
Tailoring the Luminescence Properties of Strontium Aluminate Phosphors for Unique Smartphone Detectable Optical Tags

 , ,
, ,  , and
, and
Abstract
1. Introduction
2. Materials and Synthesis
2.1. Materials and Methods
2.2. Synthesis Procedure
- Cation solution: A mixture of strontium nitrate and aluminum nitrate (metal ion 0.2 M concentrate), along with Eu2+, Dy3+, and B3+ dopants in ratios of 1%, 2%, and 15%.
- Anion solution: Each precursor (KOH, LiOH, NaOH, HMTA, K2CO3, Na2CO3, or (NH4)2CO3) was dissolved in 20 mL of DI water, maintaining a Sr2+ to OH− ratio of 1:2 or Sr2+ to CO32− ratio of 1:1.
3. Characterization Techniques and Results
3.1. X-Ray Diffraction (XRD)
3.2. Morphology (SEM—Scanning Electron Microscope)
3.3. Energy Dispersive Spectroscopy (EDS)
3.4. X-Ray Photoelectron Spectroscopy XPS
3.5. TSL Analysis
4. Smartphone Measurements
4.1. Smartphone-Based Contactless Sensing
4.2. Spectral Measurements
4.2.1. Measurement Equipment
4.2.2. Smartphone Characterization
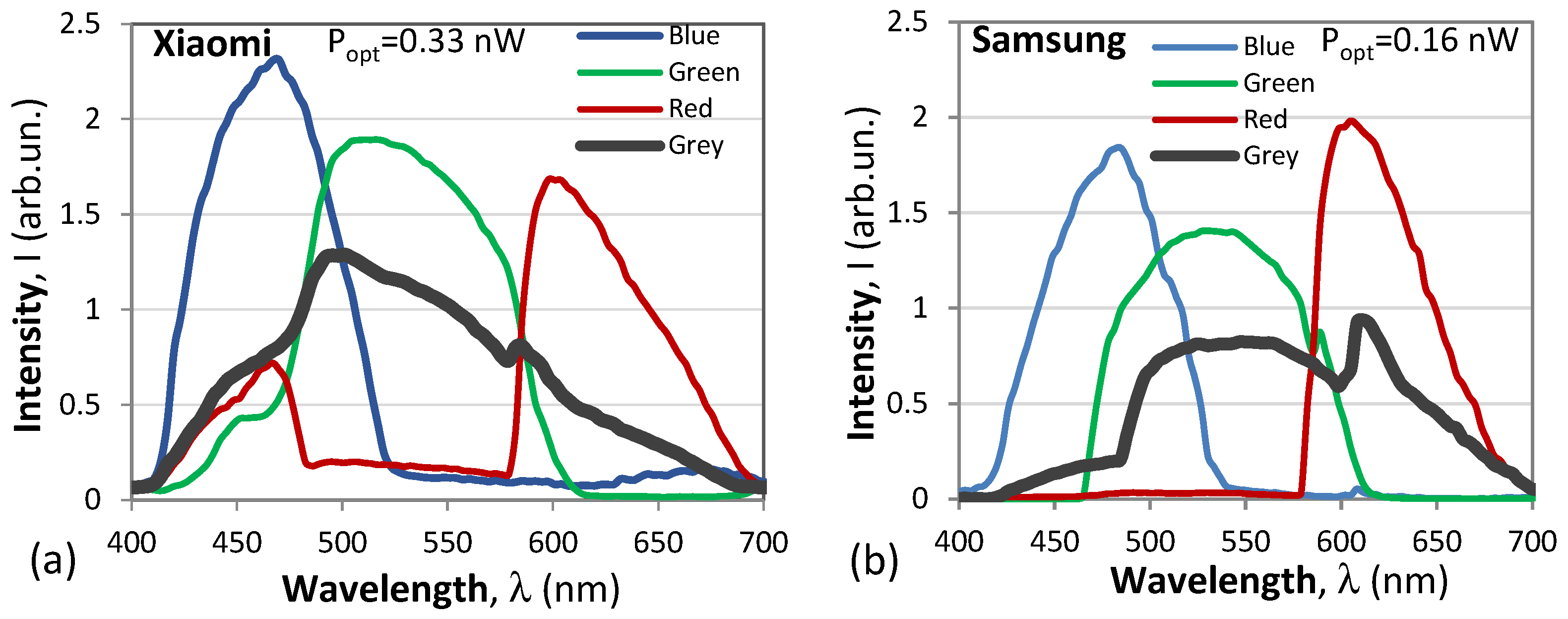
4.2.3. Smartphone Spectral Measurements
4.3. Time-Dependent Responses
4.3.1. Smartphone Measurements of the Time Responses
4.3.2. Experimental Results
- (1)
- The relative share of phosphorescence ΔuOFF of the samples are clearly different and, in all cases, rise with the duty cycle, i.e., with the excitation duration τ.
