Abstract
Nerolidol (REL), a sesquiterpene with cis and trans isomers, exhibits diverse bioactive and sensory properties. In this study, we integrate single-crystal X-ray diffraction (SC-XRD), molecular docking, molecular dynamics (MD) simulations, and MM/GBSA binding free energy calculations to investigate its inclusion behavior in β-cyclodextrin (β-CD). Crystallization from a cis/trans mixture yielded a complex containing exclusively the trans isomer, forming a 2:1 host–guest assembly where a head-to-head β-CD dimer encapsulates one trans-REL molecule in an extended conformation. Computational models of cis-REL (bent c1 and extended c8 conformers) also stabilized within the β-CD cavity, with the extended conformer showing the most favorable dynamics. The computed binding affinities for all complexes differed by less than the estimated MM/GBSA uncertainty, indicating no statistically significant preference. Since cis/trans separation of nerolidol and related long-chain terpenoids is of considerable interest, our findings suggest that crystallization selectivity in β-CD inclusion complexes cannot be rationalized solely by binding affinity; instead, it likely arises from crystal packing forces and conformational preferences that govern the solid-state assembly.
1. Introduction
Nerolidol (REL) is an acyclic sesquiterpene alcohol (C15H26O) naturally occurring in the essential oils of various plants, with significant levels reported in Melaleuca leucadendra (76–91%) [1], Cinnamomum camphora nerolidol chemotype (16–57%), and several Piper species such as Piper aduncum (~10–25%) [2] and Piper claussenianum (nerolidol-rich major constituent) [3]. It also found at moderate levels (often <20%) in more commonly used aromatic herbs and flowers, including neroli (Citrus aurantium), ginger, jasmine, lavender, tea tree, lemongrass, basil, coriander, thyme, cardamom, and lemon verbena. E-nerolidol (64.0%) is also in abundance in propolis extracted from black poplar (Populus balsamifera L.) buds [4]. Nerolidol exhibits notable antimicrobial, antioxidant, anti-inflammatory, and sedative properties [5,6], and it is widely used in cosmetics, fragrances, and pharmaceuticals, both as an active agent and a skin penetration enhancer [7]. However, its low aqueous solubility and structural complexity present challenges for formulation and selective utilization.
Structurally, nerolidol exists as a mixture of four stereoisomers, which arise from geometric (cis/trans) isomerism around the C6–C7 double bond and chiral (R/S) centers at the C3 position (Figure 1a). The most commonly encountered isomers are (S)-(+)-trans-nerolidol and (R)-(−)-cis-nerolidol, though commercial preparations typically contain a mixture of the geometric and chiral forms. The stereochemistry of nerolidol significantly influences its interaction with biological receptors and sensory perception, making its selective recognition and separation highly relevant for industrial applications [8].
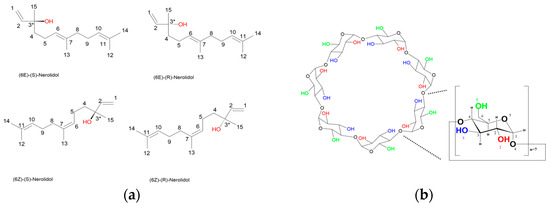
Figure 1.
(a) The four possible stereoisomers of nerolidol are shown, each differing in the configuration at C3 (chiral center) and at the C6–C7 double bond. Top left: (6E)-(S)-Nerolidol; top right: (6E)-(R)-Nerolidol; bottom left: (6Z)-(S)-Nerolidol; bottom right: (6Z)-(R)-Nerolidol. (b) The molecular structure of β-cyclodextrin, composed of seven α-D-glucopyranose units linked by α-1,4-glycosidic bonds, is shown. Hydroxyl groups at the C2 and C3 positions (secondary rim) and the C6 position (primary rim) are indicated in red, blue, and green, respectively. A single glucopyranose unit is enlarged and annotated with atom numbering for clarity.
Cyclodextrins (CDs) are naturally derived cyclic oligosaccharides, with a hydrophobic internal cavity and a hydrophilic exterior, best known for their unique ability to form host–guest inclusion complexes (ICs) with a variety of hydrophobic molecules, facilitating their solubilization, stabilization, and stereoselective recognition. Due to their chiral, toroidal structure and hydrophobic interior cavity, CDs can discriminate between cis/trans isomers and enantiomers, thus serving as versatile tools in enantioseparation and conformational selection. Notably, native β-cyclodextrin (β-CD) (Figure 1b) and its derivatives have been shown to effect chiral recognition of small drug molecules by serving as mobile-phase additives in HPLC or capillary electrophoresis, enabling enantioseparation even without derivatization [9]. While many studies focused on derivatized CDs for improved resolution, these observations establish that the inherent chiral cavity and size/shape complementarity of unmodified CDs can impart meaningful stereoselectivity in suitable host–guest systems [10]. The selective encapsulation of specific nerolidol isomers by β-CD offers a unique approach to probe host–guest complementarity and to explore the factors governing geometric and enantioselective inclusion, both in the crystalline state and in solution.
The REL/β-CD IC has been extensively characterized using experimental techniques such as spectroscopy, calorimetry and scanning electron microscopy, alongside computational modeling [11,12]. These studies consistently supported a 1:1 host–guest stoichiometry for the formed IC, which has also been adopted as the basis for subsequent molecular simulations [13]. As part of our ongoing investigation into inclusion complexes of linear terpenoids with cyclodextrins, we have previously resolved the crystal structures of β-CD with several monoterpenes such as β-citronellol, geraniol, linalool, and citral, exploring their binding behavior and enantioselectivity, particularly in the case of citral [14]. However, no crystallographic data have been reported for sesquiterpene inclusion in β-CD, and the stereoselectivity of such complexes remains unexplored.
In this study, we report the crystal structure of the inclusion complex formed between a racemic mixture of cis- and trans-nerolidol isomers and β-CD as determined by single-crystal X-ray diffraction (SC-XRD). Remarkably, only the trans-nerolidol (t-REL) isomer was found to be selectively encapsulated within the cyclodextrin cavity and in a host–guest ratio of 2:1. To further explore this stereoselectivity, molecular docking simulations were performed to model the inclusion of the cis-nerolidol (c-REL) isomer in both bent and extended guest conformations. The structural stability and conformational behavior of these complexes in an aqueous solution and in the absence of crystal packing forces were investigated by molecular dynamics (MDs) simulations that were conducted for a 12 ns simulation time. Binding free energies were estimated using the MM/GBSA method to quantify relative binding affinities. The computed binding affinities for all complexes differed by less than the estimated MM/GBSA uncertainty, indicating no statistically significant preference for the trans-isomer. Similar discrepancies have been reported in cyclodextrin systems, where experimental studies revealed the preferential inclusion of specific stereoisomers (e.g., clomiphene citrate) not fully anticipated by docking models [15]. More recently, combined NMR and computational studies of isomeric ester inclusion in β-CD demonstrated differences in binding modes and selectivity that were not accurately reproduced by docking alone, underscoring the challenges in reconciling experimental and theoretical results [16]. β-CD and its derivatives have been widely employed as chiral selectors, with several cases highlighting that stereoselectivity may arise from subtle host–guest interactions and solvent effects not captured by simplified models [17]. These precedents emphasize that stereoselectivity in CD inclusion complexes can involve factors beyond intrinsic binding affinity, justifying our combined experimental and computational approach. This combined crystallographic and computational approach provides new insights into the stereoselective inclusion of linear sesquiterpenes into β-CD, with implications for the rational design of CD-based systems for selective delivery and enantioseparation.
