Abstract
In the present work, we describe the synthesis of new tetrahydroisoquinoline derivatives and the crystal structures of two of them. Density functional theory (DFT) investigations at the B3LYP/6-31+G(d,p) level provided their structural reactivity and nonlinear optical properties. The low HOMO-LUMO gaps (EH-L) suggest a soft nature and higher reactivity, while calculated global reactivity descriptors provide assessments of their reactivity and electronic stability. The calculated natural bonding molecular orbital (NBO) charges show excellent charge separation (charge transfer) and identify the donor and acceptor parts of the molecules. Density of states (DOS) analyses show the newly generated energy states and reduced band gaps, which impart higher conductive properties. For surface reactivity, 3D MESP surfaces are plotted and show electron-rich sites near the nitrogen atoms of the tetrahydroisoquinoline rings. Nonlinear optical (NLO) properties of the crystals are predicted from calculated polarizability (αo) and hyperpolarizability (βo) values. For IVb, the αo and βo values are 415.53 and 1003.44 au. The remarkable value (1003.44 au) of the hyperpolarizability (βo) shows IVb has excellent NLO properties. Structural activity relationship analysis suggests that nitric oxide synthases are better targets for both compounds, and they were further subjected to molecular docking simulations to understand the binding efficiency. In addition, ADMET analyses were carried out to understand the potential activity of the molecules as drug candidates.
1. Introduction
The family of nitric oxide synthases (NOS) plays a vital role in many pathological disorders as well as several physiological systems. However, the metals, hydroxides, their alloys, and organic compounds are not free from some disadvantages. One of which is their low resistance to the aggressive influence of environmental factors such as temperature, oxygen, and electromagnetic radiation. The oxide compounds in this sense are much more stable when used up to 1000 °C. There is such a class of materials as complex iron oxides with excellent electronic properties that are also promising for practical application [1,2,3,4]. There are three different types of NOS: neuronal NOS (nNOS, also known as NOS 1), endothelial NOS (eNOS, also known as NOS 3), and inducible NOS (iNOS, or NOS 2) [3,4]. nNOS and eNOS are both constitutively expressed. Traditionally, eNOS has been regarded as the primary isoform controlling vascular function, but recent research has revealed that nNOS is present in the vascular endothelium and significantly contributes to the upkeep of the cardiovascular system’s homeostasis [3,4].
It is well known that nNOS is also expressed in numerous non-neuronal cells, such as the endothelium and smooth muscle cells of many types of arteries in animals [5,6,7] and humans [8]. Also, nNOS is widely expressed in neurons and is related to the modulation of neuronal processes [9].
In addition to NO, nNOS creates H2O2 under physiological conditions, which contributes to endothelial-dependent vascular relaxation [10]. Endothelial nNOS-derived H2O2 production failure has been linked to atherosclerosis [11] and hypertension [12]. By incorporating NO-donors (e.g., ester nitrates, furoxans, benzofuroxans, NONOates, S-nitrosothiols, or metal complexes) into molecules that already have potential therapeutic value, hybrid compounds with an NO release advantage can be produced while keeping the activity of the native medicine. It is well known that the combination of different compounds that have excellent electronic properties leads to new composite materials that have earned great technological interest in recent years. The addition of a second phase can significantly improve the electronic properties of the resulting composite material [13,14].
Many areas of medicinal chemistry, including cancer, bacterial, fungal, viral, parasitic, ophthalmic, and inflammatory illnesses, have shown the value of this strategy. The creation of powerful and precise nitric oxide synthase inhibitors is mostly based on the structure of the enzyme. However, because of the significant similarity between the isoforms, these investigations have proven to be highly challenging. The goal of the research is to strike a compromise between keeping the natural drug activity and releasing therapeutic levels of NO, particularly at the targeted site of action [15,16]. Modern medication design places a lot of emphasis on finding novel and potent NO-donor hybrid medicines as well as selective nitric oxide synthase inhibitors to control the NO-dependent biochemical pathways that affect a variety of human organ dysfunctions [17,18,19,20].
After synthesizing new derivatives of tetrahydroisoquinoline and determining their crystal structures, we carried out a DFT study to investigate their structural reactivity and quantum chemical properties. B3LYP, along with the split valance basis set (6-31+G(d,p)), is adopted to explain their topological and electronic properties. The optical and nonlinear optical (NLO) properties are also inferred from the calculated dipole moment (µo), polarizability (αo), and hyperpolarizability (βo) values.
2. Materials and Methods
2.1. X-ray Crystallographic Analysis
Suitable crystals of IVa and IVb were mounted on polymer loops with a drop of heavy oil and placed in a cold nitrogen stream on the diffractometer. Intensity data were collected under the control of the APEX4 software [21], and the raw intensities were reduced to F2 values with SAINT [21], which also performed global least-squares refinements of the unit cell parameters. Numerical absorption corrections and merging of equivalent reflections were accomplished with SADABS [22] and the structures were solved by dual space methods (SHELXT [23]). The structures were refined by full-matrix, least-squares methods (SHELXL [24]), with hydrogen atoms attached to carbon included as riding contributions with isotropic displacement parameters tied to those of the attached atoms. Hydrogen atoms attached to nitrogen were refined with DFIX 0.91 0.01 instructions. Crystallographic details are presented in Table S1.