- (2)
- A fast rise response implies a fast drop in the luminescence, i.e., a lower ΔuOFF.
- (3)
- The sample #4 (NaOH) with the weakest luminescence also has the lowest share of phosphorescence (≈11%). Sample #3 (KOH) with the strongest luminescence has the second low share of phosphorescence (≈22%). The rest of the samples exhibit a phosphorescence share between 40% and 52%.
- (1)
- The Log[1-uON(t)] and Log[uOFF(t)] lines get closer as the duty cycle increases, which is due to the greater probability of populating the traps.
- (2)
- Most of the samples exhibit a change of the linear drop in the double Log plot, meaning that the rate of decay changes, and as the slope increases, this means that some critical time interval Δtc = 0.2 s–0.3 s (indicated in Figure 16) after switching OFF the excitation the phosphorescence decay speeds up. This critical duration is the same for both ON and OFF responses. The change implies that after this critical moment, the probability of electrons getting released increases.
- (3)
- For sample #3 (KOH) with the strongest luminescence and the lowest phosphorescence share, this change of decay rate is the weakest.
- (4)
- Sample #4 (NaOH) exhibits a specific time decay response. Initially, it drops very fast to a low level (Figure 16d) then slowly recovers to a phosphorescence level ≤3% followed by a slow decay after one second. This behavior was observed for all duty cycles and is indicative of some internal conversion dynamics.
- (1)
- As the duty cycle, and hence the excitation duration, increases, the A and α increase, which means that the relative share of phosphorescence immediately prior to switching off the excitation increases. However, after switching off, the decay rate increases with the duty cycle.
- (2)
- For the rise response, the opposite is true because of the periodic excitation. The longer the duration τ (duty cycle increases), the smaller the relative share of phosphorescence to saturation level, i.e., the less traps remain to be populated.
- (1)
- The product of the luminescence intensity L (in counts) and the relative share of phosphorescence ΔuOFF yield the phosphorescence intensity P immediately after switching off the excitation source
- (2)
- The phosphorescent intensity of the samples is shown on the last line so we can rank them as follows:#5 (4890), #6 (4060), #2 (3570), #7 (3400) #3 (2350), #1 (1970), and #4 (155)
- (3)
- Looking at their decay rate as expressed by α, we can arrange them in the following from slowest to fastest order#3 (−0.094), #7 (−0.173), #5 (−0.185), #6 (−0.192), #4 (−0.218), #2 (−0.268), #1 (−0.334)
- (4)
- The three samples that appear among the first four in the above two arrangements are: #5 (K2CO3), #6 (Na2CO3), #7 (NH4)2CO3). With Reference to Table 2, we find that these samples have comparable trapping center depths.
4.4. Temperature Dependent Responses
- (1)
- The overall intensity of luminescence decreases with temperature (see Equation (A1)). For the normalized responses, the decrease is ΔuT over a temperature interval ΔT from which the temperature sensitivities are calculated and presented in Table 5.
- (2)
- As temperature increases both the rise and decay responses slow down i.e., α0 and α diminish.
- (3)
- The relative share of phosphorescence ΔuOFF tends to increase
5. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A. Power Law Approximation for the Time Responses
Appendix A.1. The Power Law Approximations

Appendix A.2. Physical Meaning of the Power Law Parameters
- A.
