
Seven New Irbesartan Salts: Significantly Improved Dissolution, Excellent Hygrothermal Stability, and Characteristic Supramolecular Synthons

 , ,
, ,
Abstract

1. Introduction
2. Experimental Section
2.1. Materials
2.2. Screening Methods
2.3. Preparation of IRB Salts
2.3.1. Preparation of Single Crystals
2.3.2. Preparation of Powders
2.4. Accelerated Stability Study
2.5. Dissolution Study
2.6. In Situ Raman Spectrometer Monitoring of the Phase Transformation Process
2.7. Characterization
2.7.1. Powder X-Ray Diffraction (PXRD)
2.7.2. Single-Crystal X-Ray Diffraction (SCXRD)
2.7.3. Differential Scanning Calorimetry (DSC) and Thermogravimetric Analysis (TGA)
2.7.4. Nuclear Magnetic Resonance (NMR) Spectroscopy
2.7.5. Crystal Morphology Analysis
2.7.6. Dynamic Vapor Sorption (DVS) Analysis
2.8. Computational Details
2.8.1. Full Interaction Map (FIM) Analysis
2.8.2. Supramolecular Synthon Analysis
2.8.3. Conformation Similarity Analysis
2.8.4. Hirshfeld Surface (HS) Analysis
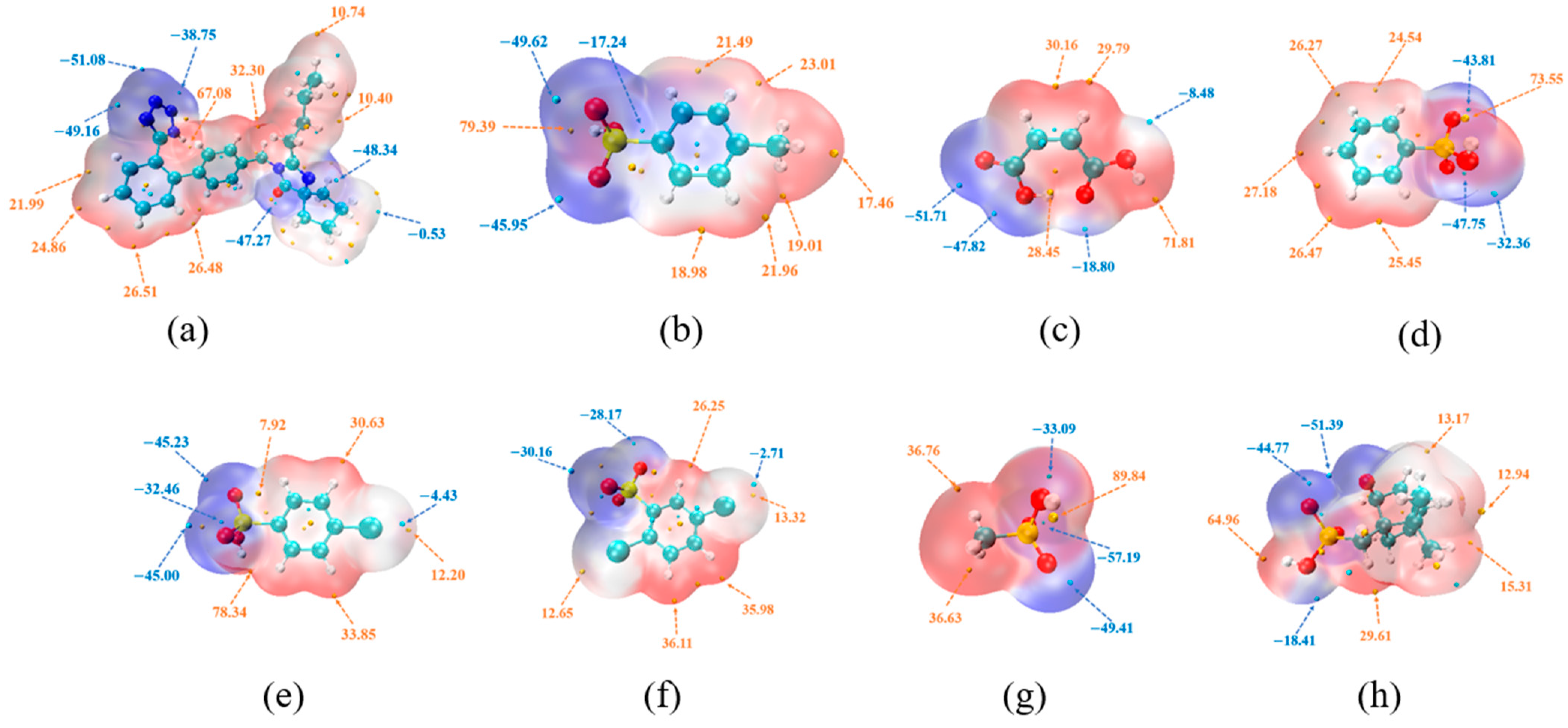
2.8.5. Molecular Electrostatic Potential Surface (MEPS) Analysis
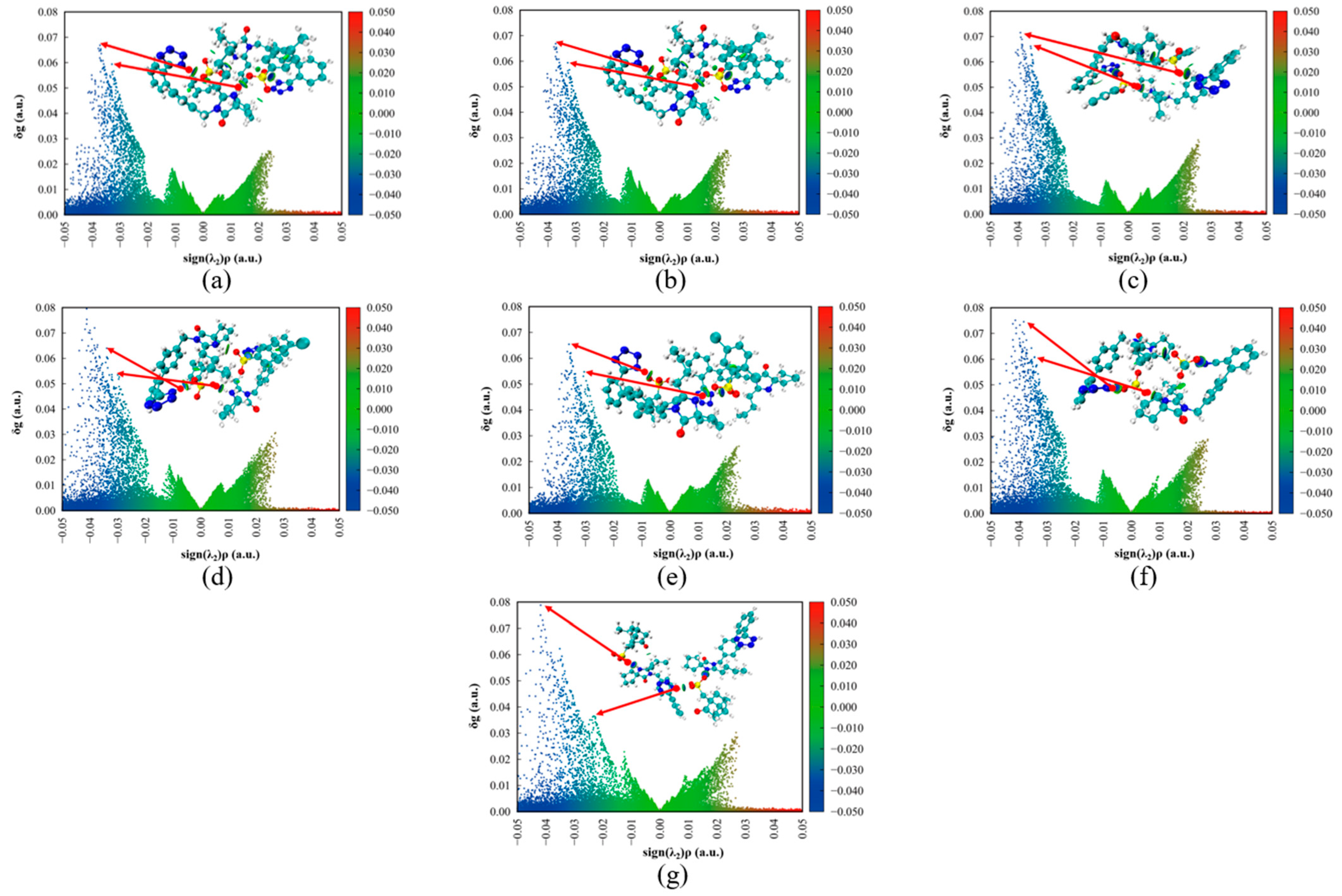
2.8.6. Atoms in Molecules (AIM) and Independent Gradient Model Based on Hirshfeld Partition (IGMH) Analysis
3. Results and Discussion
3.1. FIM Analysis
3.2. Screening Results
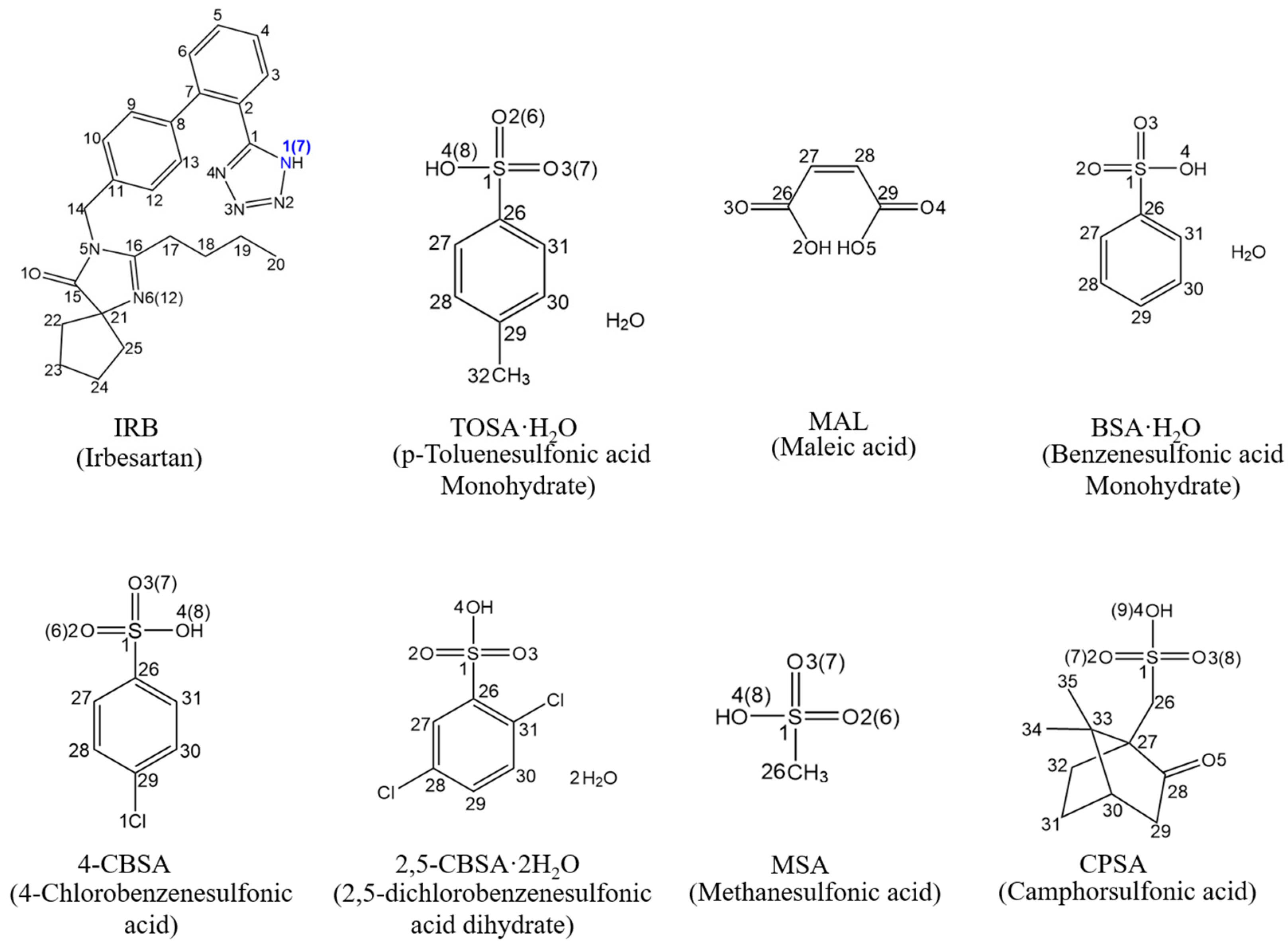
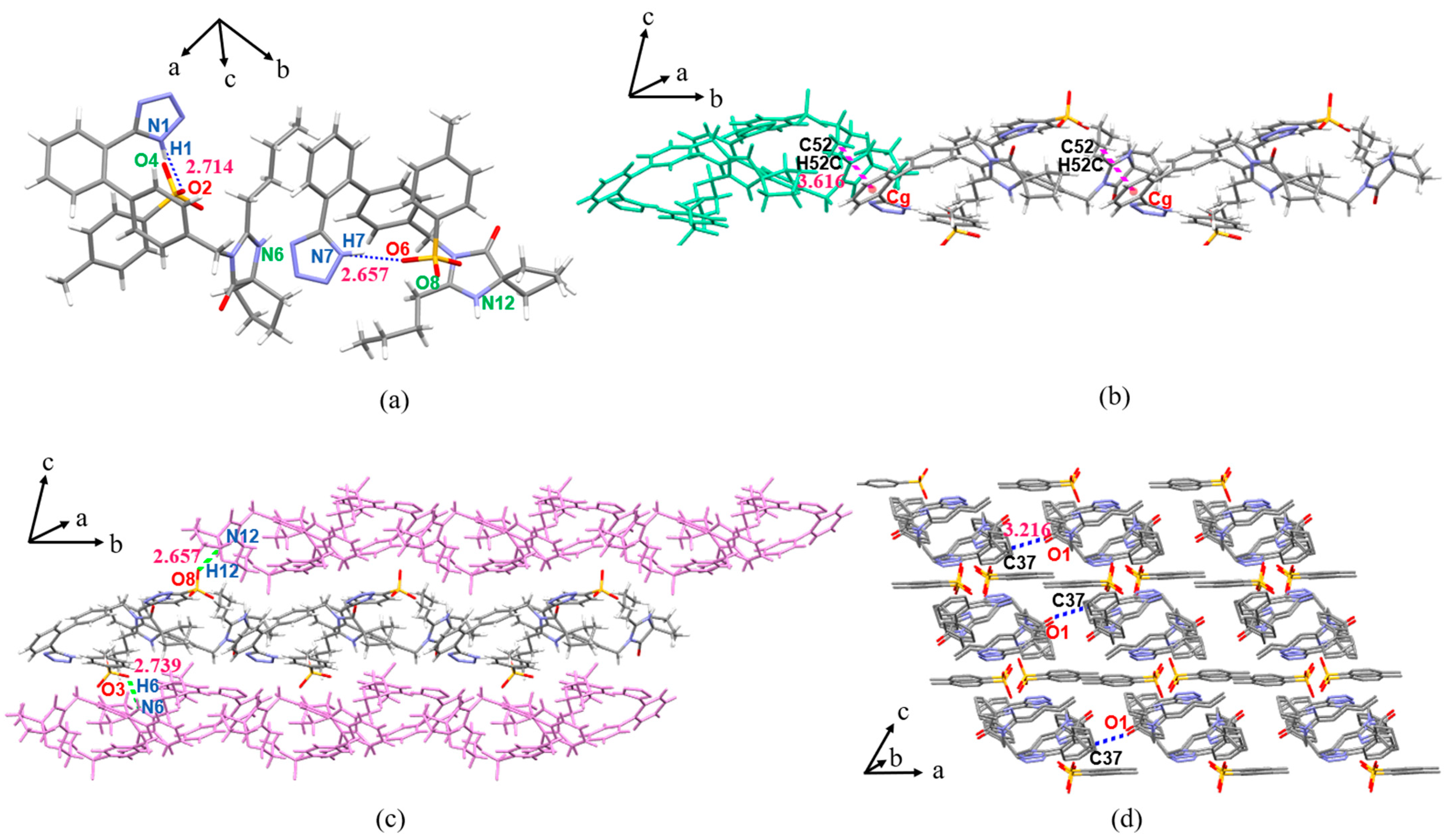
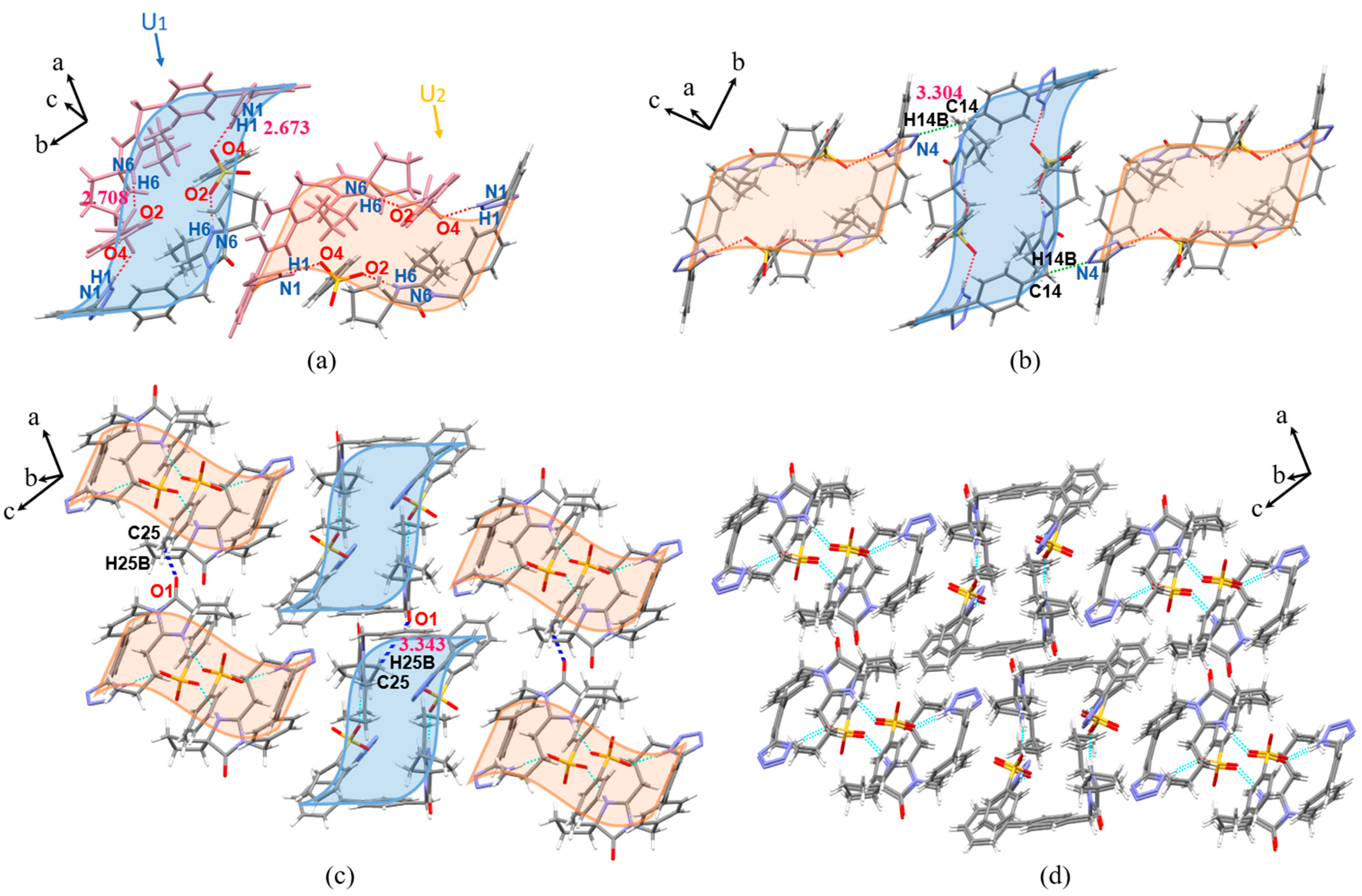
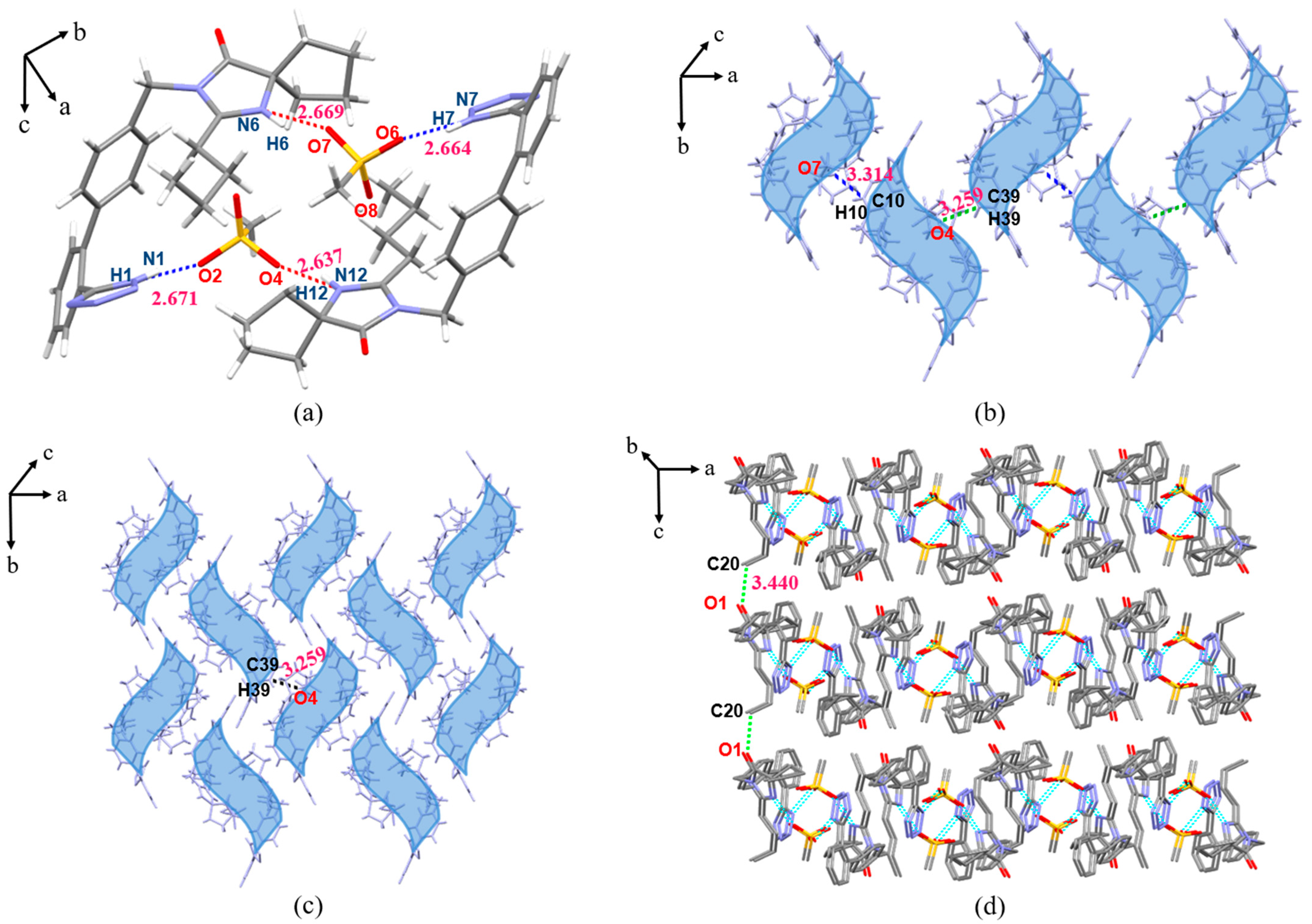
3.3. Crystal Structure Analysis
3.3.1. SCXRD Analysis
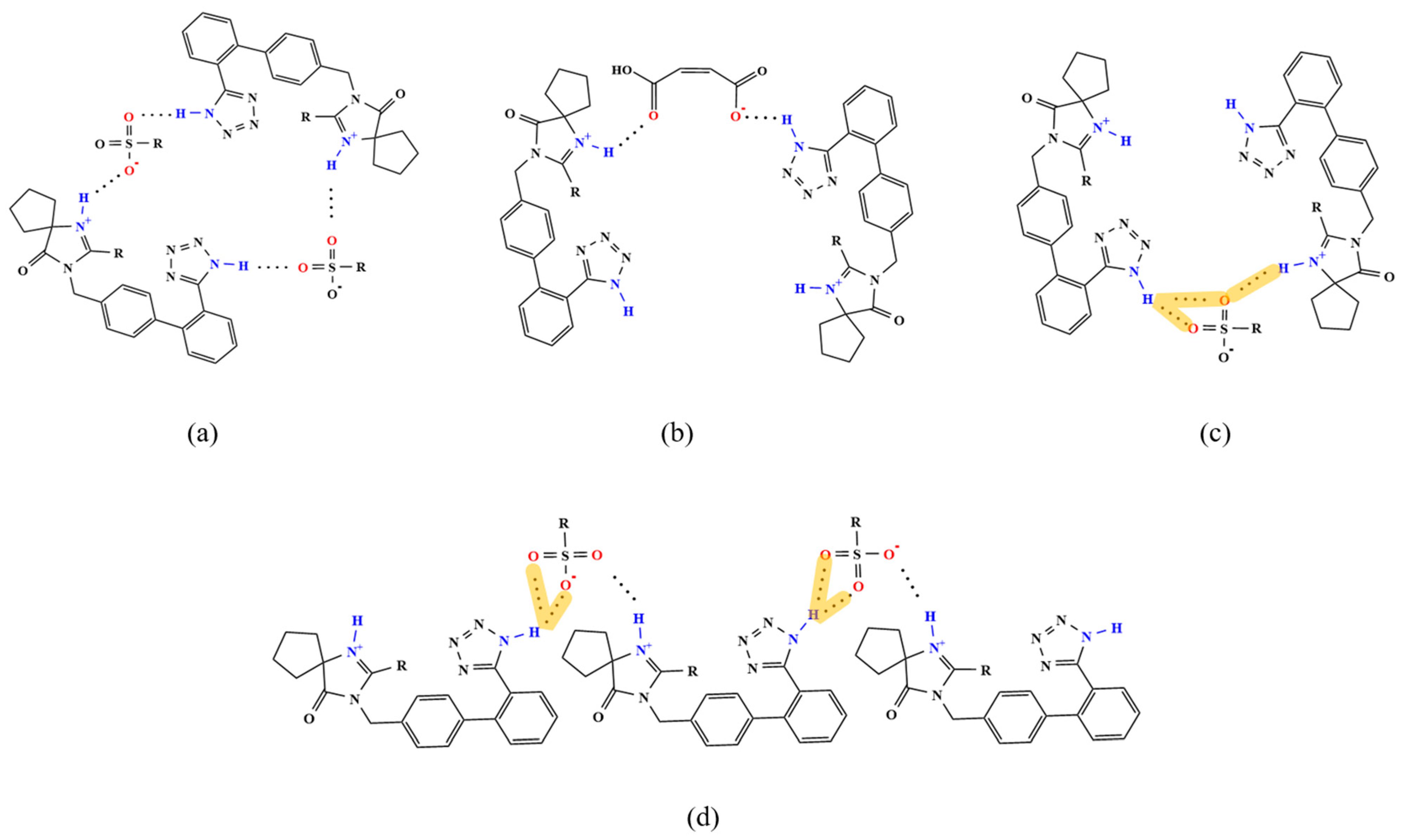
3.3.2. Analysis of Supramolecular Synthons
3.3.3. NMR Analysis
3.3.4. ΔDC–O/S–O Analysis
3.3.5. Analysis of Conformation Similarity
3.4. Crystal Characterization and Property Analysis
3.4.1. Thermal Analysis
3.4.2. Crystal Morphology Analysis
3.4.3. Hygroscopicity and Moisture–Thermal Stability Analysis
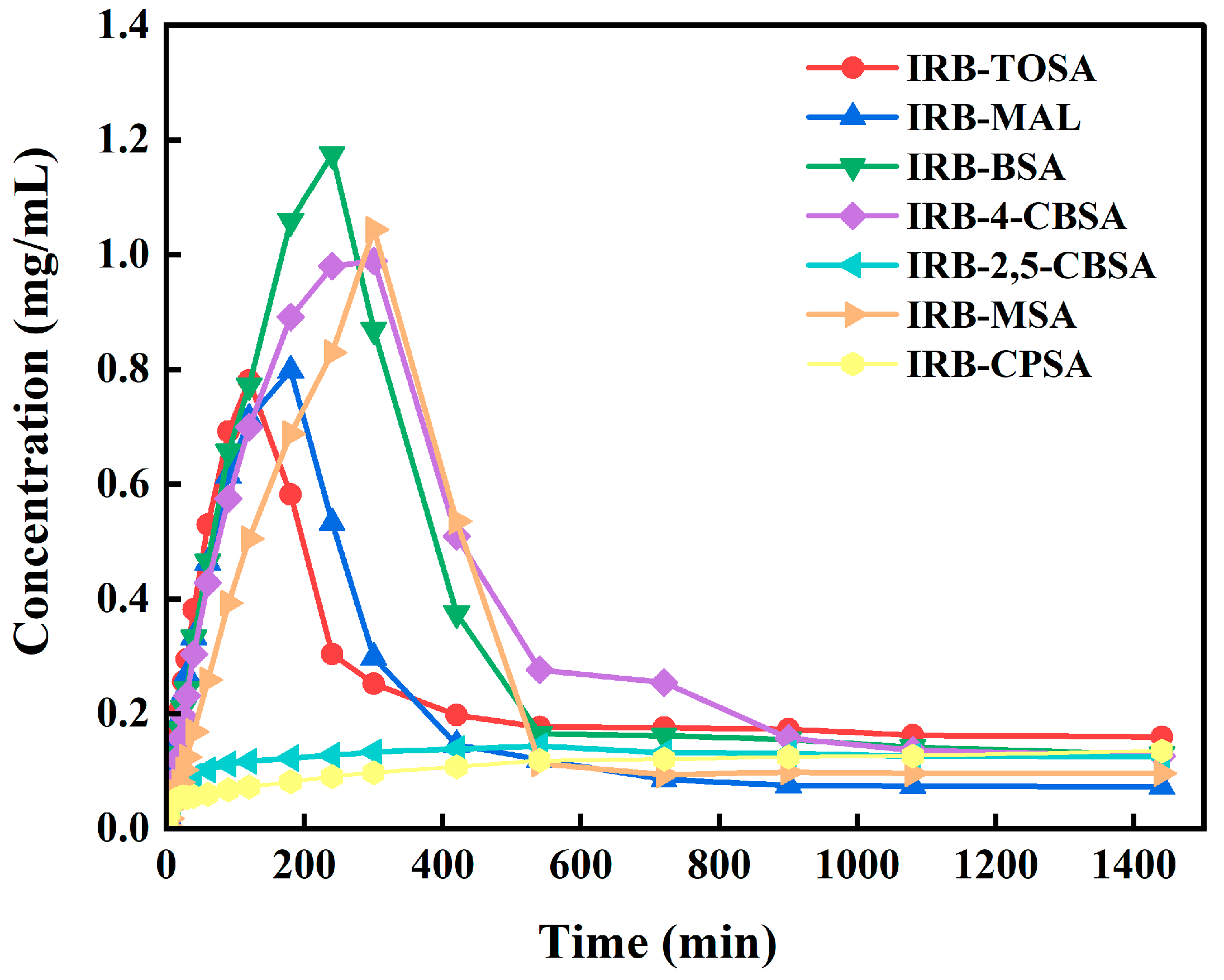
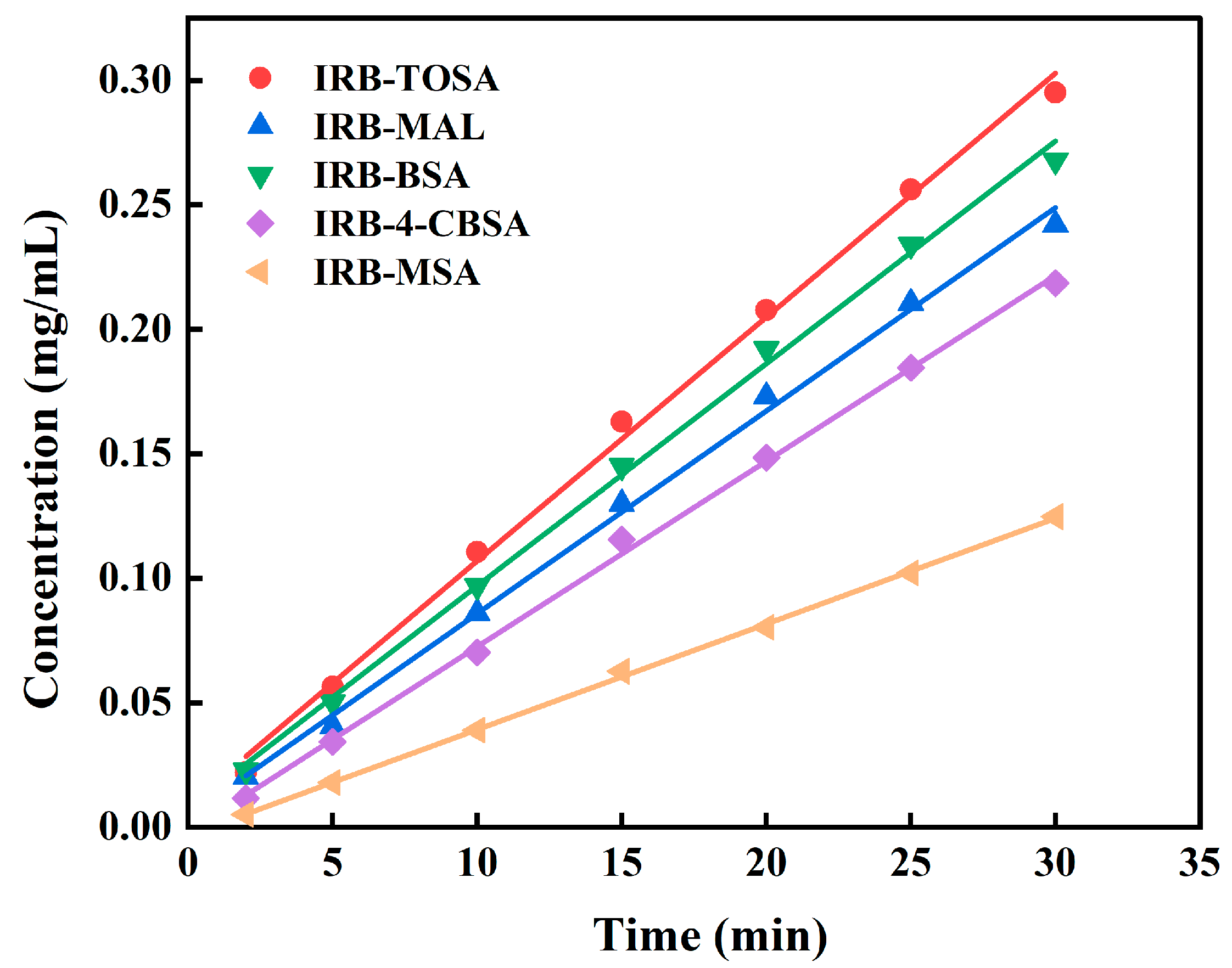
3.4.4. Dissolution and IDR Analysis
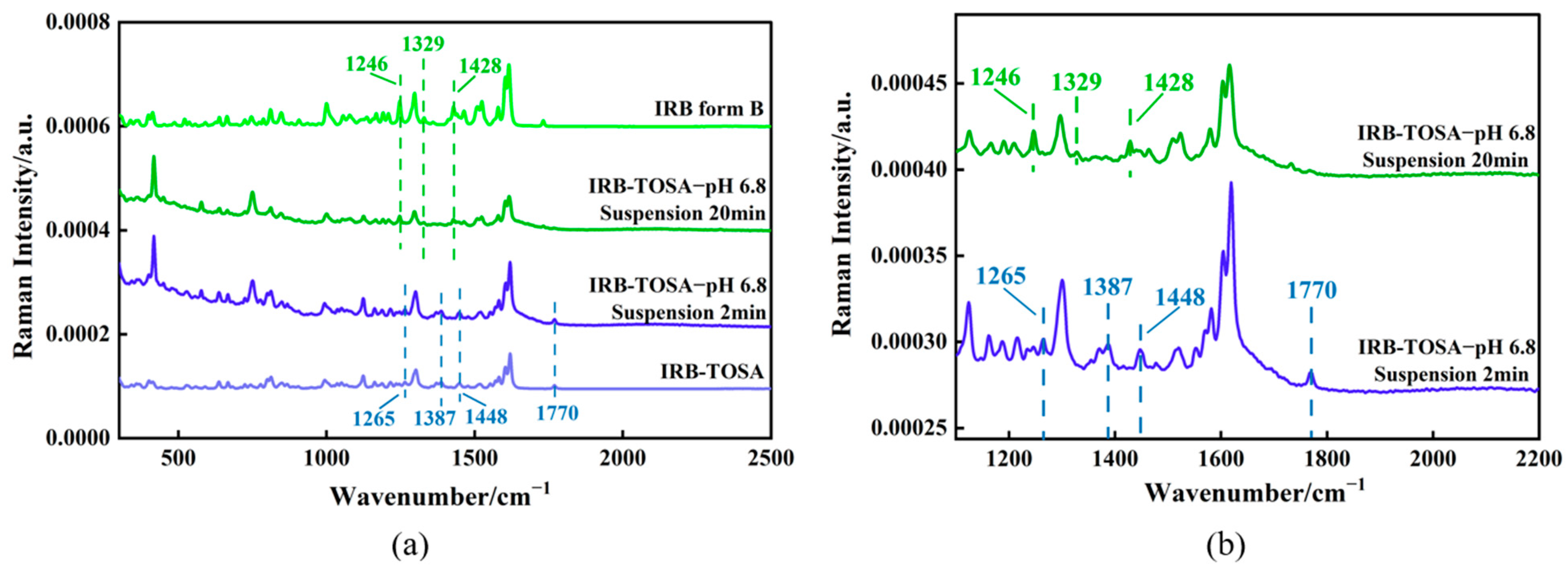
3.4.5. Analysis of the Phase Transformation Process
3.5. Computational Analysis
3.5.1. HS Analysis
3.5.2. MEPS Analysis
3.5.3. AIM and IGMH Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peng, B.; Wang, J. Advances in drug-drug complexes based on the crystal engineering design. Yao Xue Xue Bao 2020, 55, 2358–2367. [Google Scholar]
- Shi, Q.; Chen, H.B.; Wang, Y.A.; Xu, J.; Liu, Z.Y.; Zhang, C. Recent advances in drug polymorphs: Aspects of pharmaceutical properties and selective crystallization. Int. J. Pharm. 2022, 45, 121320. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, H.; Chen, Z.; Meng, J.; Li, Y.; Qi, L.; Zhang, S.; Chen, X.; Lei, M. A drug-drug cocrystal and a co-amorphous form, prepared from honokiol and ligustrazine, inspired by Chinese patent medicine. Acta Crystallogr. Sect. B 2023, 79, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Li, J.; Mu, C.; Liu, D.; Yang, Y.; Li, Y.; Liu, T.; Wang, X.; Liu, F. Hepatoprotective Pyrazinamide–Baicalein Cocrystal with a Rare Ratio of 7:3. Cryst. Growth Des. 2023, 23, 885–891. [Google Scholar] [CrossRef]
