
First-Principles Investigation: Effects of Molybdenum Substitution on the Elastic Properties of Uranium Dioxide

Abstract
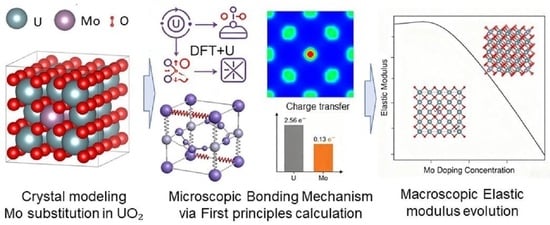
1. Introduction
2. Computational Methods
2.1. DFT Framework and Parameters
2.2. Supercell Modeling of Mo
2.3. Calculation of Elastic Constants and Moduli
3. Results and Discussion
3.1. Structural Properties and Thermodynamics Stability
3.2. Elastic Constants
3.3. Macroscopic Elastic Moduli and Anisotropy
3.4. Electronic Structure and Bonding Analysis
3.5. Thermal Properties and Vickers Hardness
4. Conclusions
- Mo substitution induces local lattice contraction due to the smaller ionic radius of Mo4+ and the formation of shorter, stronger Mo–O bonds. Negative formation energies for low Mo concentrations indicate that doping is energetically favorable, but substitution becomes less favorable with increasing Mo content.
- The influence of Mo on elastic moduli is non-monotonic. At Mo1, the shear modulus (G) and Young’s modulus (E) increase by approximately 16% and 14%, respectively, indicating a pronounced stiffening effect. At Mo2-doped and Mo3-doped UO2, both G and E decrease significantly, leading to material softening.
- The initial stiffening at low Mo concentration arises from enhanced directional Mo 4d–O 2p bonds, supported by a reduction in Cauchy pressure, an increase in Debye temperature, and a corresponding increase in Vickers hardness. The subsequent softening at higher concentrations is attributed to increased lattice distortion, symmetry reduction, and enhanced bond delocalization, as evidenced by the sharp increase in Cauchy pressure, decrease in Debye temperature, and drop in hardness to approximately 3.8–4.8 GPa.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goodson, M.; Noble, G.; Li, L.; Cai, L.; Pavlov, T.; Munro, T. Review of Thermal Gap Conductance Models and Measurement Approaches Toward an Understanding Beyond UO2 and Zirconium-Alloy Interfaces. Int. J. Thermophys. 2025, 46, 112. [Google Scholar] [CrossRef]
- Ge, Z.; Yan, D.; Lei, P.; Yun, D. A Comprehensive Review of High Burn-Up Structure Formation in UO2: Mechanisms, Interactions, and Future Directions. Nanomaterials 2025, 15, 325. [Google Scholar] [CrossRef]
- Koivuranta, S. Gamma Spectrometry and Gamma and X-Ray Tomography of Nuclear Fuel. Master’s Thesis, Lappeenranta University of Technology, Lappeenranta, Finland, 2009. [Google Scholar]
- Huang, Z.; Ma, L.; Zhang, J.; Zhou, Q.; Yang, L.; Wang, H. First-principles study of elastic and thermodynamic properties of UO2, γ-UO3 and α-U3O8. J. Nucl. Mater. 2022, 572, 154084. [Google Scholar] [CrossRef]
- Szuta, M. Total surface area change of Uranium dioxide fuel in function of burn-up and its impact on fission gas release during neutron irradiation for small, Intermediate and High Burn-up. In Proceedings of the 9th International Conference on WWER Fuel Performance, Modelling and Experimental Support, Burgas, Bulgaria, 17–24 September 2011. [Google Scholar]
- Lei, Y.; Huang, H.; Yu, C.; Yang, J.; Liu, Y. Effect of Zr-doping on Structure and Mechanical Properties of UO2 by First-principles Calculations. J. Mater. Sci. Eng. 2014, 32, 126–153. [Google Scholar] [CrossRef]