- The rise phase. Prior to the rise phase when the excitation is ON, no luminescence exists as the Eu ions are not excited. As the excitation is ON, the first luminescence to be observed within durations less than a millisecond is the fluorescence, which is caused by transitions form the excited Eu2+* to the ground state. With some probability, excited electrons absorb additional thermal energy kT and pass to the conduction zone where they are captured by traps of random location and energy depth. After some time delay, trapped electrons are released by thermal fluctuations and after encountering an ionized Eu center relax to ground level releasing a photon. The delayed emission constitutes the phosphorescence share. At the first moment t0 that luminescence of the sample is measured, the level uON,0 = uON(t0) is mostly due to fluorescence. As fluorescence is a much faster process, at a constant excitation rate (rectangular pulses), its share remains constant. Therefore, we can consider that the remaining 1-uON,0 is actually the relative share of phosphorescence. This level is indicated in Figure 15 and determined from Equation (A3a) as:
- B.
- The decay phase. At the end of the ON phase, a certain proportion of the traps in the material have captured electrons from the conduction band that are still not released. Since no excitation is present, we have only a process of electron release and a delayed emission—i.e., phosphorescence. Immediately after the switching OFF of the source, the fast drop to the level D0 = D(t0) is caused by fluorescence, which means that the quantity 1-uOFFN,0 can be used as a measure of the relative share of fluorescence, and correspondingly the relative share of phosphorescence ΔuOFF is:
Appendix B. Rietveld Refinement Results

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Phase | Space Group | Crystal System | a (Å) | b (Å) | c (Å) | Weight Fraction (%) |
|---|---|---|---|---|---|---|---|
| KOH | Sr4Al14O25 | Pmma (No. 51) | Orthorhombic | 4.8823 | 8.4806 | 24.7697 | 0.48 |
| KOH | SrAl2O4 | P21 (No. 4) | Monoclinic | 5.1621 | 8.8156 | 8.4535 | 12.85 |
| KOH | Sr3Al2O6 | Pa-3 (No. 205) | Cubic | 15.9999 | 15.9999 | 15.9999 | 86.68 |
| NH4CO3 | Sr4Al14O25 | Pmma (No. 51) | Orthorhombic | 4.8823 | 8.4806 | 24.7697 | 8.87 |
| NH4CO3 | SrAl2O4 | P21 (No. 4) | Monoclinic | 5.1621 | 8.8156 | 8.4535 | 52.08 |
| NH4CO3 | Sr3Al2O6 | Pa-3 (No. 205) | Cubic | 15.9999 | 15.9999 | 15.9999 | 39.05 |
| LiOH | Sr4Al14O25 | Pmma (No. 51) | Orthorhombic | 4.8823 | 8.4806 | 24.7697 | 3.7 |
| LiOH | SrAl2O4 | P21 (No. 4) | Monoclinic | 5.1621 | 8.8156 | 8.4535 | 60.08 |
| LiOH | Sr3Al2O6 | Pa-3 (No. 205) | Cubic | 15.9999 | 15.9999 | 15.9999 | 36.22 |
| Na2CO3 | Sr4Al14O25 | Pmma (No. 51) | Orthorhombic | 4.8823 | 8.4806 | 24.7697 | 0.0 |
| Na2CO3 | SrAl2O4 | P21 (No. 4) | Monoclinic | 5.1621 | 8.8156 | 8.4535 | 9.91 |
| Na2CO3 | Sr3Al2O6 | Pa-3 (No. 205) | Cubic | 15.9999 | 15.9999 | 15.9999 | 90.09 |
| NaOH | Sr4Al14O25 | Pmma (No. 51) | Orthorhombic | 4.8823 | 8.4806 | 24.7697 | 17.32 |
| NaOH | SrAl2O4 | P21 (No. 4) | Monoclinic | 5.1621 | 8.8156 | 8.4535 | 18.4 |
