
Multiple Slater Determinants and Strong Spin-Fluctuations as Key Ingredients of the Electronic Structure of Electron- and Hole-Doped Pb10−xCux(PO4)6O


 , and
, and
Abstract
1. Introduction
2. Results and Discussion
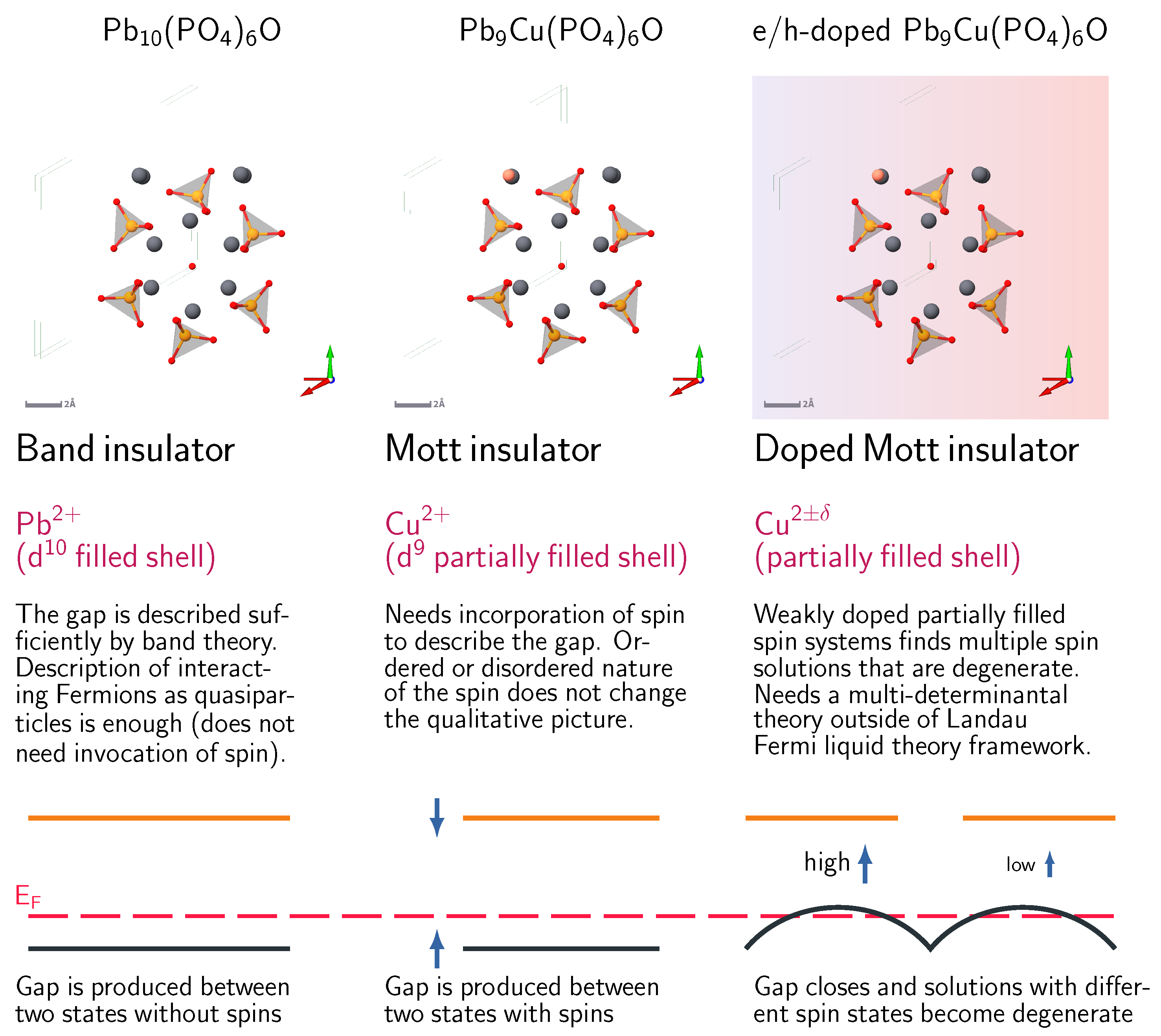
2.1. Lattice Structure
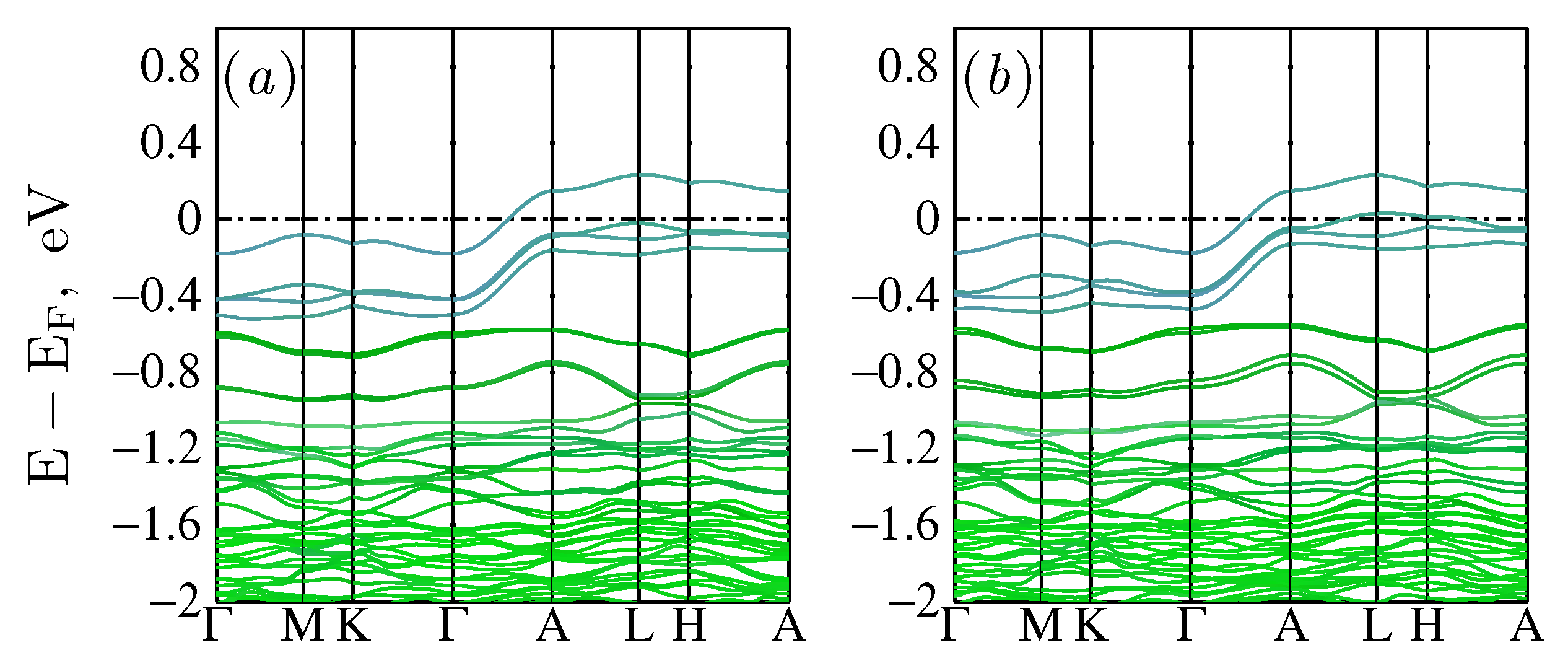
2.2. Energy Band Structure of the Parent LK-99 Compound
2.3. Doping
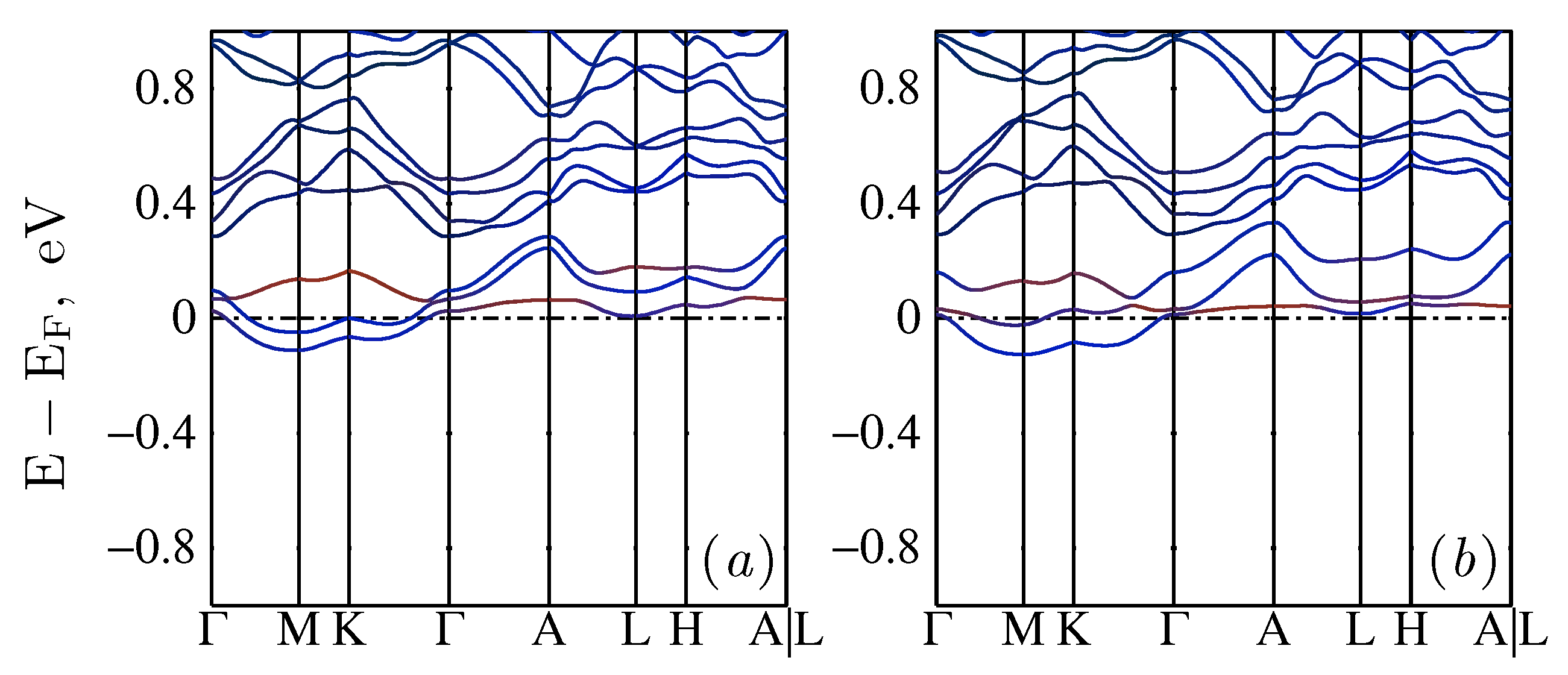
2.4. Hole-Doped Case
2.5. Electron-Doped Case
3. Conclusions
4. Methods
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, S.; Kim, J.; Kim, H.-T.; Im, S.; An, S.; Auh, K.H. Superconductor Pb10−xCux(PO4)6O showing levitation at room temperature and atmospheric pressure and mechanism. arXiv 2023, arXiv:2307.12037. [Google Scholar]
- Lee, S.; Kim, J.-H.; Kwon, Y.-W. The first room-temperature ambient-pressure superconductor. arXiv 2023, arXiv:2307.12008. [Google Scholar]
- Lee, S.; Kim, J.; Im, S.; An, S.; Kwon, Y.-W.; Ho, A.K. Consideration for the development of room-temperature ambient-pressure superconductor (LK-99). J. Korean Cryst. Growth Cryst. Technol. 2023, 33, 61. [Google Scholar]
- Guo, K.; Li, Y.; Jia, S. Ferromagnetic half levitation of LK-99-like synthetic samples. Sci. China Phys. Mech. Astron. 2023, 66, 107411. [Google Scholar] [CrossRef]
- Timokhin, I.; Chen, C.; Wang, Z.; Yang, Q.; Mishchenko, A. Synthesis and characterisation of LK-99. arXiv 2023, arXiv:2308.03823v2. [Google Scholar]
- Liu, L.; Meng, Z.; Wang, X.; Chen, H.; Duan, Z.; Zhou, X.; Yan, H.; Qin, P.; Liu, Z. Semiconducting Transport in Pb10−xCux(PO4)6O Sintered from Pb2SO5 and Cu3P. Adv. Funct. Mater. 2023, 33, 2308938. [Google Scholar] [CrossRef]