2.2. DFT Studies
Density functional theory (DFT) calculations were performed in the gas phase approximation using the Gaussian 09 software [12] to investigate the electronic and optical properties of IVa and IVb. The DFT study used the hybrid B3LYP functional with the double zeta split valence basis set (6-31+G(d,p)) method for optimization and property calculations, as this method is considered suitable for calculating the electronic and nonlinear optical properties of molecules and clusters. Gauss View 5.0 was used for visualizing the optimized geometries generated. The choice of this hybrid method (B3LYP) is reliable for organic molecules and conjugated systems due to its long-range non-covalent interactions and the extent to which it captures electron correlations. This method produces reliable and comparable results to experimental studies for geometry optimization, electronic properties, and spectroscopic properties [25,26,27,28,29,30,31,32]. Additionally, frequency calculations were carried out to obtain stable geometries on the potential energy surface.
Frontier molecular orbital (FMO) analyses were performed to obtain measures of the reactivity and kinetic stability of IVa and IVb, while NBO charges were calculated at the same theoretical level to assess the charge transfer and reactivity of the two molecules. For a deeper insight into the electronic properties and reactivity surfaces of IVa and IVb, we calculated global reactivity parameters including ionization energy (IE), electron affinity (EA), chemical hardness, chemical potential, electronegativity, and electrophilicity indices. Additionally, the 3D molecular electrostatic potential (MESP) surfaces and density of states (DOS) spectra are obtained for locating reactive sites and conductive properties. DOS spectra were plotted with the GaussSum software. For optical and nonlinear optical (NLO) properties, the polarizability (αo) and hyperpolarizability (βo) values were calculated with the same DFT method.
2.3. Molecular Docking and ADMET Analysis
The protein’s crystal structure was obtained from the Protein Data Bank [33] under the PDB ID: 6PNA. The protein structure was initially pre-treated to eliminate crystal water molecules and het atoms. The structure was then optimized and minimized using the OPLS2005 force field and the protein wizard panel in the Schrodinger package [34,35]. Simultaneously, the ligand molecules IVa and IVb and the native ligand molecule were energetically minimized and independently integrated into the protein’s native ligand site. The induced fit docking panel from the Schrodinger software was used for the molecular docking simulation. In addition to this ADMET, analyses were carried out using SWISS ADME (http://www.swissadme.ch/, accessed on 9 June 2022) and the Pro Tox Web II server (https://tox-new.charite.de/protox_II/index.php?site=compound_input/, accessed on 9 June 2022).
3. Results and Discussion
3.1. Synthesis
The tetrahydroisoquinoline derivatives IVa and IVb were synthesized following the literature [36] (Figure 1) and confirmed by single-crystal X-ray diffraction analysis.
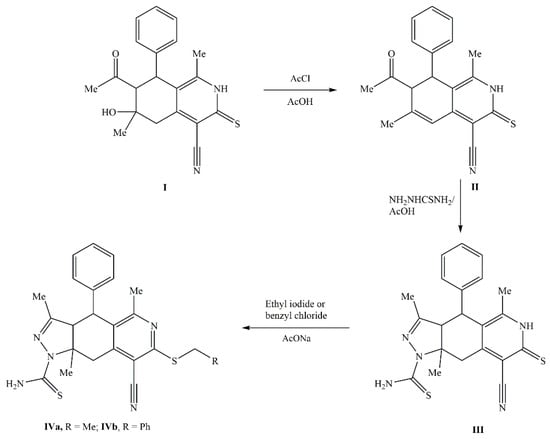
Figure 1.
Synthesis of compounds IVa and IVb.
To a mixture of compound III (2.04 g, 5 mmol) and ethyl iodide or benzyl chloride (5 mmol) in ethanol (50 mL), sodium acetate trihydrate (0.75 g, 7 mmol) was added. The reaction mixture was heated under reflux for 2 h and then allowed to cool. The product that formed was collected and recrystallized from methanol to give white needles of compound IVa or IVb, respectively.
8-Cyano-7-ethylthio-3,5,9a-trimethyl-4-phenyl-3a,4,9,9a-tetrahydro-1H-pyrazolo[3,4-g]isoquinoline-1-carbothioamide (IVa)
It was synthesized by using ethyl iodide; m.p.: 241–243 °C; Yield: 78%. IR (cm−1): 3448, 3251, 3131 (NH2); 3030 (C-H, sp2); 2967, 2910 (C-H, sp3); 2227 (C≡N); 1640 (C=N); 1587 (C=C). 1H NMR: δ 7.31–7.35 (m, 3H, Ar-H); 6.97–6.99 (d, J = 8 Hz, 2H, Ar-H); 6.74 (s, 1H, NH); 5.85 (s, 1H, NH); 5.33–5.37 (d, J = 8 Hz, 1H, CH); 4.63 (s, 1H, CH); 3.94 (s, 1H, CH); 3.28–3.32 (q, J = 8 Hz, 2H, SCH2); 2.51 (s, 3H, CH3); 2.35–2.39 (d, J = 16 Hz, 1H, CH); 2.08 (s, 3H, CH3); 1.85 (s, 3H, CH3); 1.41–1.43 (t, J = 8 Hz, 3H, SCH2CH3). Anal. Calcd. For C23H25N5S2 (435.16): C, 63.42; H, 5.78; N, 16.08%. Found: C, 63.21; H, 6.02; N, 16.31%.
