
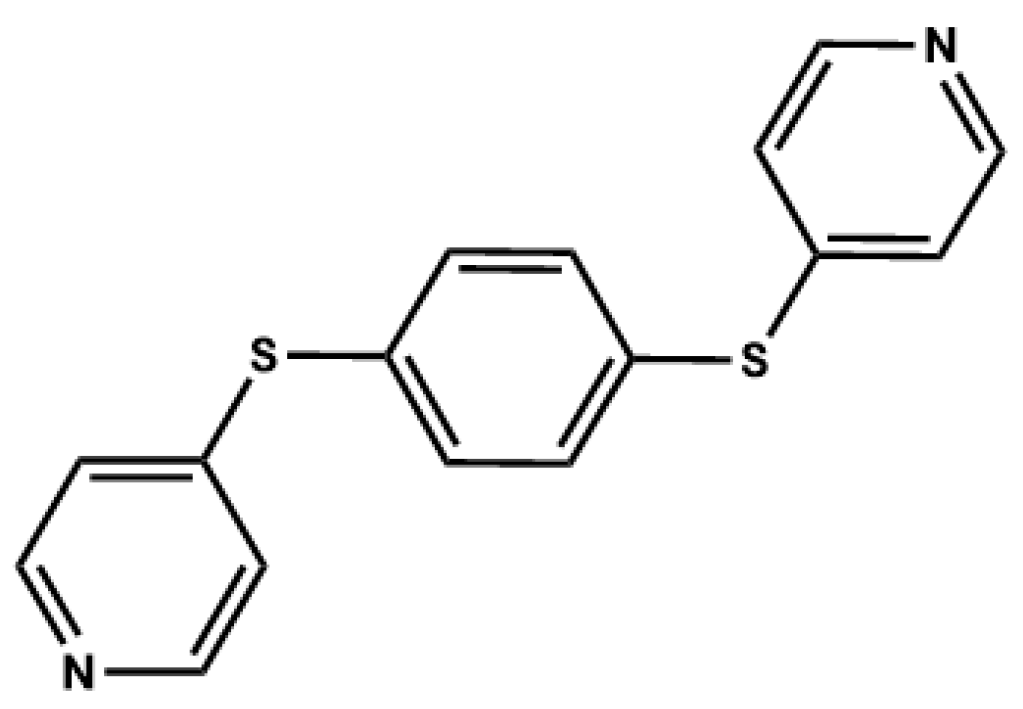
Polymorphism and the Phenomenon of Whole-Molecule Disorder Revealed in a Novel Dipodal Thiopyridine Ligand



Abstract

1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Measurements
2.3. Computational Studies
2.4. Structure Determination
3. Results and Discussion
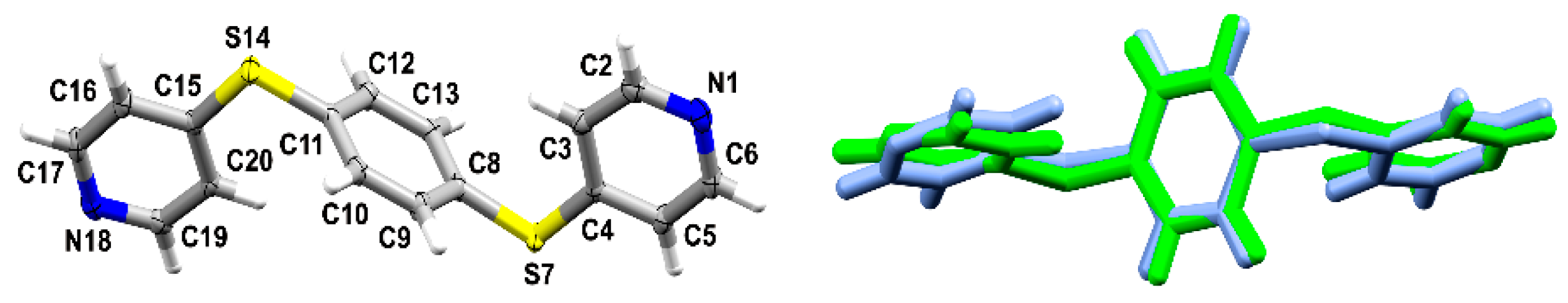
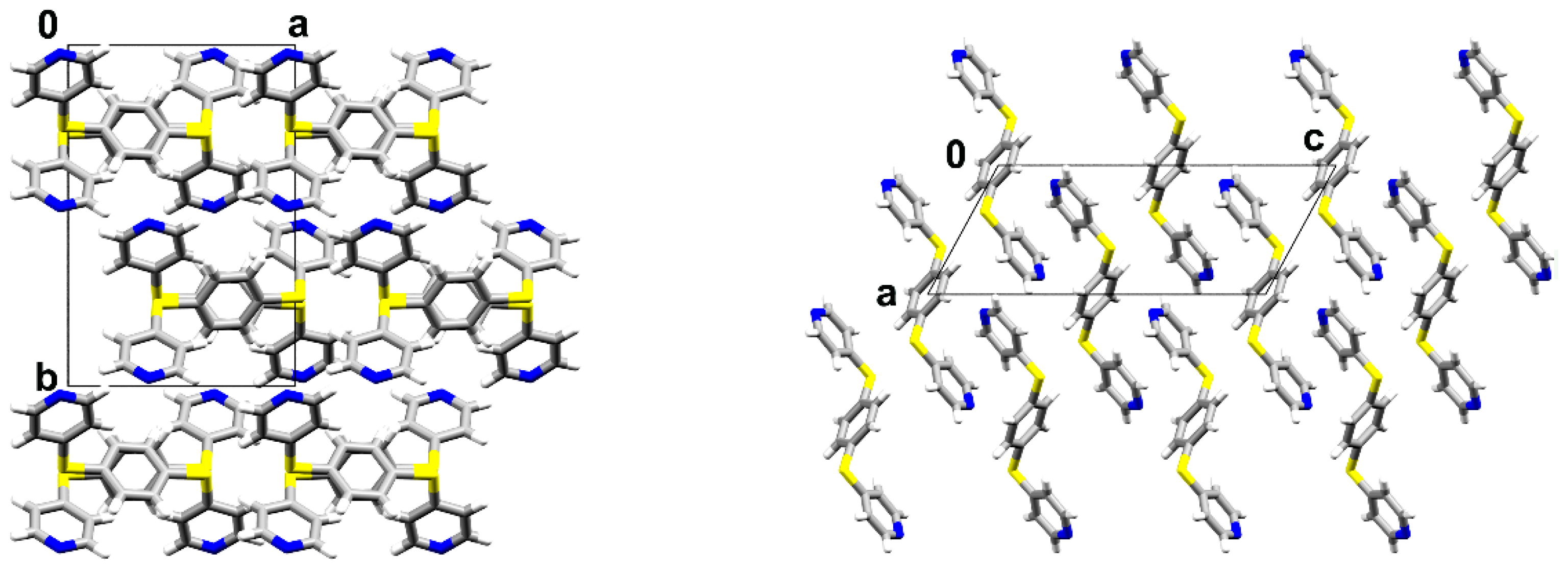
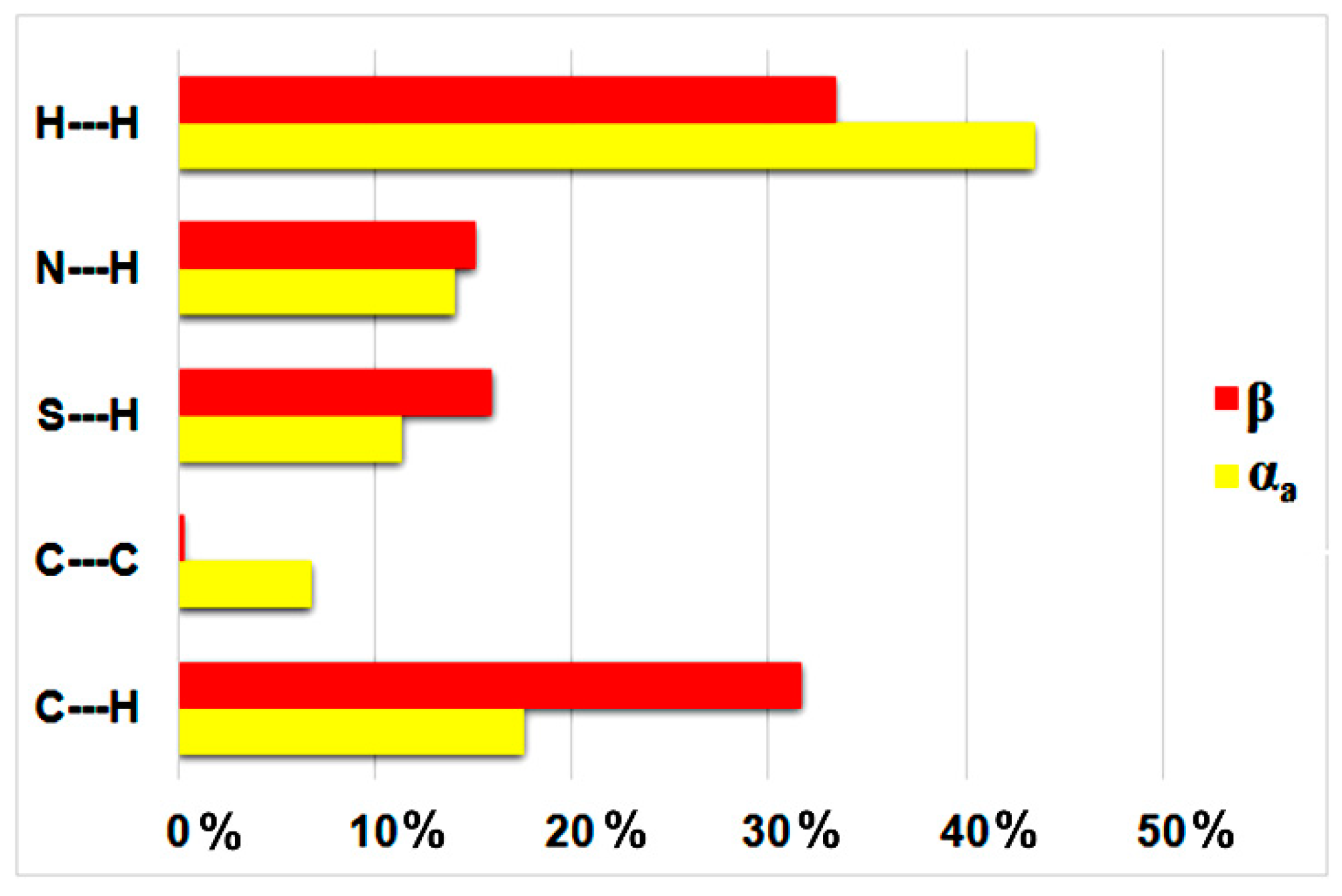
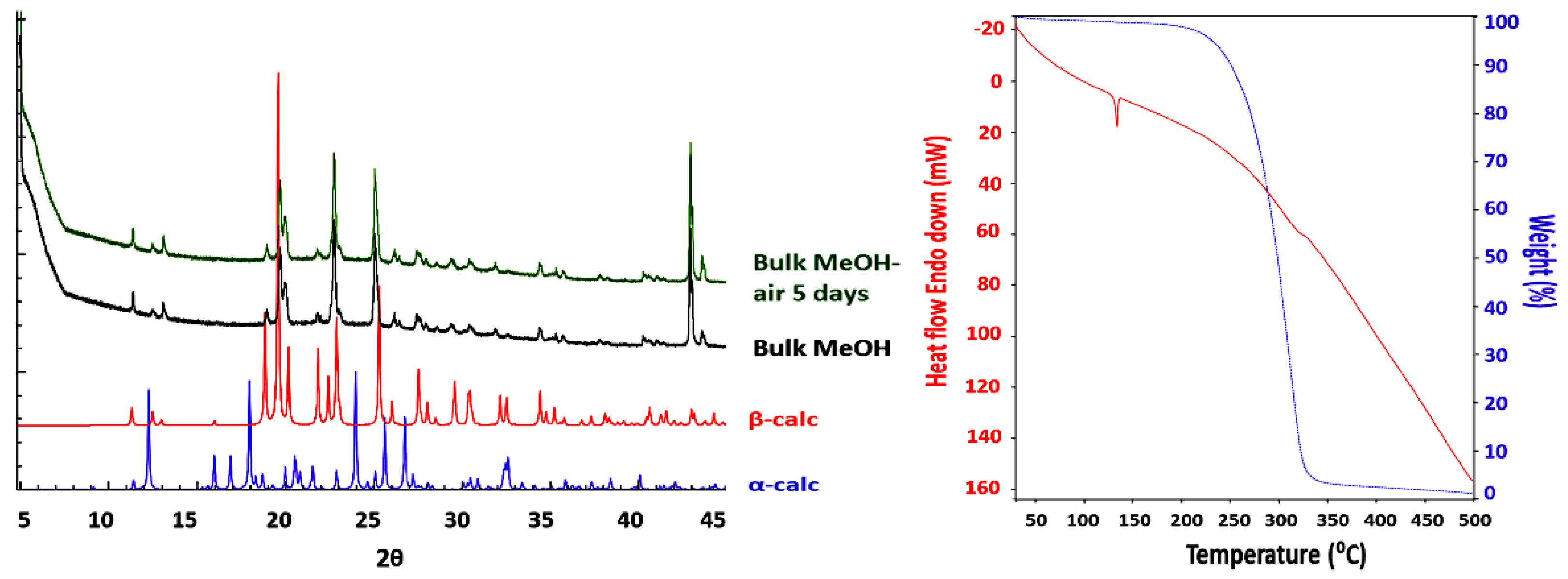
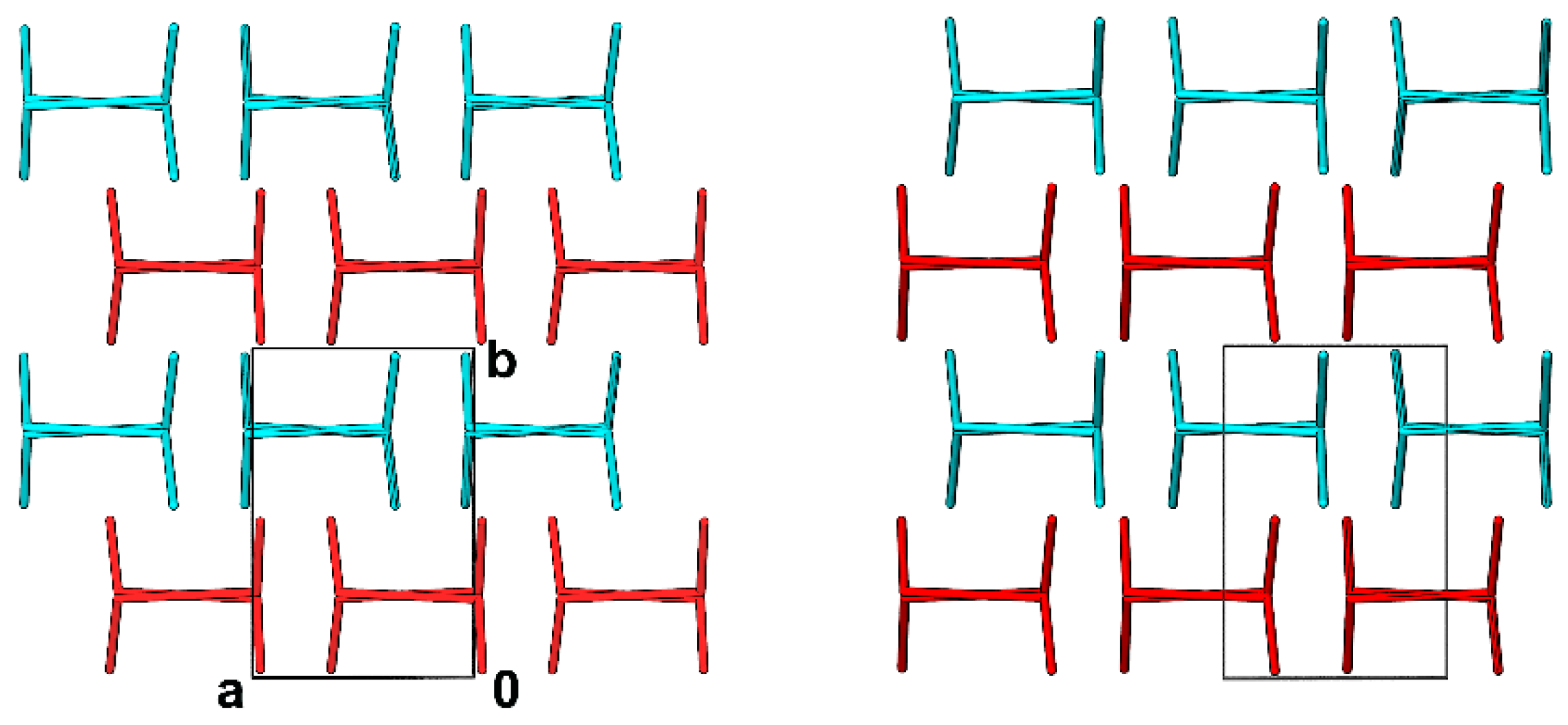
3.1. Crystal Structures and Studies on Bulk
3.2. Polymorph Stability
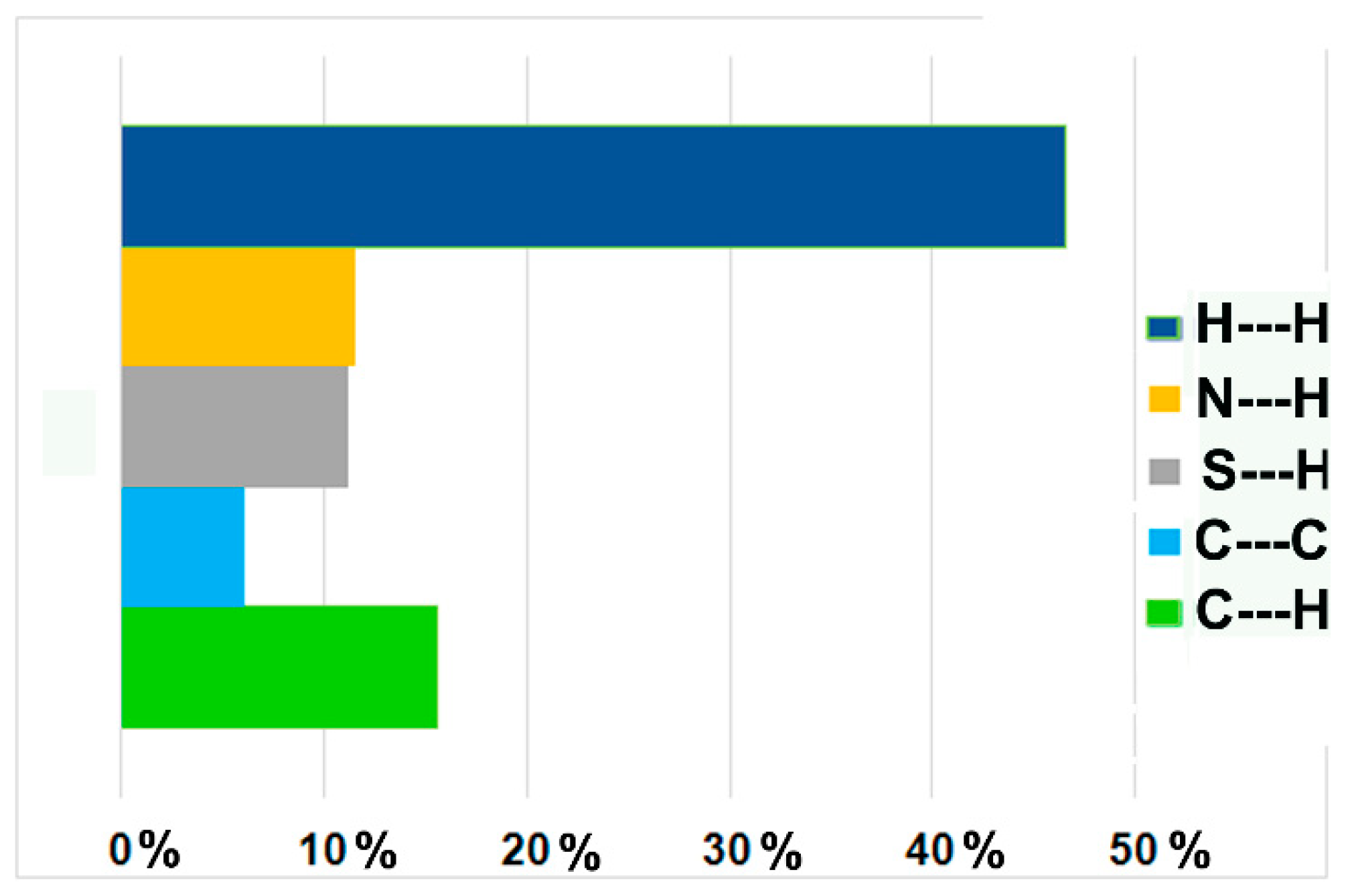
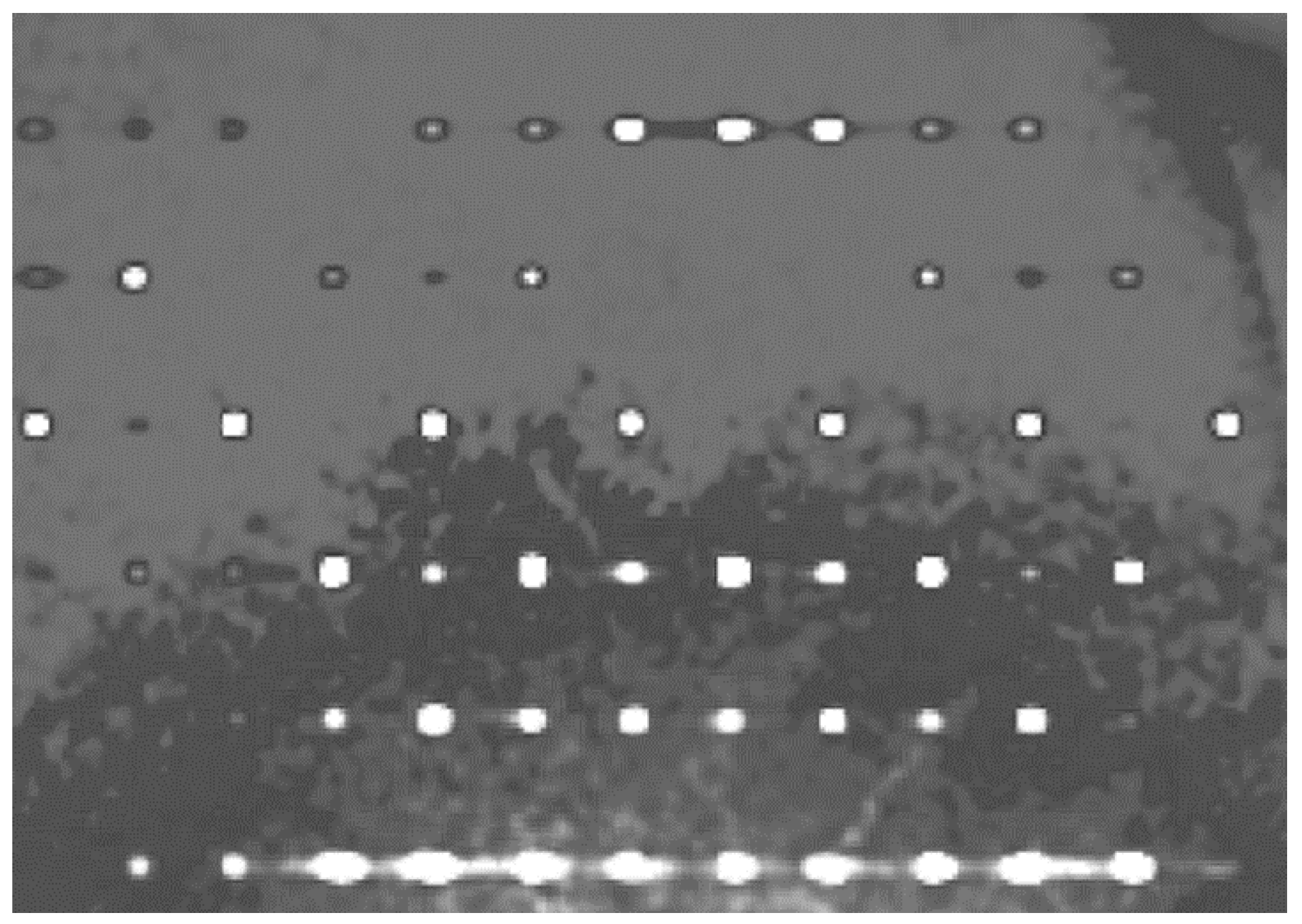
3.3. Whole-Molecule Disorder
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cruz-Cabeza, A.J.; Reutzel-Edens, S.M.; Bernstein, J. Facts and Fictions about Polymorphism. Chem. Soc. Rev. 2015, 44, 8619–8635. [Google Scholar] [CrossRef]
- Nangia, A. Conformational Polymorphism in Organic Crystals. Acc. Chem. Res. 2008, 41, 595–604. [Google Scholar]
- Cruz-Cabeza, A.J.; Bernstein, J. Conformational Polymorphism. Chem. Rev. 2014, 114, 2170–2191. [Google Scholar]
- Sengupta, B.; Beasley, M.; Mason, B. Conformational Polymorphism in Organic Crystals: Structural and Functional Aspects—A Review. Curr. Res. Mater. Chem. 2019, 1, 104. [Google Scholar]
- Purohit, R.; Venugopalan, P. Polymorphism: An Overview. Resonance 2009, 14, 882–893. [Google Scholar]