2. Materials and Methods
2.1. X-Ray Crystallography
2.1.1. Materials
Nerolidol (98% pure, MW = 222.37 g/mol, d = 0.875 g/mL at 25 °C) as a colorless to light yellow liquid mixture of cis/trans isomers was purchased from Merck KGaA (Damstadt, Germany). β-CD of pharmaceutical-grade quality was obtained as a white powder from Cyclolab Ltd. (Budapest, Hungary). Double-distilled water was utilized for the preparation of all the examined solutions.
2.1.2. Crystallization
Single crystals of REL/β-CD IC were prepared by the slow-cooling method. More specifically, 0.09 g of β-CD (0.08 mmoles) was weighted into vials, and 5 mL of distilled water was added. An equimolar quantity of REL (20 μL, 0.08 mmoles) was added dropwise, and the mixture was stirred for about 4 h at 343 K until a clear solution was obtained. The solution was then cooled gradually to ambient temperature over a period of one week. Colorless prism-like crystals were formed and harvested for SC-XRD analysis.
2.1.3. Data Collection and Processing
Data collection for the REL/β-CD crystal was carried out using a Bruker D8 Venture diffractometer (Bruker AXS GmbH, Karlsruhe, Germany) equipped with a PHOTON 50 detector (CMOS type) and a Cu source, IμS Microfocus X-ray Source, using the APEX3 software suite. The experiment was conducted under cryogenic conditions at 120 K. A total of 1186 frames were recorded and consequently indexed and integrated with the Bruker SAINT software package Version 8.34A [18] using a narrow-frame algorithm. The integration of the data, using a monoclinic unit cell, yielded a total of 63,527 reflections to a maximum θ angle of 64.86° (0.85 Å resolution), of which 11,260 were independent (average redundancy = 5.642, completeness = 99.5%, Rint = 3.50%, Rsig = 2.14%) and 10,528 (93.50%) were greater than 2σ(F2). The crystal was monoclinic with cell constants of a = 18.917(11) Å, b = 24.424(15) Å, c = 15.688(10) Å, β = 110.634(11)°, volume = 6783.(7) Å3, which are based on the refinement of the XYZ-centroids of 9694 reflections above 20 σ(I) with 6.048° < 2θ < 131.2°. Data were corrected for absorption effects using the multi-scan method (SADABS) [19]. The ratio of minimum to maximum apparent transmission was 0.790 (Table 1).

Table 1.
Experimental details and crystallographic parameters.
2.1.4. Structure Determination and Refinement
The structure was solved using SHELXT [20], and all structure refinements were conducted with SHELXL-2014 [21] through the graphical interface SHELXLE [22]. To achieve the optimal refinement of the structures, several restraints were applied on bond lengths and angles of the host and guest molecules where necessary. These restraints were implemented using the SHELXL commands DFIX, DANG, SIMU, and FLAT, and were adjusted along with their standard deviations according to the SHELXL manual [23]. Specifically, the primary restraints used involved 1,2 bond distances (DFIX) and 1,3 bond distances (DANG). The SIMU command was primarily applied to the guest atoms to ensure similar thermal displacement parameters among them. Hydrogen atoms were added computationally using the HFIX command, following the rigid model (i.e., riding on their parent atoms), at distances dependent on the experimental temperature (defined with the TEMP command), and with U = 1.200. The ANIS command was applied to the non-disordered host atoms to refine them anisotropically.
Water molecules were introduced where necessary, based on peaks in the difference Fourier maps (Fo–Fc), following successive refinement cycles. Final structure refinement was performed using the least-squares method on F2. The final anisotropic full-matrix least-squares refinement on F2 with 805 variables converged at R1 = 7.7% for the observed data and wR2 = 22.2% for all data. The goodness of fit (S) was 1.05. The largest peak in the final difference electron density synthesis was 0.74 e−/Å3 and the largest hole was −0.42 e−/Å3. On the basis of the final model, the calculated density was 1.33 g/cm3 and F(000) was 2880.0 e−. A solvent correction (SWAT) was applied to deal with the diffuse electron density arising from disordered water molecules, and 13 reflections showing poor agreement with the refined model were omitted (OMIT) during the final refinement cycles.
After structure validation, the atomic coordinates along with the structure factors of the REL/β-CD IC were deposited at the CSD database under the deposition number 2475921. The images of the inclusion complex were generated using the MERCURY software v4.3.1. [24], while the geometric analysis—specifically the calculation of the geometrical parameters of the macrocyclic host—was performed using the Olex2 program [25]. All the experimental details and crystallographic parameters are quoted in Table 1.
2.2. Computational Methods
2.2.1. Molecular Docking
The crystal structure revealed that t-REL is encapsulated within β-CD, with its aliphatic tail emerging from the primary rim (Section 3.1). While the solid-state assembly corresponded to a dimer, we constructed a β-CD trimer model as the starting point for computational analyses. This trimer—derived from the crystal structure (CCDC 2475921) and consisting of three β-CD units in a head–head–tail arrangement surrounding one t-REL molecule—was employed to introduce conformational flexibility, to explore the potential for higher-order supramolecular associations, and to assess whether alternative dimerization modes, such as head–head or tail–tail contacts, might be favored. Across all simulations, however, the system consistently converged to the head–head 2:1 stoichiometry observed in the crystal (Section 3.2). Prior to the docking analysis, small modifications in the structure were applied as the t-REL guest molecule and the determined water molecules were discarded using PyMOL v 2.0.6 [26]. By using symmetry operations, the additional two CDs were generated.