- Chen, T.; Yuan, Y.; Wang, J.; Wu, J.; Wang, B.; Chen, X.; Moelans, N.; Wang, J.; Pan, F. Features and classification of solid solution behavior of ternary Mg alloys. J. Magnes. Alloys 2025, 13, 2522–2539. [Google Scholar] [CrossRef]
- Chen, T.; Gao, Q.; Yuan, Y.; Li, T.; Xi, Q.; Liu, T.; Tang, A.; Pan, F. Coupling physics in machine learning to investigate the solution behavior of binary Mg alloys. J. Magnes. Alloy 2022, 10, 2817. [Google Scholar] [CrossRef]
- Vazhappilly, T.; Kumar Pathak, A. A first principle based study on the mechanical and thermal properties of UO2: Effect of La and Dy fission product concentrations. Comp. Mater. Sci. 2020, 185, 109933. [Google Scholar] [CrossRef]
- Alharbi, F.; Ragnauth, H.; Abram, T.; Turner, J. Fabrication of UO2–Mo composite fuel pellets with enhanced thermal conductivity by using wet mixing. J. Nucl. Mater. 2026, 623, 156440. [Google Scholar] [CrossRef]
- Ha, Y.; Kim, J.; Park, Y.; Park, S.; Song, K. Behaviors of molybdenum in UO2 fuel matrix. Nucl. Eng. Technol. 2011, 43, 309–316. [Google Scholar] [CrossRef]
- Denecke, M.A.; Petersmann, T.; Marsac, R.; Dardenne, K.; Vitova, T.; Prüßmann, T.; Borchert, M.; Bösenberg, U.; Falkenberg, G.; Wellenreuther, G. XANES characterization of UO2/Mo(Pd) thin films as models for ϵ-particles in spent nuclear fuel. J. Phys. Conf. Ser. 2013, 430, 012113. [Google Scholar] [CrossRef]
- Kim, D.; Kim, D.; Oh, J.; Jeon, S.; Kim, K.; Kim, J.; Yang, J. Enhanced Radial Thermal Conductivity of UO2 Fuel Pellets with Molybdenum Microplates. In Proceedings of the TopFuel 2018 Conference, Prague, Czech Republic, 30 September–4 October 2018; Available online: https://www.euronuclear.org/archiv/topfuel2018/fullpapers/TopFuel2018-A0060-fullpaper.pdf (accessed on 9 April 2026).
- Frazer, D.; Jadernas, D.; Bolender, N.; Madden, J.; Giglio, J.; Hosemann, P. Elevated temperature microcantilever testing of fresh U-10Mo fuel. J. Nucl. Mater. 2019, 526, 151746. [Google Scholar] [CrossRef]
- Gong, H.; Xiao, H.; Wu, H.; Meng, F.; Ren, Q.; Liao, Y.; Zhang, G. The comparable structural, elastic anisotropic and thermophysical properties of advanced U–Si fuel to baseline UO2: A DTF method. Eur. Phys. J. B 2022, 95, 121. [Google Scholar] [CrossRef]
- Hadi, M.; Dahlqvist, M.; Christopoulos, S.-R.; Naqib, S.; Chroneos, A.; Islam, A. Chemically stable new MAX phase V2 SnC: A damage and radiation tolerant TBC material. RSC Adv. 2020, 10, 43783–43798. [Google Scholar] [CrossRef]
- Wang, K.; Xiang, H.; Xu, L.; Feng, A.; Qu, S.; Wang, H.; Chen, D. The Effect of Nb Doping on the Properties of Ti-Al Intermetallic Compounds Using First-Principles Calculations. Materials 2024, 17, 358. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 48, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Huang, Z.; Ma, L.; Zhang, J.; Zhou, Q.; Yang, L.; Wang, H. A first-principles simulation study on solubility of La, Nd, Zr and Mo in UO2 and U3O8. J. Nucl. Mater. 2025, 603, 155414. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Yamazaki, T.; Kotani, A. Systematic Analysis of 4f Core Photoemission Spectra in Actinide Oxides. J. Phys. Soc. Jpn. 1991, 60, 49–52. [Google Scholar] [CrossRef]