- Kumar, K.; Karn, N.K.; Kumar, Y.; Awana, V. Absence of superconductivity in LK-99 at ambient conditions. ACS Omega 2023, 8, 41737. [Google Scholar] [CrossRef]
- Kumar, K.; Karn, N.; Awana, V. Synthesis of possible room temperature superconductor LK-99: Pb9Cu (PO4)6O. Supercond. Sci. Technol. 2023, 36, 10LT02. [Google Scholar] [CrossRef]
- Wu, H.; Yang, L.; Xiao, B.; Chang, H. Successful growth and room temperature ambient-pressure magnetic levitation of LK-99. arXiv 2023, arXiv:2308.01516. [Google Scholar]
- Jain, P.K. Superionic phase transition of copper (I) sulfide and its implication for purported superconductivity of LK-99. J. Phys. Chem. C 2023, 127, 18253. [Google Scholar] [CrossRef]
- Griffin, S.M. Origin of correlated isolated flat bands in copper-substituted lead phosphate apatite. arXiv 2023, arXiv:2307.16892. [Google Scholar]
- Lai, J.; Li, J.; Liu, P.; Sun, Y.; Chen, X.-Q. First-principles study on the electronic structure of Pb10−xCux(PO4)6O (x = 0, 1). J. Mater. Sci. Technol. 2023, 171, 66–70. [Google Scholar] [CrossRef]
- Kurleto, R.; Lany, S.; Pashov, D.; Acharya, S.; van Schilfgaarde, M.; Dessau, D.S. Pb-apatite framework as a generator of novel flat-band CuO based physics, including possible room temperature superconductivity. arXiv 2023, arXiv:2308.00698. [Google Scholar]
- Si, L.; Held, K. Electronic structure of the putative room-temperature superconductor Pb9Cu (PO4)6O. Phys. Rev. B 2023, 108, L121110. [Google Scholar] [CrossRef]
- Cabezas-Escares, J.; Barrera, N.F.; Lavroff, R.H.; Alexandrova, A.N.; Cardenas, C.; Munoz, F. Electronic structure and vibrational stability of copper-substituted lead apatite LK-99. Phys. Rev. B 2024, 109, 144515. [Google Scholar] [CrossRef]