The docking simulation was performed by choosing the centroid of the 3 β-CDs as the center for the docking grid. Cis-nerolidol molecule (c-REL) from PubChem was docked on the CD “trimer” using AutoDock Vina [27], with a default scoring function and a hybrid global–local search algorithm. The receptor was treated as rigid, while the ligand was considered fully flexible, allowing full conformational sampling of its eight rotatable bonds. The docking search was conducted within a predefined grid box centered at coordinates x = 7.856, y = 10.979, and z = 3.371, with dimensions of 70 × 40 × 60 Å and a grid-point spacing of 0.375 Å established for each dimension. The exhaustiveness parameter, which modulates the depth of the conformational search, was set to 10. Docking was performed using eight parallel CPU threads, with a randomly generated seed to ensure reproducibility and statistical robustness of the results. Ultimately, the docked structure with the lowest binding energy was selected as the starting point for MD simulations for the c1-REL/β-CD case. Additionally, the 8th ranked pose (c8-REL/β-CD case), in which the guest adopts an extended conformation, strongly resembling the one of t-REL as determined from the SC-XRD experiment, was also used for MD calculations.
2.2.2. Molecular Dynamics Simulations
Molecular dynamics (MD) simulations were performed using the Amber22 software package [28]. The host β-cyclodextrin atoms were parameterized with the CLYCAM_06 carbohydrate force field [29], while nerolidol was described using the General Amber Force Field (GAFF) with AM1-BCC-derived charges, generated through the ANTECHAMBER module [30]. The systems were solvated in an explicit TIP3P water environment [31] using an octahedral periodic box that extended at least 10 Å beyond any solute atom. System preparation, including addition of hydrogens in the three hosts and construction of the water shell, was carried out with xLEaP.
The SANDER engine was employed for both the minimization and MD simulations. Long-range electrostatics were treated with the particle mesh Ewald (PME) method, and a 10 Å cutoff was applied for short-range nonbonded interactions. Temperature and pressure regulation followed a Berendsen-type weak coupling scheme, using a relaxation time of 0.5 ps during equilibration and 1.0 ps in production stages.
The equilibration protocol consisted of several steps:
- (a)
- Selective energy minimization of hydrogen atoms;
- (b)
- A 50 ps relaxation of the solvent molecules in the NVT ensemble, applying 50 kcal·mol−1·Å−2 restraints on the host–guest atoms;
- (c)
- Full minimization without restraints;
- (d)
- Gradual heating of the restrained system from 5 K to 300 K with positional restraints of 10 kcal·mol−1·Å−2;
- (e)
- Gradual release of restraints at 300 K;
- (f)
- A 250 ps density equilibration in the NPT ensemble;
- (g)
- A 400 ps unrestrained equilibration at 300 K and 1 atm.
Following equilibration, production trajectories of 12 ns were generated under constant pressure and temperature (NPT).
Simulations were carried out for 12 ns, a timescale commonly employed in β-CD inclusion studies, as this duration is sufficient to capture the stable accommodation of guest molecules.
Simulation data were processed and analyzed using the CPPTRAJ utility. In addition, the binding free energies (ΔG) of the host–guest complexes were estimated by the molecular mechanics/generalized Born surface area (MM/GBSA) approach [32].
2.2.3. MMGB/SA Analysis
In the MM/GBSA approach, the term ΔH is calculated as a sum of MM-based electrostatics energy term and two solvation-related energy terms, Equation (1). ΔGpol corresponds to the generalized Born (GB) approximation of the Poisson–Boltzmann equation, which in turn describes the electrostatic environment of the solute in a solvent containing ions. ΔGnonpol relates to the Solvent Accessible Surface Area (SASA), which is an implicit approach for describing the relationship between ΔG and surface area of a solute molecule.
ΔH = ΔEMM + ΔGpol + ΔGnonpol,
The entropy term T∙ΔS was obtained from the normal mode analysis with a constant temperature using the respective module of the Amber 22 suite and added to the ΔG term according to Equation (2). The entropy term was calculated by taking snapshots every 100 frames of the production run. As this estimation is approximate and highly method-dependent, the entropy terms are reported qualitatively and should be interpreted with caution.
ΔG = ΔH + T·ΔS,
3. Results
3.1. Crystal Structure
3.1.1. Description of the Structure
The inclusion complex of REL/β-CD crystallizes in a monoclinic system and in the C2 space group. The asymmetric unit contains one β-CD host molecule, one REL guest with an occupancy factor of 0.5, and seven water molecules distributed over ten sites (Figure 2a). Only the trans-isomer of nerolidol was found included inside the CD cavity.
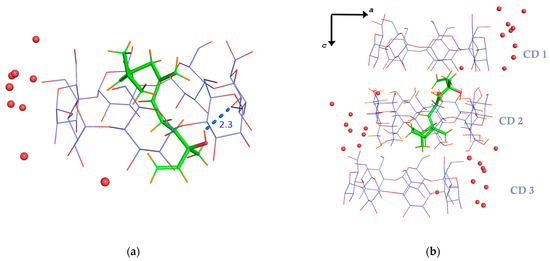
Figure 2.
(a) The asymmetric unit of the crystal structure of the trans-nerolidol/β-CD IC comprises one nerolidol molecule with an occupancy factor of 0.5, one CD molecule, and eight water molecules. The guest is tethered inside the host’s cavity by an intermolecular H-bond between its hydroxyl group and an etheric O atom of the host. (b) Three successive β-CD molecules along the crystallographic c-axis host only one REL molecule, as stereochemical constraints prevent the inclusion of a second guest molecule either in CD1 or in CD3 cavities of the formed “CD trimer”. Consequently, the host-to-guest molecular ratio is always 2:1.
The inclusion mode of nerolidol—a sesquiterpene (15 carbon atoms)—in β-cyclodextrin differs from that of other linear monoterpene (10-carbon) inclusion complexes, such as linalool, β-citronellol, geraniol, and citral/β-CD [14,33,34,35]. For monoterpene inclusion complexes, a 2:2 stoichiometry is typically observed, with the guest molecules accommodated within the cavity of a β-CD dimer. In contrast, in the nerolidol/β-CD complex, nerolidol adopts a vertical orientation within the cavity of CD2. Its hydroxyl group is positioned near the secondary rim. The hydrophobic tail extends from the primary rim of CD2 and partially inserts into the cavity of CD3 through its primary side (Figure 2b). A second cyclodextrin, CD1, contributes to the formation of a classical head-to-head dimer with CD2. This dimer is stabilized by intermolecular hydrogen bonds between their O3n-H hydroxyl groups. Additional stabilization arises from intramolecular hydrogen bonds between the O2n-H and O3(n+1)-H hydroxyls of adjacent glucose units within each host molecule. Due to the extended length of the nerolidol molecule, only one guest molecule is accommodated within the head-to-head CD1–CD2 dimer. Notably, the polar end (hydroxyl group) resides within the center of the dimeric cavity (Figure 2b). This contrasts with monoterpene/β-CD ICs, where the polar group protrudes from the narrow CD rim and the aliphatic tail occupies the center of the dimeric cavity.