- Lee, E.H. A Practical Guide to Pharmaceutical Polymorph Screening & Selection. Asian J. Pharm. Sci. 2014, 9, 163–175. [Google Scholar]
- Price, S.L. Computational Prediction of Organic Crystal Structures and Polymorphism. Int. Rev. Phys. Chem. 2008, 27, 541–568. [Google Scholar]
- Wu, E.J.; Kelly, A.W.; Iuzzolino, L.; Lee, A.Y.; Zhu, X. Unprecedented Packing Polymorphism of Oxindole: An Exploration Inspired by Crystal Structure Prediction. Angew. Chem. Int. Ed. 2024, 63, e202406214. [Google Scholar]
- Beran, G.J.O. Frontiers of Molecular Crystal Structure Prediction for Pharmaceuticals and Functional Organic Materials. Chem. Sci. 2023, 14, 13290–13312. [Google Scholar] [CrossRef]
- Dudek, M.K.; Drużbicki, K. Along the Road to Crystal Structure Prediction (CSP) of Pharmaceutical-like Molecules. CrystEngComm 2022, 24, 1665–1678. [Google Scholar]
- Nikhar, R.; Szalewicz, K. Reliable Crystal Structure Predictions from First Principles. Nat. Commun. 2022, 13, 3095. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, S.; Kędziera, D.; Rafiński, Z.; Dobrzańska, L. Solvent-Induced Polymorphism in Dipodal N-Donor Ligands Containing a Biphenyl Core. RSC Adv. 2023, 13, 30625–30632. [Google Scholar]
- Milić, D.; Alešković, M.; Matkovic-Calogovic, D.; Mlinarić-Majerski, K. (1-Adamantyl)Methyl Esters: Whole-Molecule Disorder in the Crystal Structure of (1-Adamantyl)Methyl-1-Adamantanecarboxylate. Croat. Chem. Acta 2009, 82, 833–839. [Google Scholar]