| NaOH | Sr3Al2O6 | Pa-3 (No. 205) | Cubic | 15.9999 | 15.9999 | 15.9999 | 64.27 |
References
- Liepina, V.; Millers, D.; Smits, K. Tunneling luminescence in long lasting afterglow of SrAl2O4:Eu,Dy. J. Lumin. 2017, 185, 151–154. [Google Scholar] [CrossRef]
- Vitola, V.; Millers, D.; Bite, I.; Smits, K.; Spustaka, A. Recent progress in understanding the persistent luminescence in SrAl2O4:Eu,Dy. Mater. Sci. Technol. 2019, 35, 1661–1677. [Google Scholar] [CrossRef]
- Prasanth, D.; Sunitha, D.V.; Kumar, P.R.; Darshan, G.P. Design strategies, luminescence mechanisms, and solid-state lighting applications of lanthanide-doped phosphorescent materials. ChemPhysMater 2025, 4, 108–123. [Google Scholar] [CrossRef]
- Sharma, V.; Nigam, S.; Sudarsan, V.; Vatsa, R.K. High temperature stabilization of Y2Sn2O7: Eu luminescent nanoparticles—A facile synthesis. J. Lumin. 2016, 179, 248–253. [Google Scholar] [CrossRef]
- Laganovska, K.; Vitola, V.; Einbergs, E.; Bite, I.; Zolotarjovs, A.; Leimane, M.; Tunens, G.; Smits, K. Impact of Europium and Niobium Doping on Hafnium Oxide (HfO2): Comparative Analysis of Sol–Gel and Combustion Synthesis Methods. Ceramics 2024, 7, 15-28. [Google Scholar] [CrossRef]
- Gao, P.; Wang, J.; Wu, J.; Xu, Q.; Yang, L.; Liu, Q.; Qi, Y.; Li, Z. Preparation of SrAl2O4: Eu2+, Dy3+ Powder by Combustion Method and Application in Anticounterfeiting. Coatings 2023, 13, 808. [Google Scholar] [CrossRef]
- Xue, Z.; Deng, S.; Liu, Y.; Lei, B.; Xiao, Y.; Zheng, M. Synthesis and luminescence properties of SrAl2O4:Eu2+,Dy3+ hollow microspheres via a solvothermal co-precipitation method. J. Rare Earths 2013, 31, 241–246. [Google Scholar] [CrossRef]
- Doke, G.; Antuzevics, A.; Krieke, G.; Kalnina, A.; Sarakovskis, A. Novel broadband near-infrared emitting long afterglow phosphor MgGeO3: Cr3+. J. Alloys Compd. 2022, 918, 165768. [Google Scholar] [CrossRef]
- Doke, G.; Krieke, G.; Antuzevics, A.; Sarakovskis, A.; Berzina, B. Optical properties of red-emitting long afterglow phosphor Mg2Si1−xGexO4: Mn2+/Mn4+. Opt. Mater. 2023, 137, 113500. [Google Scholar] [CrossRef]
- Mushtaq, U.; Ayoub, I.; Kumar, V.; Sharma, V.; Swart, H.C.; Chamanehpour, E.; Rubahn, H.-G.; Mishra, Y.K. Persistent luminescent nanophosphors for applications in cancer theranostics, biomedical, imaging and security. Mater. Today Bio 2023, 23, 100860. [Google Scholar] [CrossRef]
- Bite, I.; Laganovska, K.; Vanags, E.; Vitola, V. Synthesis and characterization of translucent hafnia ceramics. Materialia 2023, 32, 101887. [Google Scholar] [CrossRef]
- Eftimov, T.; Kostova, I.; Arapova, A.; Patronov, G. Rise and decay time responses of Sr aluminate phosphorescent materials. J. Lumin. 2021, 235, 117985. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Q.; Wang, J.; Qi, Y.; Li, Z.; Li, J.; Zhang, Z.; Wang, X.; Li, C.; Wang, R. Study on the Luminescence Performance and Anti-Counterfeiting Application of Eu2+, Nd3+ Co-Doped SrAl2O4 Phosphor. Nanomaterials 2024, 14, 1265. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Z.; Huo, X.; Meng, X.; Wang, Y.; Suo, H.; Li, P. Anti-Counterfeiting Application of Persistent Luminescence Materials and Its Research Progress. Laser Photon. Rev. 2024, 18, 2300751. [Google Scholar] [CrossRef]