- Sun, Y.; Ho, K.-M.; Antropov, V. Metallization and spin fluctuations in Cu-doped lead apatite. Phys. Rev. Mater. 2023, 7, 114804. [Google Scholar] [CrossRef]
- Tao, K.; Chen, R.; Yang, L.; Gao, J.; Xue, D.; Jia, C. The 1/4 occupied O atoms induced ultra-flatband and the one-dimensional channels in the Pb9Cux(PO4)6O4 (x= 0, 0.5) crystal. APL Mater. 2024, 12, 021120. [Google Scholar] [CrossRef]
- Jiang, Y.; Lee, S.B.; Herzog-Arbeitman, J.; Yu, J.; Feng, X.; Hu, H.; Călugăru, D.; Brodale, P.S.; Gormley, E.L.; Vergniory, M.G.; et al. Pb9Cu(PO4)6(OH)2: Phonon bands, localized flat-band magnetism, models, and chemical analysis. Phys. Rev. B 2023, 108, 235127. [Google Scholar] [CrossRef]
- Van Schilfgaarde, M.; Kotani, T.; Faleev, S. Quasiparticle Self-Consistent GW Theory. Phys. Rev. Lett. 2006, 96, 226402. [Google Scholar] [CrossRef]
- Pashov, D.; Acharya, S.; Lambrecht, W.R.L.; Jackson, J.; Belashchenko, K.D.; Chantis, A.; Jamet, F.; van Schilfgaarde, M. Questaal: A package of electronic structure methods based on the linear muffin-tin orbital technique. Comp. Phys. Comm. 2020, 249, 107065. [Google Scholar] [CrossRef]
- Puphal, P.; Akbar, M.; Hepting, M.; Goering, E.; Isobe, M.; Nugroho, A.; Keimer, B. Single crystal synthesis, structure, and magnetism of Pb10−xCux(PO4)6O. APL Mater. 2023, 11, 101128. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Sun, J.; Ruzsinszky, A.; Perdew, J.P. Strongly constrained and appropriately normed semilocal density functional. Phys. Rev. Lett. 2015, 115, 036402. [Google Scholar] [CrossRef] [PubMed]