- Dorset, D.L.; McCourt, M.P. Disorder and the Molecular Packing of C60 Buckminsterfullerene: A Direct Electron-Crystallographic Analysis. Acta Crystallogr. Sect. A 1994, 50, 344–351. [Google Scholar]
- David, W.I.F.; Ibberson, R.M.; Matthewman, J.C.; Prassides, K.; Dennis, T.J.S.; Hare, J.P.; Kroto, H.W.; Taylor, R.; Walton, D.R.M. Crystal Structure and Bonding of Ordered C60. Nature 1991, 353, 147–149. [Google Scholar] [CrossRef]
- Ichharam, V.; Boeyens, J.C.A. Structural Analysis of 2-(2-Thienyl)-1-(2-Pyrazinyl)Ethene [C10H8N2S] and 2-(2-Thienyl)-1-(2-Quinoxalinyl)Ethene [C14H10N2S]. Cryst. Eng. 2001, 4, 171–178. [Google Scholar]
- Cox, P.J.; Wardell, J.L. 4,4′-Sulfonyl-Bis[N-(4-Nitro-Phenyl-Methylene)Benzen-Amine. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2003, 59, 706–708. [Google Scholar] [CrossRef]
- Seidel, R.W.; Goddard, R.; Schlegel, T.U.; Richter, A.; Rudolph, I.; Imming, P. Polymorphism and Whole-Molecule Disorder of an Antitubercular 8-Nitrobenzothiazinone. J. Chem. Crystallogr. 2024, 1–10. [Google Scholar] [CrossRef]
- Trankle, W.G.; Kopach, M.E. Green Chemical Synthesis of 2-Benzenesulfonyl-Pyridine and Related Derivatives. Org. Process Res. Dev. 2007, 11, 913–917. [Google Scholar]
- Kraus, W.; Nolze, G. POWDER CELL—A Program for the Representation and Manipulation of Crystal Structures and Calculation of the Resulting X-Ray Powder Patterns. J. Appl. Crystallogr. 1996, 29, 301–303. [Google Scholar]