7-Benzylthio-8-cyano-3,5,9a-trimethyl-4-phenyl-3a,4,9,9a-tetrahydro-1H-pyrazolo[3,4-g]isoquinoline-1-carbothioamide (IVb)
It was synthesized by using benzyl chloride; m.p.: 238–240 °C; Yield: 91%. IR (cm−1): 3437, 3253, 3134 (NH2); 3025 (C-H, sp2); 2960, 2904 (C-H, sp3); 2222 (C≡N); 1592 (C=C). 1H NMR: δ 7.45–7.47 (m, 2H, Ar-H); 7.28–7.34 (m, 6H, Ar-H); 6.96–6.98 (d, J = 8 Hz, 2H, Ar-H); 6.75 (broad s, 1H, NH); 5.83 (broad s, 1H, NH); 5.34–5.38 (d, J = 16 Hz, 1H, CH); 4.64 (s, 1H, CH); 3.93 (s, 1H, CH); 4.48–4.57 (q, J = 12 Hz, 2H, SCH2); 2.54 (s, 3H, CH3); 2.34–2.38 (d, J = 16 Hz, 1H, CH); 2.08 (s, 3H, CH3); 1.85 (s, 3H, CH3). 13C NMR: δ 176.08, 160.92, 158.63, 154.77, 150.61, 137.25, 137.19, 129.28, 128.51, 127.55, 127.32, 126.63, 126.32, 114.19, 105.70, 77.41, 77.09, 76.77, 69.00, 64.77, 40.46, 34.70, 28.38, 22.03, 14.26 ppm. Anal. Calcd. For C28H27N5S2 (497.17): C, 67.57; H, 5.47; N, 14.07%. Found: C, 67.53; H, 5.43; N, 13.86%.
3.2. Single Crystal X-ray Diffraction
3.2.1. Crystal Structure of IVa
The molecule adopts a “pincer”-type conformation (Figure 2), with the C4···C7 axis forming the hinge. A puckering analysis of the C5/C6/C13/N4/N3 ring gave the parameters Q(2) = 0.0866(8) Å and φ(2) = 131.4(5)°, with the conformation described as a twist on C6—C5. For the C4···C8 ring, the puckering parameters are Q = 0.6388(8) Å, θ = 94.60(7)°, and φ = 46.34(7)°. The dihedral angle between the mean planes of the C15···C20 and C1/C2/C3/C8/C9/N1 rings is 78.14(2)°, while the ethylthio substituent is nearly coplanar with the latter ring from the dihedral angle between its mean plane and that defined by C1/S1/C22/C23 of 2.40(6)°. There also appears to be a weak interaction between H4A and S2 (Table 1). In the crystal, inversion dimers are formed by N5—H5A···S2 hydrogen bonds (Table 1), and these are connected into corrugated ribbons extending along the b-axis direction and approximately parallel to (101) by C7—H7···N2 hydrogen bonds (Table 1 and Figure 3). The ribbons are linked together by inversion-related C22—H22A···Cg4 interactions (Table 1 and Figure 4).
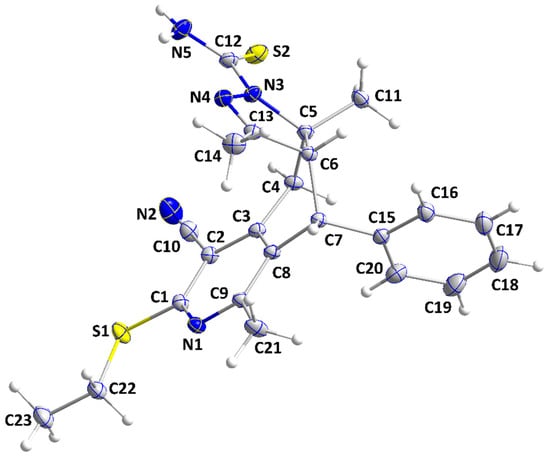
Figure 2.
Perspective view of IVa with labeling scheme and 50% probability ellipsoids.

Table 1.
Hydrogen bond geometry (Å, °) for IVa.
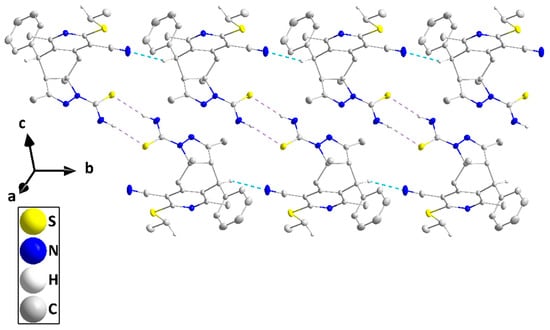
Figure 3.
View of a portion of one ribbon of IVa projected onto (101). C—H···N and N—H···S hydrogen bonds are depicted, respectively, by light purple and light blue dashed lines, while non-interacting hydrogen atoms are omitted for clarity.
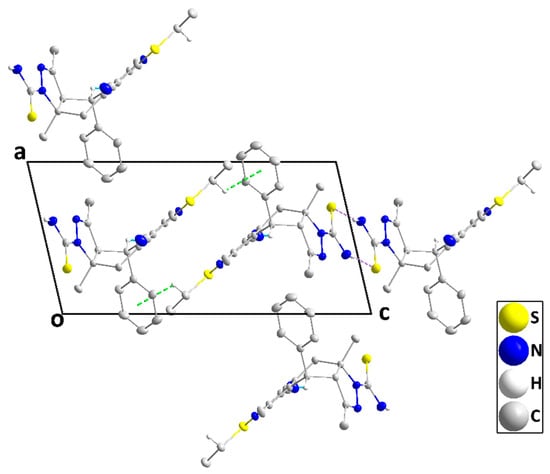
Figure 4.
Packing of IVa viewed along the b-axis direction with hydrogen bonds depicted as in Figure 2 and C—H···π(ring) interactions by green dashed lines. Non-interacting hydrogen atoms are omitted for clarity.
