
- Purewal, J.; Hwang, S.-J.; Bowman, J.R.C.; Ronnebro, E.; Fultz, B.; Ahn, C. Hydrogen sorption behavior of the ScH2-LiBH4 system: Experimental assessment of chemical destabilization effects. J. Phys. Chem. C 2008, 112, 8481–8485. [Google Scholar]
- Vajo, J.J.; Li, W.; Liu, P. Thermodynamic and kinetic destabilization in LiBH4/Mg2NiH4: promise for borohydride-based hydrogen storage. Chem. Commun. 2010, 46, 6687–6689. [Google Scholar] [CrossRef]
- Vajo, J.J.; Olson, G.L. Hydrogen storage in destabilized chemical systems. Scr. Mater. 2007, 56, 829–834. [Google Scholar] [CrossRef]
- Vajo, J.J.; Salguero, T.T.; Gross, A.F.; Skeith, S.L.; Olson, G.L. Destabilization strategies and kinetics challenges in light metal hydride systems. J. Alloys Compd. 2007, 446–447, 409–414. [Google Scholar]
- Vajo, J.J.; Skeith, S.L. Reversible storage of hydrogen in destabilized LiBH4. J. Phys. Chem. B 2005, 109, 3719–3722. [Google Scholar] [CrossRef]
- Wang, L.-L.; Graham, D.D.; Robertson, I.M.; Johnson, D.D. On the reversibility of hydrogen-storage reactions in Ca(BH4)2: Characterization via experiment and theory. J. Phys. Chem. C 2009, 113, 20088–20096. [Google Scholar]
- Wu, H.; Zhou, W.; Udovic, T.J.; Rush, J.J. Hydrogen storage in a novel destabilized hydride system, Ca2SiHx: Effects of amorphization. Chem. Mater. 2007, 19, 329–334. [Google Scholar] [CrossRef]
- Wu, H.; Zhou, W.; Udovic, T.J.; Rush, J.J. Structure and hydrogenation properties of the ternary alloys Ca2–xMgxSi (0 < x < 1). J. Alloys Compd. 2007, 446–447, 101–105. [Google Scholar]
- Majzoub, E.H.; Ozolins, V. Prototype electrostatic ground state approach to predicting crystal structures of ionic compounds: Applications to hydrogen storage materials. Phys. Rev. B 2008, 77, 104111–104115. [Google Scholar] [CrossRef]
- Majzoub, E.H.; Ronnebro, E. Crystal structures of calcium borohydride: Theory and experiment. J. Phys. Chem. C 2009, 113, 3352–3358. [Google Scholar] [CrossRef]
- Zarkevich, N.A.; Johnson, D.D. Predicting enthalpies of molecular substances: Application to LiBH4. Phys. Rev. B 2008, 100, 040601–040604. [Google Scholar] [CrossRef]
- Annual Progress Report for the DOE Hydrogen Program. Available online: http://www.hydrogen.energy.gov/annual_progress.html (accessed on 17 February 2012).
- Holladay, J.D.; Brooks, K.P.; Ronnebro, E.; Simmons, K.L.; Weimar, M.R. 2011 Annual Progress Report for the DOE Hydrogen Program. Available online: http://www.hydrogen.energy.gov/ pdfs/progress11/iv_d_6_holladay_2011.pdf (accessed on 4 May 2012).