- Hayashi, F.; Ogawa, K.; Moriya, Y.; Sudare, T.; Teshima, K. Growth of β-Li2 TiO3 Nanocrystals from LiCl and LiOH Fluxes. Cryst. Growth Des. 2019, 19, 1377–1383. [Google Scholar] [CrossRef]
- Kimijima, T.; Zettsu, N.; Onodera, H.; Yubuta, K.; Oishi, S.; Teshima, K. Low-temperature growth of idiomorphic cubic-phase Li7La3 Zr2O12 crystals using LiOH flux. CrystEngComm 2015, 17, 3487–3492. [Google Scholar] [CrossRef]
- Jones, R.; Burgess, S.; Pinard, P. Enhanced Compositional Mapping on the SEM Through Combined EDS-WDS Mapping in AztecWave. Microsc. Microanal. 2022, 28, 546–547. [Google Scholar] [CrossRef]
- Vitola, V.; Bite, I.; Millers, D.; Zolotarjovs, A.; Laganovska, K.; Smits, K.; Spustaka, A. The boron effect on low temperature luminescence of SrAl2O4:Eu,Dy. Ceram. Int. 2020, 46, 26377–26381. [Google Scholar] [CrossRef]
- Kim, D.; Kim, S.-C.; Bae, J.-S.; Kim, S.; Kim, S.-J.; Park, J.-C. Eu 2+-Activated Alkaline-Earth Halophosphates, M5(PO4)3X:Eu2+ (M = Ca, Sr, Ba; X = F, Cl, Br) for NUV-LEDs: Site-Selective Crystal Field Effect. Inorg. Chem. 2016, 55, 8359–8370. [Google Scholar] [CrossRef]
- Laganovska, K.; Bite, I.; Zolotarjovs, A.; Smits, K. Niobium enhanced europium ion luminescence in hafnia nanocrystals. J. Lumin. 2018, 203, 358–363. [Google Scholar] [CrossRef]
- Vercaemst, R.; Poelman, D.; Fiermans, L.; Van Meirhaeghe, R.L.; Laflère, W.H.; Cardon, F. A detailed XPS study of the rare earth compounds EuS and EuF3. J. Electron Spectrosc. Relat. Phenom. 1995, 74, 45–56. [Google Scholar] [CrossRef]
- Aitasalo, T.; Hölsä, J.; Jungner, H.; Krupa, J.-C.; Lastusaari, M.; Legendziewicz, J.; Niittykoski, J. Effect of temperature on the luminescence processes of SrAl2O4:Eu2+. Radiat. Meas. 2004, 38, 727–730. [Google Scholar] [CrossRef]
- Bite, I.; Krieke, G.; Zolotarjovs, A.; Laganovska, K.; Liepina, V.; Smits, K.; Auzins, K.; Grigorjeva, L.; Millers, D.; Skuja, L. Novel method of phosphorescent strontium aluminate coating preparation on aluminium. Mater. Des. 2018, 160, 794–802. [Google Scholar] [CrossRef]
- Wang, X.; Seto, T.; Zhao, Z.; Li, Y.; Wu, Q.; Lia, H.; Wang, Y. Preparation of Sr1−xCaxLiAl3N4:Eu2+ Solid Solutions and Their Photoluminescence Properties. J. Am. Ceram. Soc. 2016, 99, 3273–3279. [Google Scholar]
- Wolde, S.; Chauhan, D.; Somvanshi, D.; Unil Perera, A.G.; Li, L.H.; Chen, L.; Khanna, S.P.; Linfield, E.H. Accuracy of activation energy from Arrhenius plots and temperature-dependent internal photoemission spectroscopy. Infrared Phys. Technol. 2019, 102, 103026. [Google Scholar] [CrossRef]
- Yang, L.; Liu, Q.; Zheng, H.; Zhou, S.; Zhang, W. The decay model of Eu2+ and Eu2+, Dy3+ substituted SrAl2O4 prepared by high temperature solid phase method. J. Phys. Chem. Solids 2019, 124, 151–156. [Google Scholar] [CrossRef]