- Questaal Code Website. Available online: https://www.questaal.org (accessed on 29 June 2025).
- Kotani, T.; van Schilfgaarde, M.; Faleev, S.V. Quasiparticle self-consistent GW method: A basis for the independent-particle approximation. PRB 2007, 76, 165106. [Google Scholar] [CrossRef]
- Ismail-Beigi, S. Justifying quasiparticle self-consistent schemes via gradient optimization in Baym–Kadanoff theory. J. Phys. Condens. Matter 2017, 29, 385501. [Google Scholar] [CrossRef]
- Cunningham, B.; Gruning, M.; Pashov, D.; Van Schilfgaarde, M. QSGW: Quasiparticle self-consistent GW with ladder diagrams in W. Phys. Rev. B 2023, 108, 165104. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef]
- Acharya, S.; Pashov, D.; Rudenko, A.N.; Rosner, M.; van Schilfgaarde, M.; Katsnelson, M.I. Importance of charge self-consistency in first-principles description of strongly correlated systems. npj Comput. Mater. 2021, 7, 208. [Google Scholar] [CrossRef]
- Maksimov, E.G.; Mazin, I.I.; Savrasov, S.Y.; Uspenski, Y.A. Excitation spectra of semiconductors and insulators: A density-functional approach to many-body theory. J. Phys. Condens. Matter 1989, 1, 2493. [Google Scholar] [CrossRef]
- Vidal, J.; Botti, S.; Olsson, P.; Guillemoles, J.-F.; Reining, L. Strong Interplay between Structure and Electronic Properties in CuIn(S,Se)2: A First-Principles Study. Phys. Rev. Lett. 2010, 104, 056401. [Google Scholar] [CrossRef]
- Korotin, D.M.; Novoselov, D.Y.; Shorikov, A.O.; Anisimov, V.I.; Oganov, A.R. Electronic correlations in promising room-temperature superconductor Pb9Cu(PO4)6O: A DFT+DMFT study. Phys. Rev. B 2023, 108, L241111. [Google Scholar] [CrossRef]