3.2.2. Crystal Structure of IVb
The molecule adopts a “pincer” conformation with the C3···C6 axis as the hinge (Figure 5). The orientation of the carbothioamide group, approximately in the plane of the N2/N3/C5/C4/C17 ring (N2—N3—C19—S2 torsion angle = −177.79(9)°), is partly determined by the intramolecular C6—H6B···S2 and C20—H20C···S2 interactions (Table 2 and Figure 5). A puckering analysis [21] of the C2···C7 ring gave the parameters Q = 0.6743(13) Å, θ = 86.80(10)°, and φ = 64.82(11)°. The mean planes of the N2/N3/C5/C4/C17 and the C11···C16 rings are inclined to that of the N1/C1/C2/C7/C8/C9 ring by 69.03(4) and 83.78(5)°, respectively, while the dihedral angle between the mean planes of the N1/C1/C2/C7/C8/C9 and C23···C28 rings is 63.13(5)°. However, the remainder of the benzylthio substituent is close to being coplanar with the N1/C1/C2/C7/C8/C9 ring (N1—C9—S1—C22 torsion angle is −5.53(12)°). In the crystal, inversion dimers are created by paired N4—H4A···S2 hydrogen bonds (Table 2) and are connected by weak C18—H18B···Cg5 interactions (Cg5 is the centroid of the C23···C28 ring; H18B···Cg5 = 3.03 Å, C18—H18B···Cg5 = 121°) into zigzag chains parallel to the ac plane and extending along its principal diagonal (Figure 6). The chains are packed with normal van der Waals interactions (Figure 7).
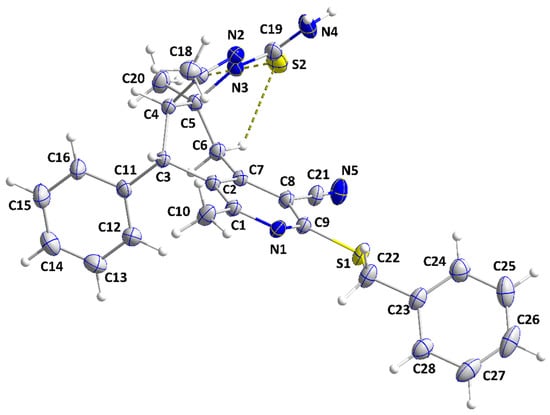
Figure 5.
Perspective view of IVb with labeling scheme and 50% probability ellipsoids. The C—H···S interactions are depicted by dashed lines.
Table 2.
Hydrogen bond geometry (Å, °) for IVb.
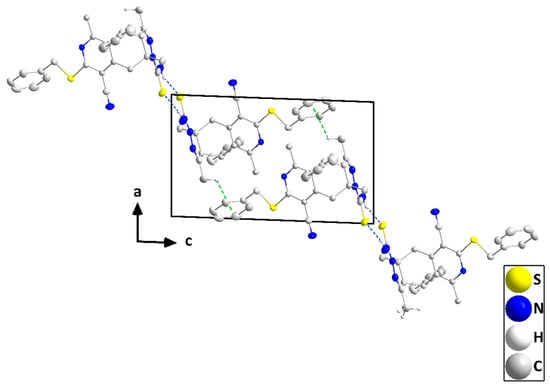
Figure 6.
A portion of one chain viewed along the b-axis direction with N—H···S hydrogen bonds and C—H···π(ring) interactions depicted, respectively, by blue and green dashed lines. Non-interacting hydrogen atoms are omitted for clarity.
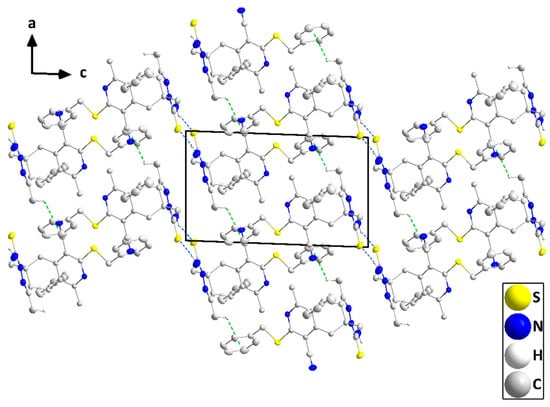
Figure 7.
Packing viewed along the b-axis direction with intermolecular interactions depicted in Figure 5. Non-interacting hydrogen atoms are omitted for clarity.
3.3. DFT Calculations
3.3.1. Optimized Geometries and Thermal Stabilities
The optimized geometries of IVa and IVb were obtained with the B3LYP/6-31+G(d,p) functional and are depicted in Figure 8. There is no imaginary frequency associated with either structure, which indicates these to be true minima on their potential energy surface (PES), and no transition or intermediate (saddle point) state was found. After having stable ground state structures, we further analyzed the reactivity of compounds. The calculated total energies are −1961.714 and −2153.46 Hartree, respectively, for IVa and IVb (Table 3). Furthermore, the dipole moments are significant and are 10.152 and 9.92 Debye, respectively, for IVa and IVb, indicating considerable asymmetric charge distribution and extent of polarity. Compound IVa has a slightly higher dipole moment as compared to compound IVb, which may be attributed to greater charge transfer and separation of charges within the structure. The higher dipole moment values also indicate the better electronic properties and reactivity of compounds IVa and IVb.