- Doebelin, N.; Kleeberg, R. Profex: A Graphical User Interface for the Rietveld Refinement Program BGMN. J. Appl. Crystallogr. 2015, 48, 1573–1580. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld Surface Analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A Program for Hirshfeld Surface Analysis, Visualization and Quantitative Analysis of Molecular Crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Gavezzotti, A. Calculation of Intermolecular Interaction Energies by Direct Numerical Integration over Electron Densities. 2. An Improved Polarization Model and the Evaluation of Dispersion and Repulsion Energies. J. Phys. Chem. B 2003, 107, 2344–2353. [Google Scholar] [CrossRef]
- Gavezzotti, A. Non-Conventional Bonding between Organic Molecules. the “halogen Bond” in Crystalline Systems. Mol. Phys. 2008, 106, 1473–1485. [Google Scholar] [CrossRef]
- Reeves, M.G.; Wood, P.A.; Parsons, S. MrPIXEL: Automated Execution of Pixel Calculations via the Mercury Interface. J. Appl. Crystallogr. 2020, 53, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- MacRae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Gelbrich, T.; Hursthouse, M.B. A Versatile Procedure for the Identification, Description and Quantification of Structural Similarity in Molecular Crystals. CrystEngComm 2005, 7, 324–336. [Google Scholar] [CrossRef]