- Si, L.; Wallerberger, M.; Smolyanyuk, A.; di Cataldo, S.; Tomczak, J.M.; Held, K. Pb10−xCux(PO4)6O: A Mott or charge transfer insulator in need of further doping for (super)conductivity. J. Phys. Condens. Matter 2023, 36, 065601. [Google Scholar] [CrossRef] [PubMed]
- Gruning, M.; Marini, A.; Rubio, A. Density functionals from many-body perturbation theory: The band gap for semiconductors and insulators. J. Chem. Phys. 2006, 124, 154108. [Google Scholar] [CrossRef] [PubMed]
- Neaton, J.B.; Hybertsen, M.S.; Louie, S.G. Renormalization of molecular electronic levels at metal-molecule interfaces. Phys. Rev. Lett. 2006, 97, 216405. [Google Scholar] [CrossRef]
- Dingle, R. Luminescent transitions associated with divalent copper impurities and the green emission from semiconducting zinc oxide. Phys. Rev. Lett. 1969, 23, 579. [Google Scholar] [CrossRef]
- Lany, S.; Zunger, A. Polaronic hole localization and multiple hole binding of acceptors in oxide wide-gap semiconductors. Phys. Rev. B 2009, 80, 085202. [Google Scholar] [CrossRef]
- Grzeszczyk, M.; Acharya, S.; Pashov, D.; Chen, Z.; Vaklinova, K.; van Schilfgaarde, M.; Watanabe, K.; Taniguchi, T.; Novoselov, K.; Katsnelson, M.; et al. Strongly Correlated Exciton-Magnetization System for Optical Spin Pumping in CrBr3 and CrI3. Adv. Mater. 2023, 35, 2209513. [Google Scholar] [CrossRef]
- Acharya, S.; Pashov, D.; Weber, C.; van Schilfgaarde, M.; Lichtenstein, A.I.; Katsnelson, M.I. A theory for colors of strongly correlated electronic systems. Nat. Commun. 2023, 14, 5565. [Google Scholar] [CrossRef]