- Tang, X.; Ehler, E.; Brost, E.; Mathew, D. Evaluation of SrAl2O4:Eu, Dy phosphor for potential applications in thermoluminescent dosimetry. J. Appl. Clin. Med. 2021, 22, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Yuan, Z.; Mao, D. Eu2+ activated long persistent strontium aluminate nano scaled phosphor prepared by precipitation method. J. Alloys Compd. 2006, 415, 220–222. [Google Scholar] [CrossRef]
- Romero, M.; Castaing, V.; Lozano, G.; Miguez, H. Trap Depth Distribution Determines Afterglow Kinetics: A Local Model Applied to ZnGa2O4:Cr3+. J. Phys. Chem. Lett. 2024, 15, 9129–9135. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, Y.; Hu, P.; Zhang, Y.; Zi, Y.; Zhang, Y.; Yang, Z.; Jiang, J. Hydrolytic-Resistance Long-Persistent Luminescence SrAl2O4:Eu2+,Dy3+ Ceramics for Optical Information Storage. Adv. Funct. Mater. 2025, 35, 2414872. [Google Scholar] [CrossRef]
- Eftimov, T.A.; Kostova, I.; Peltekov, A.; Hristova, K.; Brabant, D.; Fouzar, S.; Nikolov, K. A Study of Strontium Aluminates for All Optical Contactless Sensing Applications Using Smartphone Interrogation. Opt. Laser Technol. 2024, 176, 110965. [Google Scholar] [CrossRef]
- Fouzar, S.; Kostova, I.; Eftimov, T.; Benmounah, A.; Ouchabane, M.; Lakhssassi, A. Excitation and spectral dependence of the rise and decay time responses of Eu2+ and Dy3+ doped strontium aluminates. J. Mater. Sci Mater. Electron. 2022, 33, 20962–20980. [Google Scholar] [CrossRef]
- Wang, L.; Shang, Z.; Shi, M.; Cao, P.; Yang, B.; Zou, J. Preparing and testing the reliability of long after glow SrAl2O4:Eu2+, Dy3+ phosphor flexible films for temperature sensing. RSC Adv. 2020, 10, 11418. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, W.; Shang, Z.; Cao, P.; Shi, M.; Yang, B.; Zou, J. Optical and reliability properties studies of Sr4Al14O25:Eu2+ film for high-CRI white LEDs. J. Mater. Sci. Mater. Electron. 2020, 31, 13561–13569. [Google Scholar] [CrossRef]
- Fouzar, S.; Eftimov, T.; Kostova, I.; Benmounah, A.; Lakhssassi, A. Effects of temperature on the time responses of strontium aluminates. Opt. Mater. 2021, 122, 111619. [Google Scholar] [CrossRef]
- Eftimov, T.; Kostova, I.; Fouzar, S.; Brabant, D.; Nikolov, K.; Vladev, V. Smartphone-Readable Optical-Fiber Quasi-Distributed Phosphorescent Temperature Sensor. Photonics 2024, 11, 694. [Google Scholar] [CrossRef]
- Ramalho, J.F.C.B.; António, L.C.F.; Correia, S.F.H.; Fu, L.S.; Pinho, A.S.; Brites, C.D.S.; Carlos, L.D.; André, P.S.; Ferreira, R.A.S. Luminescent QR codes for smart labelling and sensing. Opt. Laser Technol. 2018, 101, 304–311. [Google Scholar] [CrossRef]
- Ramalho, J.F.C.B.; Correia, S.F.H.; Fu, L.; Dias, L.M.S.; Adão, P.; Mateus, P.; Ferreira, R.A.S.; André, P.S. Super modules-based active QR codes for smart trackability and IoT: A responsive-banknotes case study. NPJ Flex. Electron. 2020, 4, 11. [Google Scholar] [CrossRef]
- Hristova, K.; Kostova, I.P.; Eftimov, T.A.; Patronov, G.; Tsoneva, S. Synthesis and Characterization of Smartphone-Readable Luminescent Lanthanum Borates Doped and Co-Doped with Eu and Dy. Photonics 2025, 12, 171. [Google Scholar] [CrossRef]
- Yakunin, S.; Chaaban, J.; Benin, B.M.; Cherniukh, I.; Bernasconi, C.; Landuyt, A.; Shynkarenko, Y.; Bolat, S.; Hofer, C.; Romanyuk, Y.E.; et al. Radiative lifetime-encoded unicolour security tags using perovskite nanocrystals. Nat. Commun. 2021, 12, 981. [Google Scholar] [CrossRef]