- Wathoni, N.; Sari, W.A.; Elamin, K.M.; Mohammed, A.F.A.; Suharyani, I. A Review of Coformer Utilization in Multicomponent Crystal Formation. Molecules 2022, 27, 8693. [Google Scholar] [CrossRef]
- Bolla, G.; Sarma, B.; Nangia, A.K. Crystal Engineering of Pharmaceutical Cocrystals in the Discovery and Development of Improved Drugs. Chem. Rev. 2022, 122, 11514–11603. [Google Scholar] [CrossRef]
- Jiang, L.; Hu, X.; Cai, L. A Drug–Drug Multicomponent Crystal of Metformin and Dobesilate: Crystal Structure Analysis and Hygroscopicity Property. Molecules 2022, 27, 3472. [Google Scholar] [CrossRef]
- Li, M.; Sun, J.; Kuang, W.; Zhou, L.; Han, D.; Gong, J. Drug–Drug Multicomponent Crystals of Epalrestat: A Novel Form of the Drug Combination and Improved Solubility and Photostability of Epalrestat. Cryst. Growth Des. 2022, 22, 5027–5035. [Google Scholar] [CrossRef]
- Zhao, C.; Su, X.; Fang, L.; Shang, Z.; Li, Z.; Gong, J.; Wu, S. Multivariate Analysis of a Highly Effective Drug Combination Tablet Containing the Antiepileptic Drug Gabapentin to Enhance Pharmaceutical Properties with a Multicomponent Crystal Strategy. Cryst. Growth Des. 2022, 22, 7234–7247. [Google Scholar] [CrossRef]
- Fang, Z.; Zhang, B.; Xing, W.; Yu, H.; Xing, C.; Gong, N.; Lu, Y.; Du, G. An Evolving Role of Aqueous Piperazine to Improve the Solubility of Non-Steroidal Anti-Inflammatory Drugs. J. Pharm. Sci. 2022, 111, 2839–2847. [Google Scholar] [CrossRef]
- Hamideh, A.; Rahman, Z.; Dharani, S.; Khuroo, T.; Mohamed, E.M.; Nutan, M.T.H.; Reddy, I.K.; Khan, M.A. Preparation and characterization of dicarboxylic acids salt of aripiprazole with enhanced physicochemical properties. Pharm. Dev. Technol. 2021, 26, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Yuan, Z.; Zhang, M.; Ming, Y.; Huang, X.; Wang, C.; Sun, C.C. Direct compression tablet formulation of trimetazidine through systematic screening of oxalate salts. Int. J. Pharm. 2025, 48, 125255. [Google Scholar] [CrossRef]
- Niu, Y.; Wang, L.; Yu, Y.; Li, Y.; Wu, Z.; Yan, C. Molecular adduct of amantadine ferulate presents a pathway for slowing in vitro/vivo releases and raising synergistic antiviral effects via dual optimization salification strategy. Crystengcomm 2021, 23, 4389–4401. [Google Scholar] [CrossRef]
- Ainurofiq, A.; Putro, D.S.; Ramadhani, D.A.; Putra, G.; Do Espirito Santo, L. A review on solubility enhancement methods for poorly water-soluble drugs. J. Rep. Pharm. Sci. 2021, 10, 137–147. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal Engineering: The Design of Organic Solids; Elsevier: Amsterdam, The Netherlands, 1989; Volume 54. [Google Scholar]