- Reshak, A.H. Electronic structure and optical properties of β-RbSm(MoO4)2 from spin polarization calculations DFT+U. Mater. Chem. Phys. 2017, 192, 260–267. [Google Scholar] [CrossRef]
- Solovyev, I. Magnetic interactions in transition-metal oxides. arXiv 2003, arXiv:cond-mat/0305668. [Google Scholar] [CrossRef]
- Wang, V.; Xu, N.; Liu, J.; Tang, G.; Geng, W. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Jaime, M.; Saul, A.; Salamon, M.; Zapf, V.S.; Harrison, N.; Durakiewicz, T.; Lashley, J.C.; Andersson, D.A.; Stanek, C.R.; Smith, J.L.; et al. Piezomagnetism and magnetoelastic memory in uranium dioxide. Nat. Commun. 2017, 8, 99. [Google Scholar] [CrossRef]
- Sanati, M.; Albers, R.; Lookman, T.; Saxena, A. Elastic constants, phonon density of states, and thermal properties of UO2. Phys. Rev. B Condens. Matter Mater. Phys. 2011, 84, 014116. [Google Scholar] [CrossRef]
- Varshney, D.; Shriya, S. Pressure and temperature dependent elastic, mechanical and thermodynamical properties of nuclear fuel: UO2 and UN2. J. Nucl. Mater. 2013, 440, 344–365. [Google Scholar] [CrossRef]
- Behera, R.K.; Deo, C.S.; Xu, H. Effect of the substitution of f-electron elements on the structure and elastic properties of UO2. J. Nucl. Mater. 2013, 433, 504–513. [Google Scholar] [CrossRef]
- Nandi, C.; Phatak, R.; Shafeeq, M.; Kesari, S.; Rao, R.; Prakash, A. Phase relations and lattice parameter trends in Nd3+−substituted UO2 system under reducing and oxidizing conditions. J. Nucl. Mater. 2023, 579, 154396. [Google Scholar] [CrossRef]
- Debernardi, A. First principles simulations of microscopic mechanisms responsible for the drastic reduction of electrical deactivation defects in Se hyperdoped silicon. Phys. Chem. Chem. Phys. 2021, 23, 24699–24710. [Google Scholar] [CrossRef]
- Chen, T.; Yuan, Y.; Mi, X.; Wu, J.; Tang, A.; Wang, J.; Moelans, N.; Pan, F. Interaction of elements in dilute Mg alloys: A DFT and machine learning study. J. Mater. Res. Technol. 2022, 21, 4512–4525. [Google Scholar] [CrossRef]
- Pathak, A.; Sankarasubramanian, R. First-principle calculations of Co2MSi (M = Cr, Mn, Fe) Heusler alloys. J. Appl. Res. Technol. 2022, 20, 355–368. [Google Scholar] [CrossRef]
- Ranganathan, S.I.; Ostoja-Starzewski, M. Universal elastic anisotropy index. Phys. Rev. Lett. 2008, 101, 055504. [Google Scholar] [CrossRef]
- Balasubramanian, K.; Khare, S.V.; Gall, D. Valence electron concentration as an indicator for mechanical properties in rocksalt structure nitrides, carbides and carbonitrides. Acta Mater. 2018, 152, 175–185. [Google Scholar] [CrossRef]
- Wang, M.; Zhou, Y.; Chen, T.; Yang, H.; Tan, J.; Chen, X.; Pan, F. Tailoring the structure, mechanical, electronic, and thermodynamic properties of Al8Mn5 by doping Ti atom with different atomic site configurations. J. Mater. Res. Technol. 2023, 26, 4469–4482. [Google Scholar] [CrossRef]