The guest molecule is arranged perpendicularly with its linear axis (formed by atoms C1–C2–C3–C5–C6–C7–C8–C9–C11–C12–C13, see Figure 1a) spanning the hydrophobic cavities of CD1, CD2, and CD3. The guest is highly disordered with an occupancy factor of 0.5. It is stabilized inside the cavity with one H-bond and many H-H closed-shell interactions (Supplementary Table S1). Its accommodation within the dimeric host cavity sterically prevents a second molecule from entering. As a result, the host-to-guest ratio consistently remains 2:1. Furthermore, the portion of the guest protruding from the primary rim of the dimeric host enforces the repetition of the same complex unit in consecutive head-to-head dimers along the channels. A guest in CD2 cannot coexist with a guest in CD3 of the adjacent complex unit due to steric hindrance (see Figure 2b).
Water molecules bridge neighboring inclusion complexes by forming hydrogen bonds with oxygen atoms of adjacent cyclodextrins (Figure 2b). Main intra- and intermolecular interactions between the host, guest, and water molecules are quoted in Supplementary Table S1.
The geometric features of the host molecules related to their spatial arrangement are presented in Supplementary Table S2. The glucosidic O4n atoms in the host molecule form a nearly regular heptagon, which is essentially planar. This is reflected in their distances from their approximate centroids (dK), the distance between adjacent O4n atoms (d), and their deviations (dev) from the O4n mean plane. This symmetry is illustrated by the radar plots of the geometrical features in Supplementary Figure S1. It is reflected in the equal distances from the respective centroids and the small deviations from the corresponding mean O4n planes. This indicates that the macrocyclic host adopts a circular conformation. This conformation arises from both inclusion complex formation and the intramolecular hydrogen bonding between adjacent glucosidic units. The glucosidic units display positive dihedral angles τ, defined by O4n–1–C1n–C4n–O4n and the plane of the O4 atoms. This confirms that all glucose units retain the characteristic conical geometry of the cyclodextrin molecule.
Furthermore, analysis of the O5n–C5n–C6n–O6n torsion angles (τ1) in each glucose revealed that five out of seven units adopt the gauche–gauche (gg) conformation. In these units, the O6 atoms point outward from the truncated cone structure of the macrocycle. One glucose adopts the gauche–trans (gt) conformation, with its O6 atom pointing toward the cavity. Another unit, in which the O6 atom is disordered over two positions, exhibits both gg and gt conformations. These details are shown in Supplementary Table S2 and Supplementary Figure S1.
3.1.2. Molecular Packing
The nerolidol/β-CD inclusion complex crystallizes in the space group C2 and forms channels along the c-axis (Figure 3a). The mean plane of the O4n atoms in the dimer forms an angle of 89.56° with the crystallographic c-axis. The offset between two neighboring dimers is 2.76 Å, indicating that the inclusion complexes crystallize in a channel packing mode (CH).
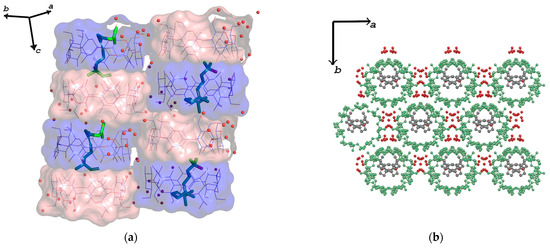
Figure 3.
(a) Crystal molecular arrangement along the c-axis, indicating a channel-type (CH) packing mode. Water molecules (red spheres) bridge adjacent dimers via H-bonds. Hydrogen atoms are omitted for clarity. (b) Crystal packing of the trans-nerolidol/β-CD IC view perpendicular to the ab plane. Hydrogen atoms are omitted for clarity. CDs are depicted as green ball-and-sticks, REL as grey ball-and-sticks and ware molecules as red spheres.
Only one nerolidol molecule is encapsulated within each dimer stacked along the c-axis (Figure 3b). Water molecules acting as water bridges are stabilizing the whole structure as well as H-bonds between hydroxyl groups of adjacent hosts (Supplementary Table S1).
To identify structurally related crystalline forms, the CellCheck v1.0. tool in the WinGX platform [36] was used with its reduced cell search module, comparing the Niggli-reduced unit cell of the present sample with entries in the Cambridge Structural Database (CSD) [37]. The search returned 38 potentially related structures (similarity: 99.24–93.87%), 36 of which are isostructural β-cyclodextrin (β-CD) inclusion complexes. Representative examples (Supplementary Figure S2a–d) include the lipoic acid complex (ccdc id: HAXJIB) [38], the β-CD/methylparaben dimer (ccdc id: AJUVEG) [39], the uranyl–benzoate adduct (ccdc: HUTKOW) [40], and the extended pseudo-rotaxane network with Igepal CO-520 (ccdc id: IXETAH) [41]. Despite wide guest diversity, these structures exhibit recurring monoclinic C-centered packing, channel-type arrangements, and characteristic hydrogen-bonding motifs, underscoring the structural adaptability and host fidelity of β-CD. The close match of our crystal to these systems supports its classification as a channel-type β-CD inclusion complex within a robust and predictable supramolecular framework.
3.2. Computational Results
3.2.1. Docking Results
Molecular docking of c-REL into the β-CD “trimer” was performed using AutoDock Vina. The docking produced nine binding modes with predicted affinities ranging from −6.1 kcal/mol to −5.3 kcal/mol. Among these, Pose 1 was identified as the top-scoring conformation, exhibiting the most favorable binding free energy of −6.1 kcal/mol (Table 2). In Pose 1 (Figure 4a), cis-nerolidol adopts a bent conformation, which enables optimal accommodation within the β-CD “trimeric” cavity. Due to its highest binding affinity and structural fit, this pose was selected as the primary case for further analysis.
Table 2.
Top-ranked binding modes of the guest with β-CD predicted by AutoDock Vina. Binding affinities are given in kcal/mol. Each mode represents a distinct guest conformation. RMSD l.b. (lower bound) and RMSD u.b. (upper bound) are root-mean-square deviations in Å from the best-scoring pose (Mode 1).

Figure 4.
Binding poses of nerolidol within the β-CD host cavity as predicted by AutoDock Vina. (a) Pose 1 (best-scoring pose, affinity = −6.1 kcal/mol) and (b) Pose 8 (affinity = −5.4 kcal/mol) are shown, visualized with UCSF Chimera. c-REL (guest) is rendered as a ball and stick model, and three β-CD molecules (host) are shown as wire and surface representations.