- Baumann, J.; Baitalow, F.; Wolf, G. Thermal decomposition of polymeric aminoborane (H2BNH2)x under hydrogen release. Thermochim. Acta 2005, 430, 9–14. [Google Scholar] [CrossRef]
- Wolf, G.; Baumann, J.; Baitalow, F.; Hoffmann, F.P. Calorimetric Process Monitoring of Thermal decomposition of B-N-H compounds. Thermochim. Acta 2000, 343, 19–25. [Google Scholar] [CrossRef]
- Stowe, A.C.; Shaw, W.J.; Linehan, J.C.; Schmid, B.; Autrey, T. Situ solid state 11B MAS-NMR studies of the thermal decomposition of ammonia borane: Mechanistic studies of the hydrogen release pathways from a solid state hydrogen storage material. Phys. Chem. Chem. Phys. 2007, 9, 1831–1836. [Google Scholar]
- Aardahl, C.L.; Autrey, T.; Camaioni, D.; Dubois, D.; Linehan, J.C.; Karkamkar, A.; Rassat, S.D.; Zheng, R.; Shaw, W.; Li, J.; et al. 2009 Annual Progress Report for the DOE Hydrogen Program. Available online: http://www.hydrogen.energy.gov/pdfs/progress09/iv_b_1d_autry.pdf (accessed on 4 May 2012).
- Stephens, F.H.; Baker, R.T.; Matus, M.H.; Grant, D.J.; Dixon, D.A. Acid initiation of ammonia-borane dehydrogenation for hydrogen storage. Angew. Chem. Int. Ed. 2007, 46, 746–749. [Google Scholar] [CrossRef]
- Himmelberger, D.W.; Yoon, C.W.; Bluhm, M.E.; Carroll, P.J.; Sneddon, L.G. Base-promoted ammonia borane hydrogen release. J. Am. Chem. Soc. 2009, 131, 14101–14110. [Google Scholar]
- Denney, M.C.; Pons, V.; Hebden, T.J.; Keinekey, D.M.; Goldberg, K.I. Efficient catalysis of ammonia borane dehydrogenation. J. Am. Chem. Soc. 2006, 128, 12048–12049. [Google Scholar] [CrossRef]
- Fulton, J.L.; Linehan, J.C.; Autrey, T.; Balasubramanian, M.; Chen, Y.; Szymczak, N.K. When is a Nanoparticle a Cluster? An operando EXAFS Study of Amine BoraneDehydrocoupling by Rh4–6 Clusters. J. Am. Chem. Soc. 2007, 129, 11936–11949. [Google Scholar]
- Sneddon, L. Amineborane-Based Chemical Hydrogen Storage. Proceedings of the 2007 U.S. DOE Hydrogen Program Annual Merit Review, Washington, DC, USA, 14–18 May 2007; Available online: http://www.hydrogen.energy.gov/pdfs/review07/st_27_sneddon.pdf (accessed on 4 May 2012).
- Wright, W.R.H.; Berkeley, E.R.; Alden, L.R.; Baker, R.T.; Sneddon, L.G. Transition metal catalysed ammonia-borane dehydrogenation in ionic liquids. Chem. Commun. 2011, 47, 3177–3179. [Google Scholar]