- Gelbrich, T.; Hursthouse, M.B. Systematic Investigation of the Relationships between 25 Crystal Structures Containing the Carbamazepine Molecule or a Close Analogue: A Case Study of the XPac Method. CrystEngComm 2006, 8, 448–460. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Bruno, I.J.; Cole, J.C.; Kessler, M.; Luo, J.; Momerwell, W.D.S.; Purkis, L.H.; Smith, B.R.; Taylor, R.; Cooper, R.I.; Harris, S.E.; et al. Retrieval of Crystallographically-Derived Molecular Geometry Information. J. Chem. Inf. Comput. Sci. 2004, 44, 2133–2144. [Google Scholar]
- Antonijević, I.S.; Janjić, G.V.; Milčić, M.K.; Zarić, S.D. Preferred Geometries and Energies of Sulfur-Sulfur Interactions in Crystal Structures. Cryst. Growth Des. 2016, 16, 632–639. [Google Scholar]
- Bondi, A. Van Der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar]
- Gelbrich, T.; Threlfall, T.L.; Hursthouse, M.B. XPac Dissimilarity Parameters as Quantitative Descriptors of Isostructurality: The Case of Fourteen 4,5′-Substituted Benzenesulfonamido-2-Pyridines Obtained by Substituent Interchange Involving CF3/I/Br/Cl/F/Me/H. CrystEngComm 2012, 14, 5454–5464. [Google Scholar] [CrossRef]
- Jelsch, C.; Ejsmont, K.; Huder, L. The Enrichment Ratio of Atomic Contacts in Crystals, an Indicator Derived from the Hirshfeld Surface Analysis. IUCrJ 2014, 1, 119–128. [Google Scholar]
- Munday, L.B.; Solares, S.D.; Chung, P.W. Generalized Stacking Fault Energy Surfaces in the Molecular Crystal RDX. Philos. Mag. 2012, 92, 3036–3050. [Google Scholar] [CrossRef]
- Ouvrard, C.; Price, S.L. Toward Crystal Structure Prediction for Conformationally Flexible Molecules: The Headaches Illustrated by Aspirin. Cryst. Growth Des. 2004, 4, 1119–1127. [Google Scholar]