- Chen, X.; Niu, H.; Li, D.; Li, Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallics 2011, 19, 1275–1281. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, B.; Zhao, Z. Microscopic theory of hardness and design of novel superhard crystals. Int. J. Refract. Met. Hard Mater. 2012, 33, 93–106. [Google Scholar] [CrossRef]
- Husainy, M.; Szenknect, S.; Podor, R.; Le Goff, X.; Kaczmarek, T.; Moisy, P.; Dacheux, N. Speciation and chemical behavior of molybdenum in uranium dioxide samples prepared by hydroxide precipitation. J. Nucl. Mater. 2024, 596, 155075. [Google Scholar] [CrossRef]
- Lopes, D.A.; Ulrich, T.; Kercher, A.; Bronikowski, M.; Samperton, K.; Scott, S.; Wellons, M.; Green, G.; Spano, T.; Harp, J.; et al. UO2 microstructural evolutions induced by Ni, Mo, and W dopants for intentional forensics. J. Nucl. Mater. 2025, 614, 155885. [Google Scholar] [CrossRef]
- Malakkal, L.; Zhou, S.; Mishra, H.; Bachhav, M.; Ke, J.H.; Jiang, C.; He, L.; Biswas, S. Xenon–metal pair formation in UO2 investigated using DFT + U. J. Appl. Phys. 2025, 137, 155001. [Google Scholar] [CrossRef]
- Gusev, A.I.; Sadovnikov, S.I. Conditions of elastic mechanical stability and elastic properties of crystal structures with different symmetry. Phys. Solid State 2022, 64, 671–675. [Google Scholar] [CrossRef]
- Gong, B.; Frazer, D.; Yao, T.; Hosemann, P.; Tonks, M.; Lian, J. Nano- and micro-indentation testing of sintered UO2 fuel pellets with controlled microstructure and stoichiometry. J. Nucl. Mater. 2019, 516, 169–177. [Google Scholar] [CrossRef]
- Gong, B.; Frazer, D.; Shaffer, B.; Lim, H.C.; Hosemann, P.; Peralta, P. Micro-cantilever beam experiments and modeling in porous polycrystalline UO2. J. Nucl. Mater. 2021, 557, 153210. [Google Scholar] [CrossRef]
- Kurosaki, K.; Saito, Y.; Uno, M.; Yamanaka, S. Characterization of simulated burnup fuel by nanoindentation. J. Nucl. Mater. 2006, 350, 203–207. [Google Scholar] [CrossRef]
- Schneider, C.; Fayette, L.; Zacharie-Aubrun, I.; Blay, T.; Sercombe, J.; Favergeon, J.; Chevalier, S. Study of the hardness and Young’s modulus at the fuel-cladding interface of a high-burnup PWR fuel rod by nanoindentation measurements. J. Nucl. Mater. 2022, 560, 153511. [Google Scholar] [CrossRef]
- Henry, R.; Zacharie-Aubrun, I.; Blay, T.; Tarisien, N.; Chalal, S.; Iltis, X.; Gatt, J.-M.; Langlois, C.; Meille, S. Irradiation effects on the fracture properties of UO2 fuels studied by micro-mechanical testing. J. Nucl. Mater. 2020, 536, 152179. [Google Scholar] [CrossRef]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Senkov, O.N.; Miracle, D.B. Generalization of intrinsic ductile-to-brittle criteria by Pugh and Pettifor for materials with a cubic crystal structure. Sci. Rep. 2021, 11, 4531. [Google Scholar] [CrossRef]
- Kleinman, L. Deformation Potentials in Silicon. I. Uniaxial Strain. Phys. Rev. 1962, 128, 2614–2621. [Google Scholar] [CrossRef]
- Ullah, W. Effects of Fault Zones and Hydrothermal Alteration on the Physical Properties of Crystalline Rocks. Master’s Thesis, Aalto University, Espoo, Finland, 2025. [Google Scholar]