To explore potential conformational diversity and compare it with experimental data, we also examined Pose 8 (Figure 4b), which, despite a slightly less favorable binding energy of −5.4 kcal/mol, presents a more extended, nearly linear conformation of nerolidol. This geometry closely resembles that of t-REL, whose inclusion complex with the β-CD “trimer” has been determined crystallographically. Pose 8 represents a distinct binding conformation, as evidenced by its high RMSD values relative to Pose 1.
This dual-pose examination allows us to correlate docking predictions with known structural data and gain insights into the conformational preferences of nerolidol isomers within cyclodextrin-based hosts.
3.2.2. Trajectory Analysis
The crystallographically determined structure of the t-REL/β-CD inclusion complex (host–guest ratio = 3:1), excluding water molecules of hydration, was used as the starting model. In addition, two ICs of cis-REL isomer, obtained from molecular docking analysis, were also selected for MD simulations. These docked ICs, denoted as c1-REL/β-CD and c8-REL/β-CD, represent the first and eighth top-ranked binding modes: one with the guest adopting a bent and another with a more extended conformation, respectively.
Prior to the MD simulations, all models were subjected to equilibration. Subsequent MD simulations were performed at 300 K in explicit water solvent for approximately 12 ns, as described in Section 2.2.2.
In the first IC case (t-REL/β-CD), the simulation reveals that the guest immediately (0.3 ns) shifts deeper into the CD1 cavity, in an effort to form the classical stable 2:1 β-CD dimer (Figure 5a). Around 3 ns, the third host, CD3, dissociates from the original trimeric assembly and diffuses independently in the solution. The guest retains its extended conformation during the most course of the simulation, optimizing its fit within the hydrophobic core of the β-CD dimer. The final “image” consists of the stable 2:1 dimer formed by CD1 and CD2, while CD3 is found moving freely in the simulated water solution.
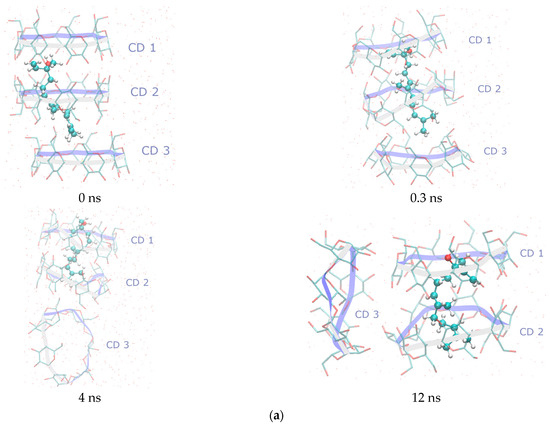
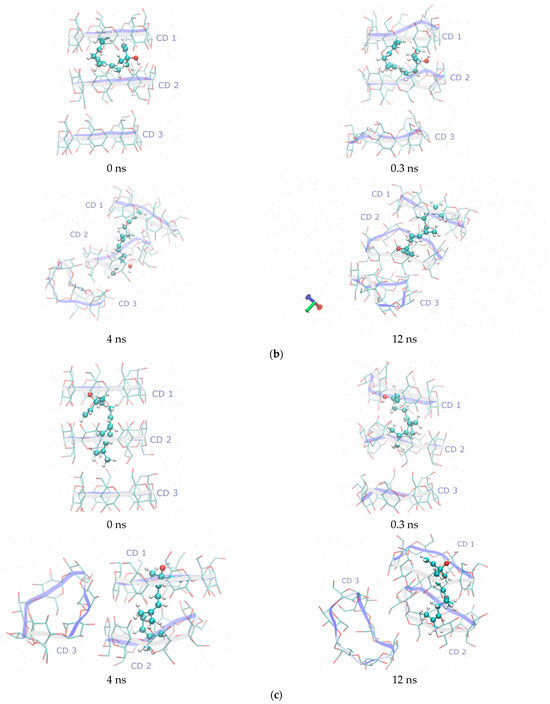
Figure 5.
Representative snapshots from MD simulations of the (a) t-REL/β-CD IC, (b) c1-REL/β-CD IC, and (c) c8-REL/β-CD IC taken at key timepoints (0, 0.3, 4, and 12 ns) to illustrate structural evolution and dynamic behavior in an aqueous solution. The selected timepoints highlight major conformational events and guest encapsulation states. All simulations were performed in explicit water at 300 K. Guest molecules are represented using the CPK coloring scheme, while the β-CD host macrocycles are depicted using a “twister” cartoon style. All molecular visualizations were generated using the VMD v1.9.3 software.
In the second case (c1-REL/β-CD), the starting docking pose, in which the hydroxyl group of the bent guest molecule is found in the interspace between CD1 and CD2 macrocycles, is initially retained. However, at the 4th ns of the simulation, c1-REL adopts the extended conformation, and the classical 2:1 β-CD dimer is formed. The extended guest now has the opposite orientation in comparison to that of the t-REL isomer as its hydroxyl group is continuously found near the O4n plane of CD2 host (Figure 5b). CD3 initially dissociates from the complex but later re-approaches CD1, reorienting to interact via its secondary face with CD1, forming a transient head-to-tail trimeric assembly. Despite these dynamic rearrangements, the guest ultimately assumes an extended conformation within the dimer cavity.
In the final case (c8-REL/β-CD), the initial docking pose placed the guest deeply embedded within the cavity of a CD1–CD2 dimer in a way that strongly resembles the position of t-REL in the dimeric cavity after the minimization procedure (0.3 ns) (Figure 5c). During the MD simulation, CD3 rapidly dissociates at 0.3 ns and remains solvated in the bulk water phase for the remainder of the trajectory. The CD1–CD2 dimer remains structurally stable, maintaining the guest in a tightly bound, linearized conformation.
The RMSD plots are indicative of this dynamic behavior. In the first case, the initial shifting of REL into the CD dimeric cavity is evident by the abrupt rise in the value of the guest (blue line). A gradually rise in the value is observed until the stabilization of the guest occurs at a distance of 20–25 Å away from its initial site (Figure 6a). In the second case, the guest rapidly departs away from its initial position, and it has also been stabilized in the CD dimeric cavity, with a stable RMSD value of 20–25 Å (Figure 6b). Finally, the RMSD value of the c8-REL guest, which adopts an extended conformation, seems to fluctuate around 15 Å and finally stabilizes at 20 Å (Figure 6c). The RMSF and B-factor values of the guests are shown in Supplementary Table S3. From these values, it is evident that the crystallographic determined position of the t-REL is not comfortable as the guest struggles to find a better fitting inside the dimeric CD cavity.