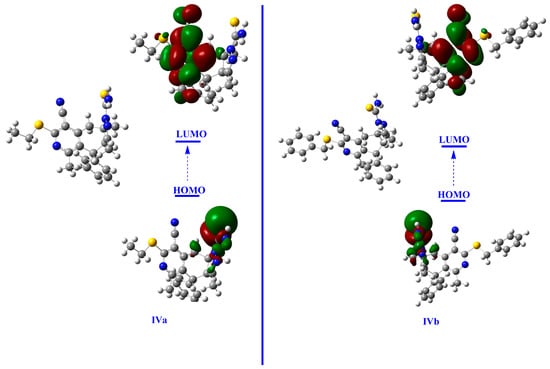
Figure 8.
The plotted HOMO and LUMO density of IVa and IVb at B3LYP/6-31+G(d,p) (iso-value 0.02).
Table 3.
Molecular symmetry, total electronic energy (in Hartree), dipole moment (µo Debye), Energies of HOMO and LUMO (in eV), HOMO-LUMO gaps (EH-L in eV), NBO charges (QX in |e|), ionization potential (IP in eV), electron affinity (EA in eV), chemical hardness (Ƞ in eV), chemical potential (µ in eV), of electronegativity (χ in eV), electrophilicity index (ω in eV), polarizability (αo in au), isotropic average polarizability volume (αv in Å3), hyperpolarizability (βo in au), and projection of hyperpolarizability on dipole moment vector (βvec in au) for IVa and IVb.
3.3.2. FMO and NBO Charge Analysis
The frontier molecular orbital (FMO) study includes the energies of the highest occupied molecular orbitals (HOMO) and the lowest unoccupied molecular orbitals (LUMO) and the HOMO-LUMO gaps (EH-L). The HOMO energy of IVa is −5.64 eV, while that of IVb is −5.63 eV. The LUMO energies of IVa and IVb are, respectively, −1.78 and −1.80 eV. The occupied orbitals show nucleophilic behavior. The HOMO-LUMO gaps (EH-L) are, respectively, 3.86 and 3.83 eV for IVa and IVb (Table 3). The small HOMO-LUMO gaps indicate the soft nature and excellent reactivity of the studied compounds. The relatively low values of the HOMO-LUMO energy gaps show that both molecules have high chemical reactivity, biological activity, and polarizability. Both compounds have comparable HOMO-LUMO gaps, and their small values suggest a facile transfer of electrons from HOMO to LUMO, leading to enhanced optical properties. Electronic density plots of the HOMOs and LUMOs are shown in Figure 8. The HOMO density lies near the nitrogen and S-atoms of the ring, while the LUMO density is spread over the central part of compounds.
3.3.3. Mulliken and NBO Charge Analysis
The Mulliken atomic charges of the optimized structure were calculated by the B3LYP/6-31+G(d,p) method and are given in Table 4. In IVa, the majority of the Mulliken atomic charges on carbon are negative and range from −0.08 to −0.83 while positive atomic charges on carbon range from +0.02 to +0.62. Hydrogens displayed a partial positive Mulliken charge, while the nitrogen atom in the six-membered ring attains a negative Mulliken charge and acts as a donor of electrons, while carbon adjacent to nitrogen acts as an acceptor. Interestingly, the two adjacent nitrogen atoms in the five-membered ring attain positive Mulliken charges, while the carbon atom has a negative charge (acting as a donor of electrons). Likewise, in IVa, the sulfur atom has a positive Mulliken charge (+0.21) while the adjacent nitrogen has a negative charge. The smaller size of carbon as compared with sulfur makes it a donor, while the S-atom acts as an acceptor. For IVb, hydrogen has a positive Mulliken charge and acts as an acceptor of electrons. Also, the majority of negative charges appear on carbon atoms, with positive charges occurring where the carbon atom is attached to an electronegative atom (nitrogen). The Mulliken charges on nitrogen are negative, and hence nitrogen acts as the donor part of the molecule.
Table 4.
Mulliken population analysis of compounds IVa and IVb.
Natural bonding orbital (NBO) analyses can be used to predict charge separation and transfer of charge within a molecule and computed NBO charges are given in Table 3. All hydrogen atoms have a net partial positive NBO charge. The majority of carbon atom bears negative NBO charges. The charge is transferred form hydrogen and carbon to electronegative nitrogen atom. After having more negative NBO charges on the nitrogen atoms, they act as donor of electron (electron-rich site) for further chemical reaction. All the nitrogen atoms carry negative NBO charges while the carbons attached to nitrogen and sulfur atoms have negative charges. The charges on the nitrogen atom range from −0.26 to −0.85 |e| while the charge in S-atom varies from positive (S-atom between two carbons) to negative at the terminal position of the ring thus, nitrogen act as an electron donor. The highest charge (−0.85 |e|) is on the nitrogen atom of IVa and IVb. The charges on the carbon atoms exhibited either positive or negative values. For IVb, the hydrogen atoms have positive NBO charges while the majority of carbon atoms have negative NBO charges which range from −0.069 to −0.75 |e|. The most negative NBO charge for carbon is on the methyl carbon (CH3) while the least negative occurs within the benzene ring.
3.3.4. Global Reactivity Paramters
The global reactivity parameters, ionization potential (IP), electron affinity (EA), chemical hardness (η), chemical potential (µ), electronegativity (χ), and electrophilicity index (ω) are theoretically calculated and are crucial to determine the reactivity of molecules. The following equations given below represent their relations;
Ionization potential (IP) = −E (HOMO)
Electron affinity (EA) = −E (LUMO)
Chemical Hardness (η) = ½ (IP − EA)
Chemical potential (µ) = −
Electronegativity (χ) = −µ
Electrophilicity index (ω) =
With the results presented in Table 3.