- Wu, C.; Xiao, Y.; Jing, Y.; Yin, Q.; Bao, Y. New Insights into the Solubilization of Multicomponent Crystals: A Case Study of Pipemidic Acid. Cryst. Growth Des. 2023, 23, 3367–3383. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, M.; Zhang, L.; Yu, F.; Hou, X.; Pan, Z.; Xie, C.; Gong, J.; Zhang, C.; Chen, W. Multicomponent Crystal Screening and Performance Testing of Sunitinib: A Combined Virtual and Experimental Study. Cryst. Growth Des. 2024, 24, 8112–8134. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, D.; Zhang, M.; Yu, F.; Bao, Y.; Xie, C.; Hou, B.; Jing, D.; Zhang, C.; Chen, W. Theoretical and experimental study of pharmaceutical salts: A case of trimethoprim. Crystengcomm 2024, 26, 3808–3822. [Google Scholar] [CrossRef]
- Zhang, M.; Hou, X.; Yu, F.; Zhang, L.; Hou, B.; Zhou, L.; Xie, C.; Wu, S.; Chen, W. Synthesis, Characterization, and Analysis of Probenecid and Pyridine Compound Salts. Crystals 2024, 14, 670. [Google Scholar] [CrossRef]
- Cho, C.; Kang, P.; Jang, C.; Lee, S.; Lee, Y.J.; Choi, C. Physiologically based pharmacokinetic (PBPK) modeling to predict the pharmacokinetics of irbesartan in different CYP2C9 genotypes. Arch. Pharm. Res. 2023, 46, 939–953. [Google Scholar] [CrossRef]
- Araya-Sibaja, A.M.; de Campos, C.E.M.; Fandaruff, C.; Vega-Baudrit, J.R.; Guillén-Girón, T.; Navarro-Hoyos, M.; Cuffini, S.L. Irbesartan desmotropes: Solid-state characterization, thermodynamic study and dissolution properties. J. Pharm. Anal. 2019, 9, 339–346. [Google Scholar] [CrossRef]
- Ochsenbein, P.; Bonin, M.; Fadaei-Tirani, F.; Lemée, M.-H.; Kieffer, J.; Görl, D.; El-Hajji, M.; SchenkJoß, K. A score and nine years of irbesartan. Crystengcomm 2024, 26, 4566–4578. [Google Scholar] [CrossRef]
- Bocskei, Z.; Simon, K.; Rao, R.; Caron, A.; Rodger, C.A.; Bauer, M. Irbesartan crystal form B. Acta Crystallogr. Sect. C-Cryst. Struct. Commun. 1998, 54, 808–810. [Google Scholar] [CrossRef]
- Gao, Y.; Tian, J. Solubility of Irbesartan Form B in an Aqueous Ethanol Mixture. J. Chem. Eng. Data 2008, 53, 535–537. [Google Scholar] [CrossRef]
- Jansook, P.; Hnin, H.M.; Praphanwittaya, P.; Loftsson, T.; Stefansson, E. Effect of salt formation on γ-cyclodextrin solubilization of irbesartan and candesartan and the chemical stability of their ternary complexes. J. Drug Deliv. Sci. Technol. 2022, 67, 102980. [Google Scholar] [CrossRef]
- Wang, X.J.; Gao, D.; Li, D.X.; Xie, Q.H.; Deng, Z.W.; Zhang, H.L. Collecting the Molecular and Ionization States of Irbesartan in the Solid State. Cryst. Growth Des. 2020, 20, 5664–5669. [Google Scholar] [CrossRef]
- Delaney, S.P.; Pan, D.H.; Galella, M.; Yin, S.X.; Korter, T.M. Understanding the Origins of Conformational Disorder in the Crystalline Polymorphs of Irbesartan. Cryst. Growth Des. 2012, 12, 5017–5024. [Google Scholar] [CrossRef]