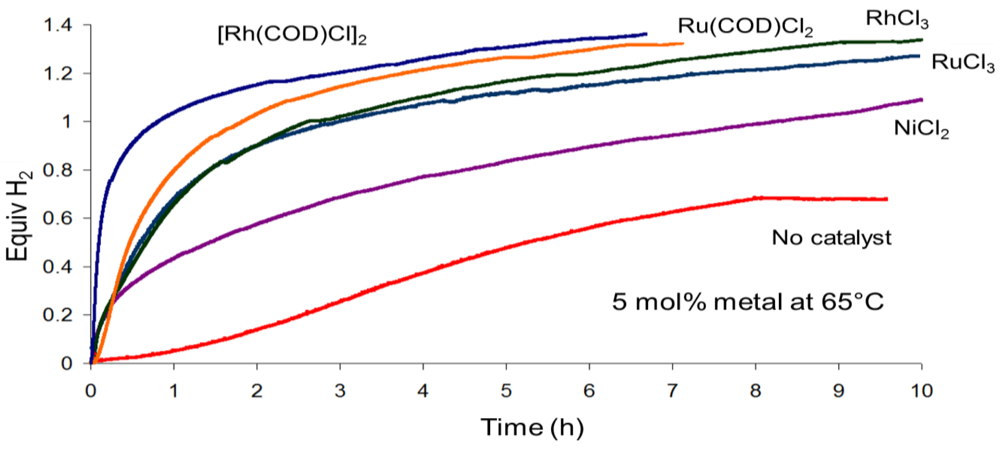
- Keaton, R.J.; Blacquiere, J.M.; Baker, R.T. Base metal catalyzed dehydrogenation of ammonia-borane for chemical hydrogen storage. J. Am. Chem. Soc. 2007, 129, 1844–1845. [Google Scholar] [CrossRef]
- Shrestha, R.P.; Diyabalanage, H.V.K.; Semelsberger, T.A.; Ott, K.C.; Burrell, A.K. Catalytic dehydrogenation of ammonia borane in non-aqueous medium. Int. J. Hydrog. Energy 2009, 34, 2616–2621. [Google Scholar]
- Himmelberger, D.W.; Alden, L.R.; Bluhm, M.E.; Sneddon, L.G. Ammonia boran hydrogen release in ionic liquids. Inorg.Chem. 2009, 48, 9883–9889. [Google Scholar]
- Aardahl, C.L.; Rassat, S.D. Overview of systems considerations for on-board chemical hydrogen storage. Int. J. Hydrog. Energy 2009, 34, 6676–6683. [Google Scholar] [CrossRef]
- Semelsberger, T.A.; Burrell, T.; Rockward, T.; Brosha, R.; Tafoya, J.; Purdy, G.; Nakagawa, T.; Davis, B. Chemical Hydride Rate Modeling, Validation and System Demonstration. Proceedings of the 2011 U.S. DOE Hydrogen Program Annual Merit Review, Arlington, VA, USA, 9–13 May 2011; Available online: http://www.hydrogen.energy.gov/pdfs/review11/ st007_semelsberger_2011_o.pdf (accessed on 4 May 2012).
- Davis, B.L.; Dixon, D.A.; Garner, E.B.; Gordon, J.C.; Matus, M.H.; Scott, B.; Stephens, F.H. Efficient regeneration of partially spent ammonia borane fuel. Angew. Chem. 2009, 121, 6944–6948. [Google Scholar] [CrossRef]
- Sutton, A.D.; Burrell, A.K.; Dixon, D.A.; Garner, E.B.; Gordon, J.C.; Nakagawa, K.C.; Ott, K.C.; Robinson, J.P.; Vasiliu, M. Regeneration of ammonia borane spent fuel by direct reaction with hydrazine and liquid ammonia. Science 2011, 331, 1426–1429. [Google Scholar]
- Sutton, A.D.; Davis, B.L.; Bhattacharyya, K.X.; Ellis, B.D.; Gordon, J.C.; Power, P.P. Recycle of tin thiolate compounds relevant to ammonia-borane regeneration. Chem. Commun. 2010, 46, 148–149. [Google Scholar]
- Ahluwalia, R.K.; Hua, T.Q.; Peng, J.-K.; Kumar, R. 2011 Annual Progress Report for the DOE Hydrogen Program. Available online: http://www.hydrogen.energy.gov/pdfs/progress11/iv_e_2_ahluwalia_2011.pdf (accessed on 4 May 2012).