- Vishweshwar, P.; McMahon, J.A.; Oliveira, M.; Peterson, M.L.; Zaworotko, M.J. The Predictably Elusive Form II of Aspirin. J. Am. Chem. Soc. 2005, 127, 16802–16803. [Google Scholar]
- Bond, A.D.; Boese, R.; Desiraju, G.R. On the Polymorphism of Aspirin. Angew. Chem. Int. Ed. 2007, 46, 615–617. [Google Scholar]
- Bond, A.D.; Boese, R.; Desiraju, G.R. On the Polymorphism of Aspirin: Crystalline Aspirin as Intergrowths of Two “Polymorphic” Domains. Angew. Chem. Int. Ed. 2007, 46, 618–622. [Google Scholar]
- Bond, A.D.; Solanko, K.A.; Parsons, S.; Redder, S.; Boese, R. Single Crystals of Aspirin Form II: Crystallisation and Stability. CrystEngComm 2011, 13, 399–401. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Reference | α | β |
|---|---|---|
| Chemical formula | C16H12N2S2 | C16H12N2S2 |
| Formula mass | 296.40 | 296.40 |
| Crystal system | monoclinic | monoclinic |
| Space group | P21/c | P21/c |
| a/Å | 10.5963(3) | 7.6480(2) |
| b/Å | 14.4449(3) | 5.85520(10) |
| c/Å | 10.0761(3) | 17.5179(5) |
| α/° | 90 | 90 |
| β/° | 113.933(3) | 118.578(4) |
| γ/° | 90 | 90 |
| Unit cell volume/Å3 | 1409.67(7) | 688.89(4) |
| Temperature/K | 100(2) | 100(2) |
| No. of formula units per unit cell, Z | 4 | 2 |
| Radiation type | CuKα | CuKα |
| Absorption coefficient, μ/mm−1 | 3.329 | 3.406 |
| No. of reflections measured | 14327 | 6681 |
| No. of independent reflections | 2904 | 1385 |