- Bartolucci, G.; Bruni, B.; Di Vaira, M.; Giannellini, V. 2-Butyl-4-oxo-3-{[2’-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl}-3-aza-1-azoniaspiro[4.4]non-1-ene chloride 1.69-hydrate (irbesartan hydrochloride 1.69-hydrate). Acta Crystallogr. Sect. E-Crystallogr. Commun. 2007, 63, O1529–O1531. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, L.; Bao, Y.; Wang, J. 2-n-Butyl-3-[2’-(1H-tetrazol-5-yl) biphenyl-4-ylmethyl]-1-azonia-3-azaspiro[4.4]non-1-en-4-one bromide sesquihydrate. Acta Crystallogr. Sect. Sect. E Struct. Rep. Online 2007, 63, o4933. [Google Scholar] [CrossRef]
- Araya-Sibaja, A.M.; Fandaru, C.; Guevara-Camargo, A.M.; Vargas-Huertas, F.; Zamora, W.J.; Vega-Baudrit, J.R.; Guillen-Girón, T.; Navarro-Hoyos, M.; Paoli, P.; Rossi, P.; et al. Crystal Forms of the Antihypertensive Drug Irbesartan: A Crystallographic, Spectroscopic, and Hirshfeld Surface Analysis Investigation. Acs Omega 2022, 7, 14897–14909. [Google Scholar] [CrossRef]
- Nair, A.S.; Harini, R.; Melissa, P.; Shah, K.; Nayak, U.Y.; Mahalaxmi, R.; Vamshi, K.T. Development and characterization of Irbesartan Co-Crystals. Res. J. Pharm. Technol. 2018, 11, 3932–3936. [Google Scholar] [CrossRef]
- Garg, A.; Yadav, S. Rapid Release Pharmaceutical Composition of Irbesartan Prepared by Co-Crystallization Method. IN202211051199, 2022–09–23.
- Maddileti, D.; Swapna, B.; Nangia, A. High Solubility Crystalline Pharmaceutical Forms of Blonanserin. Cryst. Growth Des. 2014, 14, 2557–2570. [Google Scholar] [CrossRef]
- Committee, N.P. Pharmacopoeia of the People’s Republic of China; China Medical Science and Technology Press: Beijing, China, 2020. [Google Scholar]
- Dokoumetzidis, A.; Papadopoulou, V.; Macheras, P. Analysis of Dissolution Data Using Modified Versions of Noyes–Whitney Equation and the Weibull Function. Pharm. Res. 2006, 23, 256–261. [Google Scholar] [PubMed]
- Kögler, M.; Heilala, B. Time-gated Raman spectroscopy—A review. Meas. Sci. Technol. 2021, 32, 012002. [Google Scholar] [CrossRef]
- Araya-Sibaja, A.M.; Urgellés, M.; Vásquez-Castro, F.; Vargas-Huertas, F.; Vega-Baudrit, J.R.; Guillén-Girón, T.; Navarro-Hoyos, M.; Cuffini, S.L. The effect of solution environment and the electrostatic factor on the crystallisation of desmotropes of irbesartan. RSC Adv. 2019, 9, 5244–5250. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Corpinot, M.K.; Bučar, D.-K. A Practical Guide to the Design of Molecular Crystals. Cryst. Growth Des. 2019, 19, 1426–1453. [Google Scholar] [CrossRef]
- Nauha, E.; Bernstein, J. “Predicting” Polymorphs of Pharmaceuticals Using Hydrogen Bond Propensities: Probenecid and Its Two Single-Crystal-to-Single-Crystal Phase Transitions. J. Pharm. Sci. 2015, 104, 2056–2061. [Google Scholar] [CrossRef]
- Issa, M.; Ferraz, H. Intrinsic Dissolution as a Tool for Evaluating Drug Solubility in Accordance with the Biopharmaceutics Classification System. Dissolut. Technol. 2011, 18, 6–13. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, L.; Wang, C.; Li, Y.; Wu, Y.; Zhang, M.; Yin, Q. Insight into the Role of Hydrogen Bonding in the Molecular Self-Assembly Process of Sulfamethazine Solvates. Cryst. Growth Des. 2017, 17, 6151–6157. [Google Scholar] [CrossRef]