The ionization potential (IP) and electron affinity (EA) are computed using Koopman’s approximations, with IP representing the negative of the HOMO energy and EA representing the negative of the LUMO energy. The substantial IP values suggest good electronic stability for IVa and IVb, while the electron affinity (EA) values indicate their electron-gaining tendency. The chemical hardness values are comparable at 1.93 and 1.91 eV, which suggest a soft nature and high reactivity. The global chemical potential (µ) describes the charge transfer within a system in the ground state, with chemical reactivity increasing with increasing μ. Both compounds have the same (highly negative) chemical potential value (−3.71 eV), and thus the same reactivity can be expected. The electrophilicity index (ω) measures the ability of an electrophile to acquire an extra amount of electron density. Mathematically, it is directly proportional to the square of chemical potential and inversely related to chemical hardness. Thus, for a good electrophile, the electrophilicity index should be high. The electrophilicity (ω) index values are 3.56 and 3.60 eV for IVa and IVb suggesting their electrophilic nature. The electrophilic nature of these compounds indicates their applications to studying organic reactions. The observed slightly larger value of ω for IVb may be attributed to its larger size compared to IVa.
3.3.5. Density of States (DOS) and MESP Analysis
Density of states (DOS) spectra were also plotted over the energy range of −20 to 20 eV and show the newly generated states per unit energy for IVa and IVb (Figure 9). The green lines show the occupied molecular orbitals, while the red lines indicate unoccupied orbitals. The narrow HOMO-LUMO gaps indicate that charge transfer from a particular HOMO to LUMO takes place easily. Furthermore, the newly born energy states can be seen for HOMO (green lines) and LUMO (red lines) in the TDOS spectra. The blue curve on the spectra shows the intensity of peaks, which is higher for LUMO.
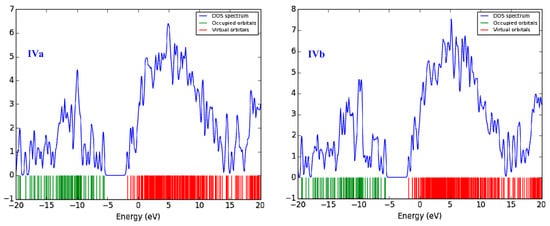
Figure 9.
Total density o states (TDOS) spectra at B3LYP of compounds IVa and IVb.
The molecular electrostatic potential (MEP) surface has frequently been used to generate the electrostatic potential (ESP) using a constant electron density for any chemical species. Three-dimensional MESP maps were generated for visualization of the reactive sites for IVa and IVb and are given in Figure 10. The red color represents an electron-rich region where an incoming electrophile can bind, while the blue color represents an electron-poor zone and a favorable place for nucleophilic attack. In IVa, the electron-rich regions lie near the nitrogen atoms of the tetrahydroisoquinoline moiety and the five-membered ring (Figure 10). For IVb, the red color also appears around the N-atom of the tetrahydroisoquinoline moiety, showing this to be the high electron density region (Figure 10).
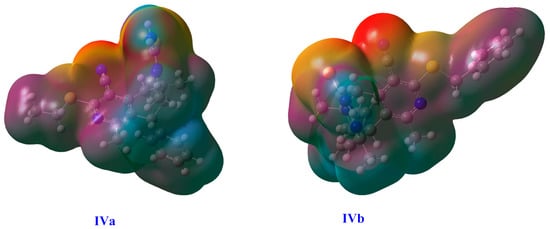
Figure 10.
The representation of 3D MESP surfaces plotted at the B3LYP/6-31+g(d,p) level.
3.3.6. Optical and Nonlinear Optical (NLO) Properties
Nonlinear optical (NLO) materials have diverse applications in interdisciplinary science, specifically in optics. Possible applications of NLO materials include laser-based endoscopy, data storage devices, and optical switching. Organic NLO materials are preferred over inorganic materials due to their ultrafast response and ease of fabrication. Also, the presence of π-conjugation networks in organic molecules further enhances the NLO properties and makes them fit for practical application. Large hyperpolarizabilities, in general, are the result of a suitable combination of various factors for a given molecular structure, such as delocalization length, donor-acceptor groups, dimensionality, and orientation. Organic moieties with delocalized pi-electron distribution have been extensively studied for their potential applications in optical signal processing, optical switching, and optical power limiting, all of which demand large and fast NLO responses. Because the effects occur primarily through electronic polarization, the NLO response of many organic materials is extremely rapid, and thus there has been a focus on the NLO properties of pi-conjugated systems. To investigate the NLO properties, the polarizability (αo), hyperpolarizability (βo), and the vector part of the hyperpolarizability (βvec) were calculated by the same DFT method.
The nonlinear optical parameters are associated with Taylor expansion when the studied system is subjected to an electric field [37].
The polarizability (αo), static hyperpolarizability (βo), and vector part of hyperpolarizability (βvec) are given by the following equations [38,39,40]:
The calculated polarizability (αo) values are 288.93 and 415.53 au for IVa and IVb, respectively, with the higher αo values indicating the asymmetric charge distribution and extent of polarization in the studied compounds. The higher value of αo also indicates the linear response to applied electric field. This characterizes the optical properties of molecules and compounds. Similarly, the calculated hyperpolarizabilities (βo) are 415.83 and 1000.23 au for IVa and IVb, respectively, showing that IVb has a significant hyperpolarizability response. The βo is a second order NLO property and shows the nonlinear change in the properties of light (electric field) after interaction with the molecule. The higher the βo value, the stronger the optical and NLO responses. The large NLO response of IVb may be attributed to higher π-conjugation, which is caused by the presence of an extra benzene ring. Thus, IVa and IVb have excellent electronic and NLO properties and can be studied for further applications based on their structural reactivity [37,38,39,40].