| Rint | 0.0311 | 0.0324 |
| Final R1 values (I > 2σ(I)) | 0.0324 | 0.0290 |
| Final wR(F2) values (I > 2σ(I)) | 0.0928 | 0.0768 |
| Final R1 values (all data) | 0.0368 | 0.0299 |
| Final wR(F2) values (all data) | 0.0958 | 0.0773 |
| Goodness of fit on F2 | 1.075 | 1.088 |
| Compound | D-H---A | H---A (Å) | D---A (Å) | D-H---A (°) |
|---|---|---|---|---|
| αa | C9-H9---N1 i | 2.79 | 3.466(2) | 129 |
| C10-H10---N1 i | 2.92 | 3.530(2) | 123 | |
| C13-H13---N1 ii | 2.67 | 3.509(2) | 148 | |
| C17-H17---N1 iii | 2.73 | 3.548(2) | 145 | |
| C6-H6---N18 iv | 2.92 | 3.642(2) | 134 | |
| C12-H12---N18 v | 2.56 | 3.390(2) | 147 | |
| β | C5-H5---N1 i | 2.99 | 3.471(2) | 112 |
| C6-H6---N1 i | 2.86 | 3.414(2) | 118 | |
| C10-H10---N1 ii | 2.57 | 3.446(2) | 153 |
| CrystalExplorer | E_ele | E_pol | E_dis | E_rep | E_tot |
|---|---|---|---|---|---|
| Tonto wavefunction calc. | |||||
| αa | −55.6 | −9.9 | −154.6 | 80.5 | −139.7 |
| β | −52.6 | −10.3 | −160.0 | 74.2 | −148.8 |
| Gaussian wavefunction calc. | |||||
| αa | −57.4 | −10.1 | −154.6 | 80.6 | −141.6 |
| β | −54.8 | −10.4 | −160.0 | 74.3 | −150.9 |
| D-H---A | H---A (Å) | D---A (Å) | D-H---A (°) |
|---|---|---|---|
| C17A-H17A---N1A i | 2.85 | 3.628(3) | 140 |
| C6A-H6A---N18A ii | 2.78 | 3.597(3) | 144 |
| C9A-H9A---N18A iii | 2.22 | 2.943(3) | 132 |
| CrystalExplorer | E_ele | E_pol | E_dis | E_rep | E_tot |
|---|---|---|---|---|---|
| Tonto wavefunction calc. | |||||