- Zhang, J.; Lu, T. Efficient evaluation of electrostatic potential with computerized optimized code. Phys. Chem. Chem. Phys. 2021, 23, 20323–20328. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.W. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring Nature and Predicting Strength of Hydrogen Bonds: A Correlation Analysis Between Atoms-in-Molecules Descriptors, Binding Energies, and Energy Components of Symmetry-Adapted Perturbation Theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, Q.X. Independent gradient model based on Hirshfeld partition: A new method for visual study of interactions in chemical systems. J. Comput. Chem. 2022, 43, 539–555. [Google Scholar] [CrossRef]
- Sajjad, M.A.; Macgregor, S.A.; Weller, A.S. A comparison of non-covalent interactions in the crystal structures of two σ-alkane complexes of Rh exhibiting contrasting stabilities in the solid state. Faraday Discuss. 2023, 244, 222–240. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W.; Carroll, M.T.; Cheeseman, J.R.; Chang, C. Properties of atoms in molecules: Atomic volumes. J. Am. Chem. Soc. 1987, 109, 7968–7979. [Google Scholar] [CrossRef]
- Yang, D.; Cao, J.; Jiao, L.; Yang, S.; Zhang, L.; Lu, Y.; Du, G. Solubility and Stability Advantages of a New Cocrystal of Berberine Chloride with Fumaric Acid. ACS Omega 2020, 5, 8283–8292. [Google Scholar] [CrossRef]
- Koch, U.; Popelier, P.L.A. Characterization of C-H-O hydrogen bonds on the basis of the charge density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Haneef, J.; Markad, D.; Chadha, R. Interaction Map Driven Cocrystallization of Ambrisentan: Structural and Biopharmaceutical Evaluation. Cryst. Growth Des. 2020, 20, 4612–4620. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J. Acid–base crystalline complexes and the pKa rule. Crystengcomm 2012, 14, 6362–6365. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, B.; Jia, L.; Wang, Y.; Wang, M.; Yang, H.; Qiao, Y.; Gong, J.; Tang, W. Tuning Physicochemical Properties of Antipsychotic Drug Aripiprazole with Multicomponent Crystal Strategy Based on Structure and Property Relationship. Cryst. Growth Des. 2020, 20, 3747–3761. [Google Scholar] [CrossRef]
- Huang, Q.; He, J.; Zhong, N.; Zheng, G. Determination of azide ion in irbesartan by column—Switching ion chromatography. Chin. J. Pharm. Anal. 2013, 33, 1431–1434. [Google Scholar]
- Ren, S.; Liu, M.; Hong, C.; Li, G.; Sun, J.; Wang, J.; Zhang, L.; Xie, Y. The effects of pH, surfactant, ion concentration, coformer, and molecular arrangement on the solubility behavior of myricetin cocrystals. Acta Pharm. Sin. B 2019, 9, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Bavishi, D.D.; Borkhataria, C.H. Spring and parachute: How cocrystals enhance solubility. Prog. Cryst. Growth Charact. Mater. 2016, 62, 1–8. [Google Scholar] [CrossRef]