- Naher, M.I.; Naqib, S.H. An ab-initio study on structural, elastic, electronic, bonding, thermal, and optical properties of topological Weyl semimetal TaX (X = P, As). Sci. Rep. 2021, 11, 5592. [Google Scholar] [CrossRef]
- Zacharie-Aubrun, I.; Henry, R.; Blay, T.; Brunaud, L.; Gatt, J.M.; Noirot, J.; Meille, S. Effects of irradiation on mechanical properties of nuclear UO2 fuels evaluated by Vickers indentation at room temperature. J. Nucl. Mater. 2021, 547, 152821. [Google Scholar] [CrossRef]
- Kurosaki, K.; Saito, Y.; Muta, H.; Uno, M.; Yamanaka, S. Nanoindentation studies of UO2 and (U,Ce)O2. J. Alloys Compd. 2004, 381, 240–244. [Google Scholar] [CrossRef]
- Spino, J.; Cobos-Sabate, J.; Rousseau, F. Room-temperature microindentation behaviour of LWR-fuels, part 1: Fuel microhardness. J. Nucl. Mater. 2003, 322, 204–216. [Google Scholar] [CrossRef]
- Xiao, H.; Wang, X.; Long, C.; Chen, H. Effect of burnup on the mechanical properties of doped UO2 ceramic pellets using the simfuel technique. J. Nucl. Mater. 2019, 524, 247–255. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Lattice Parameters (Å) | Volume/Formula Unit (Å3) | Formation Energy (eV/Atom) | Reference | ||
|---|---|---|---|---|---|---|
| a | b | c | ||||
| U4O8 | 5.471 | 5.471 | 5.471 | 40.939 | - | Exp. [30] |
| 5.569 | 5.505 | 5.567 | 42.667 | - | PBE + U [9] | |
| 5.569 | 5.569 | 5.502 | 42.659 | −3.63 | PBE + U [4] | |
| 5.539 | 5.539 | 5.539 | 42.485 | −3.716 | This work | |
| U31Mo1O64 | 11.057 | 11.057 | 11.057 | 42.248 | −3.925 | |
| U30Mo2O64 | 11.021 | 11.021 | 11.021 | 41.833 | −3.842 | |
| U29Mo3O64 | 11.015 | 10.984 | 10.984 | 41.531 | −3.742 | |
| U28Mo4O64 | 10.954 | 10.954 | 10.980 | 41.176 | −3.66 | |
| U27Mo5O64 | 10.930 | 10.930 | 10.930 | 40.805 | −3.526 | |
| System | Crystal System | C11 | C12 | C13 | C33 | C44 | C66 |
|---|---|---|---|---|---|---|---|
| Pure UO2 | Cubic | 378.99 | 103.43 | 54.46 | |||
| Mo1-doped | Cubic | 351.50 | 116.37 | 78.87 | |||
| Mo2-doped | Cubic | 353.08 | 145.16 | 54.27 | |||
| Mo3-doped | Tetragonal | 362.26 | 117.10 | 335.30 | 35.30 | 25.88 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Xu, H.; Si, J.; Lv, H.; Peng, T.; Peng, P.; Wan, X.; Chen, T.; Tang, A. First-Principles Investigation: Effects of Molybdenum Substitution on the Elastic Properties of Uranium Dioxide. Crystals 2026, 16, 378. https://doi.org/10.3390/cryst16060378
Xu H, Si J, Lv H, Peng T, Peng P, Wan X, Chen T, Tang A. First-Principles Investigation: Effects of Molybdenum Substitution on the Elastic Properties of Uranium Dioxide. Crystals. 2026; 16(6):378. https://doi.org/10.3390/cryst16060378
Chicago/Turabian StyleXu, Haixin, Jiaxuan Si, Hengheng Lv, Tao Peng, Peng Peng, Xin Wan, Tao Chen, and Aitao Tang. 2026. "First-Principles Investigation: Effects of Molybdenum Substitution on the Elastic Properties of Uranium Dioxide" Crystals 16, no. 6: 378. https://doi.org/10.3390/cryst16060378
APA StyleXu, H., Si, J., Lv, H., Peng, T., Peng, P., Wan, X., Chen, T., & Tang, A. (2026). First-Principles Investigation: Effects of Molybdenum Substitution on the Elastic Properties of Uranium Dioxide. Crystals, 16(6), 378. https://doi.org/10.3390/cryst16060378