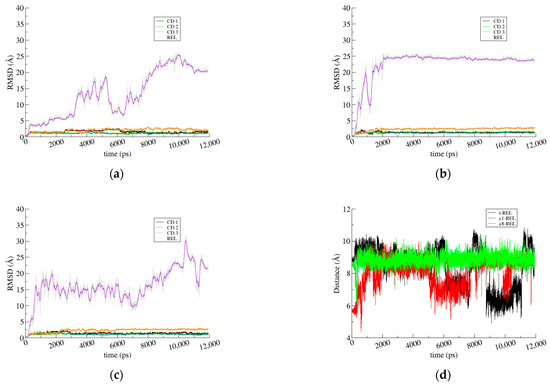
Figure 6.
(a) RMSD for the first frame as a function of the simulation time for t-REL/β-CD; (b) c1-REL/β-CD; and (c) c8-REL/β-CD IC. In all RMSD plots, both the instantaneous values (thin lines) and the running averages with a window size of 100 frames (thick lines) are shown. (d) Distance monitoring between O1 (polar end) and C13 (aliphatic tail) atoms of REL in Å reveals its preference for adopting extended conformation.
3.2.3. MM/GBSA Analysis
MM/GBSA analysis of the REL/β-CD inclusion complexes was performed as follows: the system consisting of the three β-CD hosts was used as the receptor, whereas the encapsulated guest was used as the ligand, in each one of the t-REL/β-CD, c1-REL/β-CD, and c8-REL/β-CD cases. The results indicate the formation of stable inclusion complexes mainly via van der Waals interactions in all cases as the ΔG values are similar within the error margin of the method (Table 3). By comparing these values, we end up with the following ascending order for the stability of these inclusion complexes: c1-REL/β-CD < t-REL/β-CD < c8-REL/β-CD.
Table 3.
Binding free energies and their standard deviations (kcal/mole) resulting from the MM/GBSA analysis of the inclusion complexes of t-REL/β-CD, c1-REL/β-CD, and c8-REL/β-CD (host–guest stoichiometry for all 3 cases is 3:1).
4. Conclusions
The crystallographic analysis of the β-CD inclusion complex with a mixture of cis- and trans-nerolidol showed that only the trans-isomer of nerolidol is observed within the dimer cavity and provided a clear depiction of host–guest stoichiometry, which is 2:1. In the crystal structure, two continuous β-CD molecules form a head-to-head dimer, with the hydrophobic tail of nerolidol protruding from the narrow rim of one host and partially entering a neighboring dimer. The hydroxyl group of nerolidol is located at the center of the dimer, at a depth that prevents the inclusion of a second guest molecule, thus ruling out the formation of a 2:2 stoichiometry as observed in monoterpene inclusion complexes. The inclusion complex crystallizes in a monoclinic system, the space group C2, with the dimers stacking along the c-axis, forming channels (channel-type, CH).
Molecular dynamics simulations were carried out using the crystallographically determined trans-REL/β-CD model, along with two cis-REL/β-CD models—one adopting a bent conformation (c1-REL) and the other an extended conformation (c8-REL)—derived from molecular docking studies in an aqueous environment at 300 K. To explore the structural behavior of the accommodated sesquiterpene within β-CD, we employed a 3:1 host–guest model derived from the crystal structure. This expanded stoichiometry was chosen to introduce conformational flexibility and to probe whether nerolidol could drive alternative association modes, such as head–head or tail–tail dimerization, beyond the arrangement captured in the solid state. In all cases, however, the system consistently relaxed to the head–head 2:1 stoichiometry observed crystallographically, indicating that this mode of association represents the most stable configuration.
In all cases, the guest adjusted to achieve a better fit within the head-to-head CD dimer, and the resulting 2:1 complex remained stable throughout the simulations. Within the hydrophobic cavity of the cyclodextrins, the guest molecule, regardless of isomeric form, consistently adopted an extended conformation to maximize the surface area available for hydrophobic intermolecular interactions. The computed binding affinities for all complexes differed by less than the estimated MM/GBSA uncertainty, indicating no statistically significant preference, though a slight trend toward c8-REL was observed. This discrepancy underscores an important remark: while computational approaches used in this work—molecular docking, molecular dynamics, and MM/GBSA—capture general binding modes and stability in a solution, they do not yet reliably predict stereoisomeric selectivity during crystallization. Recognizing and analyzing such mismatches are essential for improving the accuracy of predictive models of host–guest selectivity, which in turn will advance the rational design of separation strategies and selective inclusion complexes in pharmaceutical, agrochemical, and flavor/fragrance applications.
In addition to solid-state packing influences, a wealth of evidence from spectroscopic and thermodynamic investigations underscores intrinsic chiral discrimination by β-CD. For instance, Alexander et al. (2002) detailed structural asymmetries in β-CD complexes with N-acetyl-l- and d-phenylalanine, attributing different levels of order and water-mediated interactions to enantiomer-specific recognition in the solid state, and reinforcing the role of crystal lattice complementarity [42]. Furthermore, Hirata and colleagues (2020) used cryogenic ion-trap infrared spectroscopy to reveal that D-tyrosine engages more deeply and in a multi-interaction mode with permethylated β-CD compared to L-tyrosine—suggesting selective binding based on interaction multiplicity rather than cavity fit alone [43]. Thermodynamic profiling by Rekharsky and Inoue established how differences in enthalpy and entropy drive enantioselectivity across various β-CD derivatives in a solution, implying that even subtle modifications can change binding preferences significantly [44]. Studies of enhanced chiral discrimination via surface-modified β-CDs (e.g., silylated-acetylated derivatives) show that, beyond inclusion, external host–guest interactions contribute to selectivity, particularly for apolar molecules [45]. Taken together, these findings illustrate that chiral discrimination by β-CD involves a complex interplay of factors—binding geometry, hydration networks, thermodynamics, and both internal and external host–guest interactions—thus reinforcing the need for a multifaceted interpretive framework that includes both crystallographic and solution-phase perspectives.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cryst15090802/s1. Table S1: Principal intra- and intermolecular interactions (suh as distances, angles, and the corresponding estimated standard deviations) involving the β-CD host, trans-nerolidol guest, and water molecules in the crystal structure of the t-REL/β-CD inclusion complex as measured with Olex2. Table S2: Detailed conformational characteristics and geometrical parameters of the β-CD host molecule in the crystal structure of the t-REL/β-CD IC. Table S3: The RMSF (Å) and B-factor (Å2) values for the three guest molecules from three trajectory analyses. Figure S1: (a) The distances and angles as defined in Table S2. In particular, the following are presented: (a) d = O4n…O4(n+1) distances; (b) φ = O4(n-1)…O4n…O4(n+1) angles; (c) dK = distances of the approximate center K of the O4n heptagon from the O4n atoms; (d) φK = O4n…K…O4(n+1) angles; (e) dev = deviations of the O4n atoms from their least-squares plane. All distances are given in Å and angles in (°). Figure S2: The results of the isostructurality search, based on the unit cell parameters of the t-REL/β-cyclodextrin complex reported herein. (a) In the first representative isostructure [1], a 2:2 host–guest stoichiometry is observed, where the guest molecule adopts a bent conformation to accommodate itself in the outer region of the dimer cavity without disrupting the packing of the dimers along the crystallographic c-axis. (b) In the second case [2], the complex exhibits a classical “ship-in-a-bottle” inclusion mode, characterized by a 2:1 host–guest ratio. (c) In the third example [3], the presence of a uranium atom imposes a similar 2:1 stoichiometry. (d) Finally, in the fourth case [4], the rotaxane guest fully penetrates the entire β-cyclodextrin dimer, forming an extended threaded architecture.