- Li, C.; Wu, D.; Li, J.; Ji, X.; Qi, L.; Sun, Q.; Wang, A.; Xie, C.; Gong, J.; Chen, W. Multicomponent crystals of clotrimazole: A combined theoretical and experimental study. Crystengcomm 2021, 23, 6977–6993. [Google Scholar] [CrossRef]
- Wei, Y.; Zhang, L.; Wang, N.; Shen, P.; Dou, H.; Ma, K.; Gao, Y.; Zhang, J.; Qian, S. Mechanistic Study on Complexation-Induced Spring and Hover Dissolution Behavior of Ibuprofen-Nicotinamide Cocrystal. Cryst. Growth Des. 2018, 18, 7343–7355. [Google Scholar] [CrossRef]
- Deng, X.; Shi, W.; Qian, K.; Yang, J.; Yuan, S.; Li, H. Torsemide Crystalline Salts with a Significant Spring-Parachute Effect. AAPS PharmSciTech 2024, 25, 210–220. [Google Scholar] [CrossRef]
- Zhang, S.; Rasmuson, Å.C. Thermodynamics and Crystallization of the Theophylline–Glutaric Acid Cocrystal. Cryst. Growth Des. 2013, 13, 1153–1161. [Google Scholar] [CrossRef]
- Huang, M.; Shang, Z.; Zou, J.; Yang, D.; Yu, Q. Studies on the Tautomerism of Benzoylcyclohexane-1,3-dione and Its Derivatives by Ab Initio Calculation. J. Struct. Chem. 2002, 21, 678–682. [Google Scholar]
- Hu, W.; Gou, R.; Zhang, S.; Liu, Y.; Shang, F.; Chen, Y.; Bai, H. Theoretical investigation on the intermolecular interactions between 3-nitro-1,2,4-triazol-5-one and 2,6-diamino-3,5-dinitropyrazine-1-oxide using DFT methods. Chem. Pap. 2022, 76, 2747–2758. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | pKa | ΔpKa |
|---|---|---|
| IRB | 4.16 | |
| TOSA | −0.43 | 4.59 |
| MAL | 1.83 | 2.33 |
| BSA | 0.7 | 3.46 |
| 4-CBSA | −0.83 | 4.99 |
| 2,5-CBSA | −1.47 | 5.63 |
| MSA | −2.6 | 6.76 |
| CPSA | 1.17 | 2.99 |
| Compound | IRB-TOSA Salt | IRB-MAL Salt | IRB-BSA Salt | |
|---|---|---|---|---|
| Empirical formula | C25H29N6O+·C7H7O3S− | C25H29N6O+·C4H3O4− | C25H29N6O+·C6H5O3S− | |
| Formula weight | 600.73 | 544.60 | 586.70 | |
| Temperature/K | 113.15 | 113.15 | 113.15 | |
| Crystal system | triclinic | orthorhombic | monoclinic | |
| Space group | P212121 | P21/n | ||
| a/Å | 12.2685(3) | 9.27177(13) | 11.66260(10) | |
| b/Å | 15.9260(4) | 15.2804(2) | 16.1566(2) | |
| c/Å | 17.0796(3) | 19.6104(3) | 16.1328(2) | |
| α/° | 87.798(2) | 90 | 90 | |
| β/° | 71.313(2) | 90 | 102.2080(10) | |
| γ/° | 87.406(2) | 90 | 90 | |
| Volume/Å3 | 3156.98(13) | 2778.34(7) | 2971.13(6) | |
| Z, Z′ | 4, 2 | 4, 1 | 4, 1 | |
| ρcalc (g/cm3) | 1.264 | 1.302 | 1.312 | |
| F(000) | 1272.0 | 1152.0 | 1240.0 | |
| Reflections collected | 51,036 | 25,076 | 49,384 | |
| Goodness-of-fit on F2 | 1.045 | 1.047 | 1.035 | |
| I >= 2σ (I) | 0.0628 | 0.0509 | 0.0421 | |
| wR2 indexes | 0.1749 | 0.1040 | 0.1088 | |
| CCDC No. | 2407374 | 2407377 | 2407376 | |
| Compound | IRB-4-CBSA salt | IRB-2,5-CBSA salt | IRB-MSA salt | IRB-CPSA salt |
| Empirical formula | C25H29N6O+·C6H4ClO3S− | C25H29N6O+·C6H3Cl2O3S− | C25H29N6O+·CH3O3S− | C25H29N6O+·C10H15O4S− |
| Formula weight | 621.14 | 655.58 | 524.63 | 660.82 |
| Temperature/K | 138.15 | 138.15 | 138.15 | 295.15 |
| Crystal system | monoclinic | monoclinic | monoclinic | monoclinic |
| Space group | P21/c | P21/n | P21/c | P21 |