- Wu, H.; Zhou, W.; Yildirim, T. Alkali and alkaline-earth metal amidoboranes: Structure, crystal chemistry, and hydrogen storage propertie. J. Am. Chem. Soc. 2008, 130, 14834–14839. [Google Scholar]
- Xiong, Z.; Chua, Y.S.; Wu, G.; Xu, W.; Chen, P.; Shaw, W.; Karkamkar, A.; Linehan, J.C.; Smurthwaite, T.; Autrey, T. Interaction of lithium hydride and ammonia borane in THF. Chem. Commun. 2008. [Google Scholar] [CrossRef]
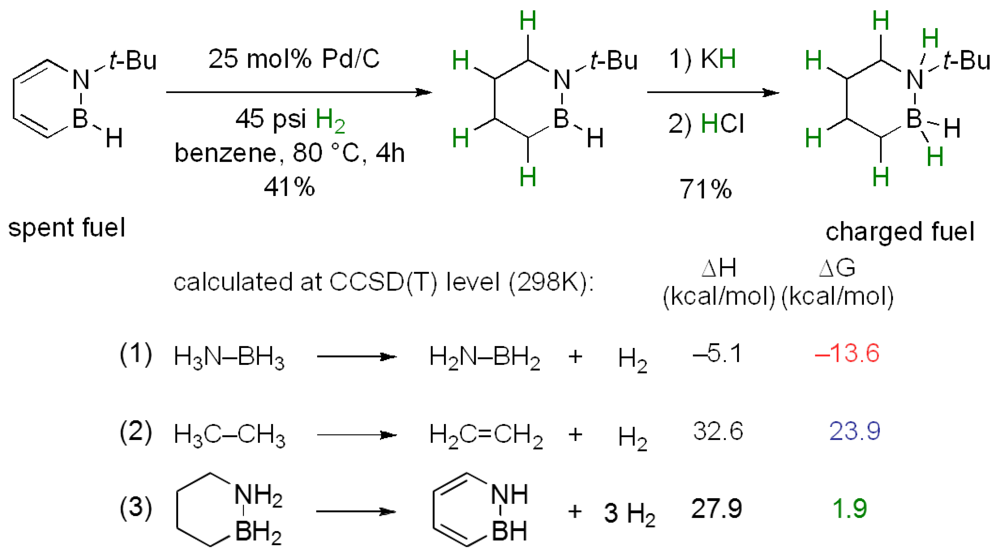
- Marwitz, A.J.V.; Matus, M.H.; Zakharov, L.N.; Dixon, D.A.; Liu, S.-Y. A hybrid organic/inorganic benzene. Angew. Chem. Int. Ed. 2009, 48, 973–977. [Google Scholar] [CrossRef]
- Arduengo, A.J.; Dixon, D.A. Main Group Element and Organic Chemistry for Hydrogen Storage and Activation. Proceedings of the 2010 U.S. DOE Hydrogen Program Annual Merit Review, Arlington, VA, USA, 7–11 June 2010; Available online: http://www.hydrogen.energy.gov/pdfs/review10/st060_dixon_2010_p_web.pdf (accessed on 4 May 2012).
- Arduengo, A.J.; Dixon, D.A. 2009 Annual Progress Report for the DOE Hydrogen Program. Available online: http://www.hydrogen.energy.gov/pdfs/progress09/iv_b_1e_dixon.pdf (accessed on 4 May 2012).
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
McWhorter, S.; O’Malley, K.; Adams, J.; Ordaz, G.; Randolph, K.; Stetson, N.T. Moderate Temperature Dense Phase Hydrogen Storage Materials within the US Department of Energy (DOE) H2 Storage Program: Trends toward Future Development. Crystals 2012, 2, 413-445. https://doi.org/10.3390/cryst2020413
McWhorter S, O’Malley K, Adams J, Ordaz G, Randolph K, Stetson NT. Moderate Temperature Dense Phase Hydrogen Storage Materials within the US Department of Energy (DOE) H2 Storage Program: Trends toward Future Development. Crystals. 2012; 2(2):413-445. https://doi.org/10.3390/cryst2020413
Chicago/Turabian StyleMcWhorter, Scott, Kathleen O’Malley, Jesse Adams, Grace Ordaz, Katie Randolph, and Ned T. Stetson. 2012. "Moderate Temperature Dense Phase Hydrogen Storage Materials within the US Department of Energy (DOE) H2 Storage Program: Trends toward Future Development" Crystals 2, no. 2: 413-445. https://doi.org/10.3390/cryst2020413
APA StyleMcWhorter, S., O’Malley, K., Adams, J., Ordaz, G., Randolph, K., & Stetson, N. T. (2012). Moderate Temperature Dense Phase Hydrogen Storage Materials within the US Department of Energy (DOE) H2 Storage Program: Trends toward Future Development. Crystals, 2(2), 413-445. https://doi.org/10.3390/cryst2020413