- Hirata, S.; Head-Gordon, M. Time-dependent density functional theory within the Tamm–Dancoff approximation. Chem. Phys. Lett. 1999, 314, 291. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Undoped (Valence) | Undoped (Conduction) | h-Doped (High Spin) | h-Doped (Low Spin) | e-Doped (High Spin) | e-Doped (Low Spin) | |
|---|---|---|---|---|---|---|
| Pb | 39 | 1 | 39 | 39 | 42 | 42 |
| Cu | 0 | 84 | 0 | 0 | 38 | 38 |
| O | 29 | 15 | 29 | 29 | 19 | 19 |
| O| | 32 | 0 | 32 | 32 | 1 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pashov, D.; Acharya, S.; Lany, S.; Dessau, D.S.; van Schilfgaarde, M. Multiple Slater Determinants and Strong Spin-Fluctuations as Key Ingredients of the Electronic Structure of Electron- and Hole-Doped Pb10−xCux(PO4)6O. Crystals 2025, 15, 621. https://doi.org/10.3390/cryst15070621
Pashov D, Acharya S, Lany S, Dessau DS, van Schilfgaarde M. Multiple Slater Determinants and Strong Spin-Fluctuations as Key Ingredients of the Electronic Structure of Electron- and Hole-Doped Pb10−xCux(PO4)6O. Crystals. 2025; 15(7):621. https://doi.org/10.3390/cryst15070621
Chicago/Turabian StylePashov, Dimitar, Swagata Acharya, Stephan Lany, Daniel S. Dessau, and Mark van Schilfgaarde. 2025. "Multiple Slater Determinants and Strong Spin-Fluctuations as Key Ingredients of the Electronic Structure of Electron- and Hole-Doped Pb10−xCux(PO4)6O" Crystals 15, no. 7: 621. https://doi.org/10.3390/cryst15070621
APA StylePashov, D., Acharya, S., Lany, S., Dessau, D. S., & van Schilfgaarde, M. (2025). Multiple Slater Determinants and Strong Spin-Fluctuations as Key Ingredients of the Electronic Structure of Electron- and Hole-Doped Pb10−xCux(PO4)6O. Crystals, 15(7), 621. https://doi.org/10.3390/cryst15070621