| a/Å | 18.09020(10) | 8.98593(3) | 16.58400(10) | 11.79510(16) |
| b/Å | 15.88270(10) | 16.63335(5) | 16.50230(10 | 12.56572(19) |
| c/Å | 22.84110(10) | 21.06129(6) | 19.74280(10) | 23.1805(3) |
| α/° | 90 | 90 | 90 | 90 |
| β/° | 108.8710(10) | 93.1611(3) | 90.3500(10) | 90.5282(12) |
| γ/° | 90 | 90 | 90 | 90 |
| Volume/Å3 | 6209.98(7) | 3143.162(15) | 5402.99(5) | 3435.52(9) |
| Z, Z′ * | 8, 2 | 4, 1 | 8, 2 | 4, 2 |
| ρcalc (g/cm3) | 1.329 | 1.385 | 1.290 | 1.278 |
| F(000) | 2608.0 | 1368.0 | 2224.0 | 1408.0 |
| Reflections collected | 62,498 | 59,377 | 98,674 | 28,594 |
| Goodness-of-fit on F2 | 1.048 | 1.053 | 1.104 | 1.036 |
| I >= 2σ (I) | 0.0485 | 0.0312 | 0.0574 | 0.0538 |
| wR2 indexes | 0.1270 | 0.0793 | 0.1671 | 0.1353 |
| CCDC No. | 2407379 | 2407390 | 2407391 | 2407392 |
| HB Type | HBA (Sulfonic Acid Group) | HBD (IRB) | IRB Salts |
|---|---|---|---|
| single HB | one oxygen | tetrazolium ring N1–H | IRB-TOSA, IRB-BSA, IRB-4-CBSA, IRB-MSA, |
| single HB | one oxygen | diazepine ring N6–H | IRB-TOSA, IRB-BSA, IRB-4-CBSA, IRB-MSA, IRB-2,5-CBSA, IRB-CPSA |
| dual-HBD bifurcated HB | one oxygen | tetrazolium ring N1–H diazepine ring N6–H | IRB-2,5-CBSA |
| dual-HBA bifurcated HB | two oxygens | tetrazolium ring N1–H | IRB-2,5-CBSA, IRB-CPSA |
| Position | Chemical Shift/(ppm) | |||||
|---|---|---|---|---|---|---|
| IRB | IRB-TOSA | IRB-BSA | IRB-4-CBSA | IRB-MSA | IRB-CPSA | |
| 3 | 7.67 (m) | 7.70 (m) | 7.68 (m) | 7.67 (m) | 7.69 (m) | 7.68 (m) |
| 4 | 7.55 (m) | 7.58 (m) | 7.55 (m) | 7.55 (m) | 7.57 (m) | 7.57 (m) |
| 5 | 7.55 (m) | 7.58 (m) | 7.55 (m) | 7.55 (m) | 7.57 (m) | 7.57 (m) |
| 6 | 7.67 (m) | 7.70 (m) | 7.68 (m) | 7.67 (m) | 7.69 (m) | 7.68 (m) |
| 9 | 7.14 (m) | 7.26 (m) | 7.26 (d, J = 7.9 Hz) | 7.26 (d, J = 7.9 Hz) | 7.28 (d, J = 7.9 Hz) | 7.29 (d, J = 7.8 Hz) |
| 10 | 7.14 (m) | 7.19 (d, J = 8.0 Hz) | 7.16 (d, J = 7.9 Hz) | 7.15 (d, J = 7.9 Hz) | 7.17 (d, J = 8.0 Hz) | 7.18 (d, J = 7.8 Hz) |
| 12 | 7.14 (m) | 7.19 (d, J = 8.0 Hz) | 7.16 (d, J = 7.9 Hz) | 7.15 (d, J = 7.9 Hz) | 7.17 (d, J = 8.0 Hz) | 7.18 (d, J = 7.8 Hz) |
| 13 | 7.14 (m) | 7.26 (m) | 7.26 (d, J = 7.9 Hz) | 7.26 (d, J = 7.9 Hz) | 7.28 (d, J = 7.9 Hz) | 7.29 (d, J = 7.8 Hz) |
| 14 | 4.76 (s) | 4.90 (s) | 4.79 (s) | 4.79 (s) | 4.96 (s) | 4.94 (s) |
| 17 | 2.40 (m) | 2.87 (t, J = 7.9 Hz) | 2.86 (t, J = 7.9 Hz) | 2.87 (t, J = 7.8 Hz) | 2.90 (t, J = 7.8 Hz) | 2.92 (t, J = 7.9 Hz) |
| 18 | 1.51 (m) | 1.61 (m) | 1.59 (m) | 1.57 (m) | 1.61 (m) | 1.62 (m) |
| 19 | 1.33 (m) | 1.41 (m) | 1.36 (m) | 1.37 (m) | 1.42 (m) | 1.42 (m) |
| 20 | 0.87 (t, J = 7.3 Hz) | 0.92 (t, J = 7.3 Hz) | 0.89 (t, J = 7.4 Hz) | 0.89 (t, J = 7.3 Hz) | 0.93 (t, J = 7.3 Hz) | 0.92 (t, J = 7.3 Hz) |
| 22 | 1.82 (m), 1.96 (m) | 2.01 (m), 2.18 (m) | 1.99 (m), 2.16 (m) | 1.99 (m), 2.16 (m) | 2.01 (m), 2.19 (m) | 2.03 (m), 2.20 (m) |
| 23 | 1.96 (m) | 2.01 (m) | 1.99 (m) | 1.99 (m) | 2.01 (m) | 2.03 (m) |