| −122.1 | −10.8 | −160.2 | 235.6 | −57.5 | |
| Gaussian wavefunction calc. | |||||
| −124.9 | −11.1 | −160.2 | 236.2 | −60.0 | |
| Pixel | −156.1 | −94 | −218.9 | 476.8 | 7.8 |
| E_ele | E_pol | E_dis | E_rep | E_tot | |
|---|---|---|---|---|---|
| ECUTUR09 | |||||
| Pixel | −178.3 | −80.5 | −264.1 | 519.7 | −3.2 |
| CrystalExplorer | −153.5 | −6.6 | −217.1 | 296.2 | −81.0 |
| NPOFNP09 | |||||
| Pixel | −137.8 | −57.4 | −218.9 | 418.3 | 4.3 |
| CrystalExplorer | −120.2 | −5.7 | −187.3 | 234.7 | −78.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaudhary, S.; Losus, R.M.; Dobrzańska, L. Polymorphism and the Phenomenon of Whole-Molecule Disorder Revealed in a Novel Dipodal Thiopyridine Ligand. Crystals 2025, 15, 289. https://doi.org/10.3390/cryst15040289
Chaudhary S, Losus RM, Dobrzańska L. Polymorphism and the Phenomenon of Whole-Molecule Disorder Revealed in a Novel Dipodal Thiopyridine Ligand. Crystals. 2025; 15(4):289. https://doi.org/10.3390/cryst15040289
Chicago/Turabian StyleChaudhary, Simran, Renny Maria Losus, and Liliana Dobrzańska. 2025. "Polymorphism and the Phenomenon of Whole-Molecule Disorder Revealed in a Novel Dipodal Thiopyridine Ligand" Crystals 15, no. 4: 289. https://doi.org/10.3390/cryst15040289
APA StyleChaudhary, S., Losus, R. M., & Dobrzańska, L. (2025). Polymorphism and the Phenomenon of Whole-Molecule Disorder Revealed in a Novel Dipodal Thiopyridine Ligand. Crystals, 15(4), 289. https://doi.org/10.3390/cryst15040289