3.4. Molecular Docking Study
The molecular docking simulation generates various alternative poses, from which we must select the optimum one based on glide energy and docking score. Each ligand molecule was made to interact with the protein at its native ligand site. Table 5 displays the ligand molecules and their respective docking scores and glide energies. The binding scores of different drugs show good binding affinity for the protein structure. The glide energies of IVa and IVb are much more negative compared to the native ligand. Compared to the native ligand, compounds IVa and IVb have much higher negative glide energies. This suggests that they form stronger interactions with the protein structure.
Table 5.
Docking score and glide energy of different small molecules.
Figure 11 represents the best-docked pose and its corresponding ligand interaction plot. Figure 11a,b display the interaction of IVa with the protein structure and its interaction plot. GLY 422 forms hydrogen bonds with the C≡N group of IVa. ARG 419 and ARG 601 are positively charged, and GLH 597 is negatively charged, while all the other residues in the protein structure are hydrophobically interacting with the ligand. Similarly, TRP A:683 forms π-π stacking with the aromatic part of IVb, while TYR A:711 forms a hydrogen bond with the amine group. Neither IVa nor IVb interacted with the native ligand molecule M16. The native ligand molecule M16 present in the protein structure was extracted separately and made to interact at the same ligand site as mentioned before. Here, the hydrophobic ligands TRP 414 and PHE 589 form π-π stacking with the aromatic ring of the ligand, and TRP 683 forms hydrogen bonds and interacts weakly with the -NH2 group. This M16 molecule mostly interacts hydrophobically with the residues present in the protein.
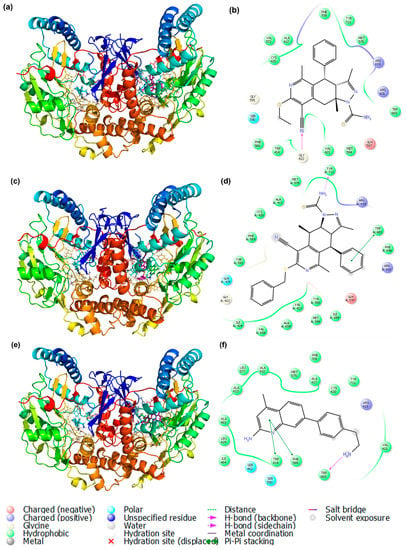
Figure 11.
Best docked poses of the protein-ligand complex and its corresponding ligand plot (a,b) with IVa, (c,d) with IVb, and (e,f) with M16 (native ligand).
This study concludes that all the ligand molecules have good binding affinity at the molecular level, and all three molecules have more hydrophobic contacts in the protein environment. Also, the native ligand M16 does not contribute to any weak or strong interactions in all three small molecules when subjected to the system.
3.5. ADMET Analysis
From Table 6, the ellipse formed by the intersection of the lipophilicity and polarity profiles indicates that Iva is moderately absorbed and IVb is poorly absorbed in the human gut. The ellipse created by the limits of WlogP between 3.0 and 5.0 and TPSA less than 142 indicates that IVa and IVb have a role in PgB. Both thus demonstrate drug-like qualities with low absorption, high polarity, and high lipophilicity, as well as excellent oral bioavailability. In addition, Table S2 indicates both compounds inhibit CY450 enzymes except for 1A2 and 2D6, so this might cause their plasma concentrations to rise and impede the elimination process. Moreover, neither molecule damages the liver due to metabolic activation since neither is an inhibitor of CYP450 2D6 because they are metabolized by O-demethylation pathways. Also, as shown in Table S2, both compounds are predicted to be in toxicity class IV with estimated LD50 values of 600 mg/kg and an immunotoxic risk with a prediction accuracy of 39%. These findings suggest that both molecules have favorable ADMET characteristics.
Table 6.
Predicted “drug-like” properties and estimated properties of absorption and distribution of 1 and 2.
4. Conclusions
To sum up, two new tetrahydroisoquinoline derivatives have been synthesized and their structures confirmed by single crystal X-ray diffraction. Their structural reactivity and nonlinear optical properties are calculated by DFT studies at the B3LYP/6-31+G(d,p) level. Small HOMO-LUMO gaps (EH-L) indicate that the compounds are soft and reactive. To determine their reactivity and electronic stability, global reactivity descriptors were calculated, and NBO charges exhibit excellent charge and donor-acceptor separation. DOS analysis reveals newly generated energy states and reduced band gaps, resulting in higher conductive properties. For surface reactivity, 3D MESP surfaces were plotted, revealing an electron-rich site near the nitrogen atom of the tetrahydroisoquinoline moiety. Crystal nonlinear optical (NLO) properties were calculated using polarizability and hyperpolarizability values. The αo and βo values of IVb are 415.53 and 1003.44 au. Molecular docking results confirm the binding efficiency of both compounds in the human neuronal nitric oxide synthase complex, with IVb having a better binding score than both the native molecule and IVa. ADMET analysis suggests that both compounds exhibit better ADMET properties.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cryst13081161/s1.