- Zaw, M.M.; Cao, P.; Ong, R.G.K.; Neo, D.C.J.; Lay, C.L. 3D Security Labels: Photostable Multimaterial Multilayered QR Codes via Two-Photon Lithography. ACS Appl. Opt. Mater. 2024, 2, 276–283. [Google Scholar] [CrossRef]

















| Sample | Eu:Dy Weight Ratio | SD | Sr:Al Weight Ratio | SD |
|---|---|---|---|---|
| #1 HTMA | 1.3 | 1.0 | 4.1 | 1.0 |
| #2 LiOH | 1.8 | 1.3 | 1.4 | 0.2 |
| #3 KOH | 0.46 | 0.02 | 1.2 | 0.6 |
| #4 NaOH | 0.7 | 0.3 | 2.0 | 0.8 |
| #5 K2CO3 | 1.7 | 0.6 | 2.7 | 1.4 |
| #6 Na2CO3 | 0.7 | 0.2 | 3.2 | 1.3 |
| #7 (NH4)2CO3 | 0.4 | 0.3 | 1.6 | 0.4 |
| Sample ID | Traping Center Depths (eV) | |
|---|---|---|
| #1—HMTA | 0.05 | 0.17 |
| #2—LiOH | 0.103 | |
| #3—KOH | 0.055 | |
| #4—NaOH | 0.161 | 0.112 |
| #5—K2CO3 | 0.117 | 0.186 |
| #6—Na2CO3 | 0.148 | |
| #7—(NH4)2CO3 | 0.150 | |
| Excitation Wavelength, λ′ Emission Wavelength, λ | #1 HTMA | #2 LiOH | #3 KOH | #4 NaOH | #5 K2CO3 | #6 Na2CO3 | #7 (NH4)2CO3 |
|---|---|---|---|---|---|---|---|
| λ′Max (nm) | 362.5 | 362.5 | 367.5 | 367.5 | 365 | 358 | 367.5 |
| λMax (nm) | 523 | 524 | 492 | 520 | 523 | 523 | 521.5 |
| #1 HTMA | #2 LiOH | #3 KOH | #4 NaOH | #5 K2CO3 | #6 Na2CO3 | #7 (NH4)2CO3 | |
|---|---|---|---|---|---|---|---|
| A | 0.1434 | 0.1748 | 0.1691 | 0.0495 | 0.1903 | 0.2816 | 0.2244 |
| α | −0.334 | −0.268 | −0.094 | −0.218 | −0.185 | −0.192 | −0.173 |
| Luminescence Intensity, L | ≈4800 | ≈8500 | ≈10,700 | ≈1400 | ≈9600 | ≈7800 | ≈8500 |
| ΔuOFF | 41% | 42% | 22% | 11% | 51% | 52% | 40% |
| Phosphorescence, P | ≈1970 | ≈3570 | ≈2350 | ≈155 | ≈4890 | ≈4060 | ≈3400 |
| #1 HTMA | #2 LiOH | #3 KOH | #4 NaOH | #5 K2CO3 | #6 Na2CO3 | #7 (NH4)2CO3 | |
|---|---|---|---|---|---|---|---|
| ΔuT | 54% | 49% | 49% | 43% | 29% | 35% | 25% |
| ΔuT/ΔT | 1.08%/K | 0.98%/K | 0.98%/K | 0.86%/K | 0.58%/K | 0.74%/K | 0.5%/K |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitola, V.; Dile, M.; Krizmane, K.; Einbergs, E.; Eftimov, T.; Nikolov, K.; Fouzar, S. Tailoring the Luminescence Properties of Strontium Aluminate Phosphors for Unique Smartphone Detectable Optical Tags. Crystals 2025, 15, 474. https://doi.org/10.3390/cryst15050474
Vitola V, Dile M, Krizmane K, Einbergs E, Eftimov T, Nikolov K, Fouzar S. Tailoring the Luminescence Properties of Strontium Aluminate Phosphors for Unique Smartphone Detectable Optical Tags. Crystals. 2025; 15(5):474. https://doi.org/10.3390/cryst15050474
Chicago/Turabian StyleVitola, Virginija, Milena Dile, Katrina Krizmane, Ernests Einbergs, Tinko Eftimov, Kristian Nikolov, and Samia Fouzar. 2025. "Tailoring the Luminescence Properties of Strontium Aluminate Phosphors for Unique Smartphone Detectable Optical Tags" Crystals 15, no. 5: 474. https://doi.org/10.3390/cryst15050474
APA StyleVitola, V., Dile, M., Krizmane, K., Einbergs, E., Eftimov, T., Nikolov, K., & Fouzar, S. (2025). Tailoring the Luminescence Properties of Strontium Aluminate Phosphors for Unique Smartphone Detectable Optical Tags. Crystals, 15(5), 474. https://doi.org/10.3390/cryst15050474