Author Contributions
Conceptualization, E.C. and K.B.; methodology, E.C., A.A. and K.B.; software, E.C., A.A., P.K. and K.B.; validation, E.C., A.A. and K.B.; formal analysis, E.C., A.A., P.K. and K.B.; investigation, E.C., A.A., P.K. and K.B.; resources, E.C., A.A. and K.B.; data curation, E.C., A.A., P.K. and K.B.; writing—original draft preparation, E.C. and K.B.; writing—review and editing, E.C. and K.B.; visualization, E.C., A.A. and K.B.; supervision, E.C. and K.B.; project administration, E.C. and K.B.; funding acquisition, E.C. and K.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Crystallographic data for the nerolidol/β-cyclodextrin inclusion complex have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under the deposition number CCDC 2475921. These data can be obtained free of charge from the CCDC via www.ccdc.cam.ac.uk/structures (accessed on 25 July 2025).
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| t-REL | trans-nerolidol |
| c1-REL | cis-nerolidol (Pose 1) |
| c8-REL | cis-nerolidol (Pose 8) |
| β-CD | beta-cyclodextrin |
| MD | molecular dynamics simulation |
| SC-XRD | single-crystal X-ray diffraction |
| IC | inclusion complex |
References
- Padalia, R.C.; Verma, R.S.; Chauhan, A.; Chanotiya, C.S. The Essential Oil Composition of Melaleuca leucadendra L. Grown in India: A Novel Source of (E)-Nerolidol. Ind. Crops Prod. 2015, 69, 224–227. [Google Scholar] [CrossRef]
- Ceole, L.F.; Cardoso, M.D.G.; Soares, M.J. Nerolidol, the Main Constituent of Piper Aduncum Essential Oil, Has Anti-Leishmania Braziliensis Activity. Parasitology 2017, 144, 1179–1190. [Google Scholar] [CrossRef]
- Curvelo, J.A.R.; Marques, A.M.; Barreto, A.L.S.; Romanos, M.T.V.; Portela, M.B.; Kaplan, M.A.C.; Soares, R.M.A. A Novel Nerolidol-Rich Essential Oil from Piper Claussenianum Modulates Candida Albicans Biofilm. J. Med. Microbiol. 2014, 63, 697–702. [Google Scholar] [CrossRef]
- Schepetkin, I.A.; Özek, G.; Özek, T.; Kirpotina, L.N.; Kokorina, P.I.; Khlebnikov, A.I.; Quinn, M.T. Neutrophil Immunomodulatory Activity of Nerolidol, a Major Component of Essential Oils from Populus Balsamifera Buds and Propolis. Plants 2022, 11, 3399. [Google Scholar] [CrossRef]
- Li, W.; Zhang, W.; Liu, Z.; Song, H.; Wang, S.; Zhang, Y.; Zhan, C.; Liu, D.; Tian, Y.; Tang, M.; et al. Review of Recent Advances in Microbial Production and Applications of Nerolidol. J. Agric. Food Chem. 2025, 73, 5724–5747. [Google Scholar] [CrossRef]
- Ephrem, E.; Najjar, A.; Charcosset, C.; Greige-Gerges, H. Use of Free and Encapsulated Nerolidol to Inhibit the Survival of Lactobacillus fermentum in Fresh Orange Juice. Food Chem. Toxicol. 2019, 133, 110795. [Google Scholar] [CrossRef]
- Chan, W.-K.; Tan, L.T.; Chan, K.-G.; Lee, L.-H.; Goh, B.-H. Nerolidol: A Sesquiterpene Alcohol with Multi-Faceted Pharmacological and Biological Activities. Molecules 2016, 21, 529. [Google Scholar] [CrossRef]
- Schubert, V.; Dietrich, A.; Ulrich, T.; Mosandl, A. The Stereoisomers of Nerolidol: Separation, Analysis and Olfactoric Properties. Z. Naturforschung C 1992, 47, 304–307. [Google Scholar] [CrossRef]
- Li, L.; Li, X.; Luo, Q.; You, T. A Comprehensive Study of the Enantioseparation of Chiral Drugs by Cyclodextrin Using Capillary Electrophoresis Combined with Theoretical Approaches. Talanta 2015, 142, 28–34. [Google Scholar] [CrossRef]
- Yu, R.B.; Quirino, J.P. Chiral Separation Using Cyclodextrins as Mobile Phase Additives in Open-Tubular Liquid Chromatography with a Pseudophase Coating. J. Sep. Sci. 2022, 45, 1195–1201. [Google Scholar] [CrossRef]
- de Souza, E.P.B.S.S.; Gomes, M.V.L.D.; dos Santos Lima, B.; Silva, L.A.S.; Shanmugan, S.; Cavalcanti, M.D.; de Albuquerque Júnior, R.L.C.; de Souza Carvalho, F.M.; Marreto, R.N.; de Lima, C.M.; et al. Nerolidol-Beta-Cyclodextrin Inclusion Complex Enhances Anti-Inflammatory Activity in Arthritis Model and Improves Gastric Protection. Life Sci. 2021, 265, 118742. [Google Scholar] [CrossRef]
- Azzi, J.; Danjou, P.-E.; Landy, D.; Ruellan, S.; Auezova, L.; Greige-Gerges, H.; Fourmentin, S. The Effect of Cyclodextrin Complexation on the Solubility and Photostability of Nerolidol as Pure Compound and as Main Constituent of Cabreuva Essential Oil. Beilstein J. Org. Chem. 2017, 13, 835–844. [Google Scholar] [CrossRef]
- Venkatesan, K.B.; Alamelu, S.; Srinivasan, M.K.; Pachaiappan, P. Nerolidol Loaded Beta Cyclodextrin Nanoparticles: A Promising Strategy for Inducing Apoptosis in Breast Cancer Cells (MCF-7). J. Biomater. Sci. Polym. Ed. 2025, 1–31. [Google Scholar] [CrossRef]
- Fourtaka, K.; Christoforides, E.; Tzamalis, P.; Bethanis, K. Inclusion of Citral Isomers in Native and Methylated Cyclodextrins: Structural Insights by X-Ray Crystallography and Molecular Dynamics Simulation Analysis. J. Mol. Struct. 2021, 1234, 130169. [Google Scholar] [CrossRef]
- Maheshwari, A.; Saraswat, H.; Upadhyay, S.K. Structural Insights into the Inclusion Complexes between Clomiphene Citrate and β-Cyclodextrin: The Mechanism of Preferential Isomeric Selection. Chirality 2017, 29, 451–457. [Google Scholar] [CrossRef]
- Peluso, P.; Dallocchio, R.; Dessì, A.; Salgado, A.; Chankvetadze, B.; Scriba, G.K.E. Molecular Modeling Study to Unravel Complexation of Daclatasvir and Its Enantiomer by β-Cyclodextrins. Computational Analysis Using Quantum Mechanics and Molecular Dynamics. Carbohydr. Polym. 2024, 346, 122483. [Google Scholar] [CrossRef]
- Napiórkowska, E.; Szeleszczuk, Ł. Review of Applications of β-Cyclodextrin as a Chiral Selector for Effective Enantioseparation. Int. J. Mol. Sci. 2024, 25, 10126. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SAINT 2013, Version 8.34A; Bruker-AXS: Madison, WI, USA, 2013.