| 24 | 1.96 (m) | 2.01 (m) | 1.99 (m) | 1.99 (m) | 2.01 (m) | 2.03 (m) |
| 25 | 1.82 (m), 1.96 (m) | 2.01 (m), 2.18 (m) | 1.99 (m), 2.16 (m) | 1.99 (m), 2.16 (m) | 2.01 (m), 2.19 (m) | 2.03 (m), 2.20 (m) |
| Compound | DS–O/DC–O (Å) | ΔDS–O/ΔDC–O (Å) | Proton Transfer |
|---|---|---|---|
| IRB-TOSA salt | 1.457, 1.446, 1.458 | 0.011, 0.001, 0.012 | yes |
| IRB-MAL salt | 1.272, 1.241 | 0.031 | yes |
| IRB-BSA salt | 1.448, 1.453, 1.444 | 0.005, 0.004, 0.009 | yes |
| IRB-4-CBSA salt | 1.447, 1.435, 1.423 | 0.012, 0.024, 0.012 | yes |
| IRB-2,5-CBSA salt | 1.439, 1.460, 1.458 | 0.021, 0.019, 0.002 | yes |
| IRB-MSA salt | 1.403, 1.437, 1.462 | 0.034, 0.059, 0.025 | yes |
| IRB-CPSA salt | 1.431, 1.451, 1.463 | 0.020, 0.032, 0.012 | yes |
| Compound | DC–O,COOH (Å) | ΔDC–O,COOH (Å) |
|---|---|---|
| IRB-MAL salt | 1.240, 1.280 | 0.040 |
| MAL | 1.202, 1.294 | 0.092 |
| 1.226, 1.306 | 0.080 |
| RH (%) | Increase in Mass (%) | |||||
|---|---|---|---|---|---|---|
| IRB-TOSA | IRB-4-CBSA | IRB-2,5-CBSA | TOSA | 4-CBSA | 2,5-CBSA | |
| 40 | 0.04 | 0.11 | 0.04 | 9.11 | 34.87 | 24.00 |
| 95 | 0.16 | 0.32 | 0.10 | 182.42 | 172.51 | 145.11 |
| Compound | H···H (%) | O···H (%) | N···H (%) | C···H (%) | Cl···H (%) | C···N (%) | C···O (%) | Others (%) |
|---|---|---|---|---|---|---|---|---|
| IRB-TOSA salt | 56.1 | 14.3 | 11.4 | 13.2 | 0 | 2.0 | 0.9 | 2.0 |
| IRB-MAL salt | 51.9 | 16.3 | 12.7 | 11.8 | 0 | 2.9 | 2.5 | 1.9 |
| IRB-BSA salt | 52.5 | 15.9 | 12.4 | 14.5 | 0 | 2.2 | 0.6 | 2.0 |
| IRB-4-CBSA salt | 50.2 | 13.5 | 13.8 | 14.8 | 3.0 | 1.6 | 0.3 | 3.0 |
| IRB-2,5-CBSA salt | 42.4 | 16.4 | 12.6 | 15.0 | 8.1 | 1.3 | 0.6 | 3.6 |
| IRB-MSA salt | 56.3 | 14.0 | 15.0 | 10.6 | 0 | 1.3 | 1.7 | 1.1 |
| IRB-CPSA salt | 55.9 | 18.4 | 13.3 | 10.7 | 0 | 0 | 1.1 | 0.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Wu, C.; Zhang, M.; Hou, L.; Chen, W.; Jing, D.; Bao, Y. Seven New Irbesartan Salts: Significantly Improved Dissolution, Excellent Hygrothermal Stability, and Characteristic Supramolecular Synthons. Crystals 2025, 15, 342. https://doi.org/10.3390/cryst15040342
Wang J, Wu C, Zhang M, Hou L, Chen W, Jing D, Bao Y. Seven New Irbesartan Salts: Significantly Improved Dissolution, Excellent Hygrothermal Stability, and Characteristic Supramolecular Synthons. Crystals. 2025; 15(4):342. https://doi.org/10.3390/cryst15040342
Chicago/Turabian StyleWang, Junxiao, Chuanhua Wu, Menglong Zhang, Lingli Hou, Wei Chen, Dingding Jing, and Ying Bao. 2025. "Seven New Irbesartan Salts: Significantly Improved Dissolution, Excellent Hygrothermal Stability, and Characteristic Supramolecular Synthons" Crystals 15, no. 4: 342. https://doi.org/10.3390/cryst15040342
APA StyleWang, J., Wu, C., Zhang, M., Hou, L., Chen, W., Jing, D., & Bao, Y. (2025). Seven New Irbesartan Salts: Significantly Improved Dissolution, Excellent Hygrothermal Stability, and Characteristic Supramolecular Synthons. Crystals, 15(4), 342. https://doi.org/10.3390/cryst15040342