- Sheldrick, G.M. SADABS 2012, Version 2008/1; Bruker-AXS: Madison, WI, USA, 2012.
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt Graphical User Interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef]
- Thorn, A.; Dittrich, B.; Sheldrick, G.M. Enhanced Rigid-Bond Restraints. Acta Crystallogr. Sect. A Found. Crystallogr. 2012, 68, 448–451. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New Features for the Visualization and Investigation of Crystal Structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Schrödinger, LLC. The PyMOL Molecular Graphics System, Version 1.8; Schrödinger, LLC.: New York, NY, USA, 2015.
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An Overview of the Amber Biomolecular Simulation Package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 3, 198–210. [Google Scholar] [CrossRef]
- Kirschner, K.N.; Yongye, A.B.; Tschampel, S.M.; Gonzalez-Outeirino, J.; Daniels, C.R.; Foley, B.L.; Woods, R.J. GLYCAM06: A Generalizable Biomolecular Force Field. Carbohydrates. J. Comput. Chem. 2008, 29, 622–655. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.; Case, D. ANTECHAMBER: An Accessory Software Package for Molecular Mechanical Calculations. J. Chem. Inf. Comput. Sci.—JCISD 2000, 222, 2001. [Google Scholar]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Miller, B.R., 3rd; McGee, T.D.J.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Fourtaka, K.; Christoforides, E.; Mentzafos, D.; Bethanis, K. Crystal Structures and Molecular Dynamics Studies of the Inclusion Compounds of β-Citronellol in β-Cyclodextrin, Heptakis(2,6-Di-O-Methyl)-β-Cyclodextrin and Heptakis(2,3,6-Tri-O-Methyl)-β-Cyclodextrin. J. Mol. Struct. 2018, 1161, 1–8. [Google Scholar] [CrossRef]
- Christoforides, E.; Fourtaka, K.; Andreou, A.; Bethanis, K. X-Ray Crystallography and Molecular Dynamics Studies of the Inclusion Complexes of Geraniol in β-Cyclodextrin, Heptakis (2,6-Di-O-Methyl)-β-Cyclodextrin and Heptakis (2,3,6-Tri-O-Methyl)-β-Cyclodextrin. J. Mol. Struct. 2020, 1202, 127350. [Google Scholar] [CrossRef]
- Ceborska, M. Structural Investigation of the β-Cyclodextrin Complexes with Linalool and Isopinocampheol—Influence of Monoterpenes Cyclicity on the Host–Guest Stoichiometry. Chem. Phys. Lett. 2016, 651, 192–197. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Rácz, C.-P.; Borodi, G.; Pop, M.M.; Kacso, I.; Sánta, S.; Tomoaia-Cotisel, M. Structure of the Inclusion Complex of β-Cyclodextrin with Lipoic Acid from Laboratory Powder Diffraction Data. Acta Crystallogr. Sect. B 2012, 68, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Caira, M.R.; de Vries, E.J.C.; Nassimbeni, L.R. Crystallization of Two Forms of a Cyclodextrin Inclusion Complex Containing a Common Organic Guest. Chem. Commun. 2003, 16, 2058–2059. [Google Scholar] [CrossRef]
- Navaza, A.; Iroulart, M.G.; Navaza, J. A Monomeric Uranyl Hydroxide System Obtained by Inclusion in the β-Cyclodextrin Cavity. J. Coord. Chem. 2000, 51, 153–168. [Google Scholar] [CrossRef]
- Guerrero-Martínez, A.; Ávila, D.; Martínez-Casado, F.J.; Ripmeester, J.A.; Enright, G.D.; Cola, L.D.; Tardajos, G. Solid Crystal Network of Self-Assembled Cyclodextrin and Nonionic Surfactant Pseudorotaxanes. J. Phys. Chem. B 2010, 114, 11489–11495. [Google Scholar] [CrossRef]
- Alexander, J.M.; Clark, J.L.; Brett, T.J.; Stezowski, J.J. Chiral Discrimination in Cyclodextrin Complexes of Amino Acid Derivatives: β-Cyclodextrin/N-Acetyl-l-Phenylalanine and N-Acetyl- d-Phenylalanine Complexes. Proc. Natl. Acad. Sci. USA 2002, 99, 5115–5120. [Google Scholar] [CrossRef]
- Hirata, K.; Mori, Y.; Ishiuchi, S.; Fujii, M.; Zehnacker, A. Chiral Discrimination between Tyrosine and β-Cyclodextrin Revealed by Cryogenic Ion Trap Infrared Spectroscopy. Phys. Chem. Chem. Phys. 2020, 22, 24887–24894. [Google Scholar] [CrossRef] [PubMed]
- Rekharsky, M.; Inoue, Y. Chiral Recognition Thermodynamics of β-Cyclodextrin: The Thermodynamic Origin of Enantioselectivity and the Enthalpy−Entropy Compensation Effect. J. Am. Chem. Soc. 2000, 122, 4418–4435. [Google Scholar] [CrossRef]
- Balzano, F.; Uccello-Barretta, G.; Sicoli, G.; Vanni, L.; Recchimurzo, A.; Aiello, F. Chiral Discrimination Mechanisms by Silylated-Acetylated Cyclodextrins: Superficial Interactions vs. Inclusion. Int. J. Mol. Sci. 2022, 23, 13169. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).