Author Contributions
Validation, writing—review and editing, Y.E.B.; Conceptualization and supervision, S.K.M. and E.A.B.; software, analysis, investigation, and resources A.A. and S.K.; data curation, S.A.; writing—original draft preparation, A.E.A.-R.; Methodology, I.S.M.; writing and data curation, J.T.M.; funding acquisition, R.A.-S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Researchers Supporting Project No. RSP-2023R353, King Saud University, Riyadh, Saudi Arabia.
Data Availability Statement
Data is available from the authors on request.
Acknowledgments
The authors extend their appreciation to the Researchers Supporting Project, King Saud University, Riyadh, Saudi Arabia, for funding this work through grant No. RSP-2023R353.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Manzoor, S.; Trukhanov, S.V.; Ansari, M.N.; Abdullah, M.; Alruwaili, A.; Trukhanov, A.V.; Khandaker, M.U.; Idris, A.M.; El-Nasser, K.S.; Taha, T.A. Flowery Ln2MnSe4 Novel Electrocatalyst Developed via Anion Exchange Strategy for Efficient Water Splitting. Nanomaterials 2022, 12, 2209. [Google Scholar] [CrossRef]
- Hassan, M.; Slimani, Y.; Gondal, M.A.; Mohamed, M.J.S.; Guener, S.; Almessiere, M.A.; Surrati, A.M.; Baykal, A.; Trukhanov, S.; Trukhanov, A. Structural Parameters, Energy States and Magnetic Properties of the Novel Se-Doped NiFe2O4 Ferrites as Highly Efficient Electrocatalysts for HER. Ceram. Int. 2022, 48, 24866–24876. [Google Scholar] [CrossRef]
- Almessiere, M.A.; Slimani, Y.; Güngüneş, H.; Baykal, A.; Trukhanov, S.V.; Trukhanov, A. V Manganese/Yttrium Codoped Strontium Nanohexaferrites: Evaluation of Magnetic Susceptibility and Mossbauer Spectra. Nanomaterials 2018, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Zdorovets, M.V.; Kozlovskiy, A.L.; Shlimas, D.I.; Borgekov, D.B. Phase Transformations in FeCo–Fe2CoO4/Co3O4-Spinel Nanostructures as a Result of Thermal Annealing and Their Practical Application. J. Mater. Sci. Mater. Electron. 2021, 32, 16694–16705. [Google Scholar] [CrossRef]
- Boulanger, C.M.; Heymes, C.; Benessiano, J.; Geske, R.S.; Lévy, B.I.; Vanhoutte, P.M. Neuronal Nitric Oxide Synthase Is Expressed in Rat Vascular Smooth Muscle Cells: Activation by Angiotensin II in Hypertension. Circ. Res. 1998, 83, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Loesch, A.; Milner, P.; Burnstock, G. Endothelin in Perivascular Nerves. An Electronimmunocytochemical Study of Rat Basilar Artery. Neuroreport 1998, 9, 3903–3904. [Google Scholar] [CrossRef]
- Schwarz, P.M.; Kleinert, H.; Förstermann, U. Potential Functional Significance of Brain-Type and Muscle-Type Nitric Oxide Synthase I Expressed in Adventitia and Media of Rat Aorta. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2584–2590. [Google Scholar] [CrossRef]
- Buchwalow, I.B.; Podzuweit, T.; Böcker, W.; Samoilova, V.E.; Thomas, S.; Wellner, M.; Baba, H.A.; Robenek, H.; Schnekenburger, J.; Lerch, M.M. Vascular Smooth Muscle and Nitric Oxide Synthase. FASEB J. 2002, 16, 500–508. [Google Scholar] [CrossRef]
- Bredt, D.S.; Hwang, P.M.; Snyder, S.H. Localization of Nitric Oxide Synthase Indicating a Neural Role for Nitric Oxide. Nature 1990, 347, 768–770. [Google Scholar] [CrossRef]
- Capettini, L.S.A.; Cortes, S.F.; Lemos, V.S. Relative Contribution of ENOS and NNOS to Endothelium-Dependent Vasodilation in the Mouse Aorta. Eur. J. Pharmacol. 2010, 643, 260–266. [Google Scholar] [CrossRef]
- Rabelo, L.A.; Cortes, S.F.; Alvarez-Leite, J.I.; Lemos, V.S. Endothelium Dysfunction in LDL Receptor Knockout Mice: A Role for H2O2. Br. J. Pharmacol. 2003, 138, 1215–1220. [Google Scholar] [CrossRef]
- Fathy, U.; Azzam, M.A.; Mahdy, F.; El-Maghraby, S.; Allam, R.M. Synthesis and in Vitro Anticancer Activity of Some Novel Tetrahydroquinoline Derivatives Bearing Pyrazol and Hydrazide Moiety. J. Heterocycl. Chem. 2020, 57, 2108–2120. [Google Scholar] [CrossRef]
- Kozlovskiy, A.L.; Zdorovets, M. V Effect of Doping of Ce4+/3+ on Optical, Strength and Shielding Properties of (0.5-x) TeO2-0.25 MoO-0.25 Bi2O3-XCeO2 Glasses. Mater. Chem. Phys. 2021, 263, 124444. [Google Scholar] [CrossRef]
- Henaish, A.M.; Hemeda, O.M.; Dorgham, A.M.; Weinstein, I.A.; Soliman, T.S.; Trukhanov, A.V.; Trukhanov, S.V.; Zhou, D.; Darwish, M.A. Structure and Optoelectronic Properties of Ferroelectric PVA-PZT Nanocomposites. Opt. Mater. 2023, 138, 113402. [Google Scholar] [CrossRef]
- Yakovenko, O.S.; Matzui, L.Y.; Vovchenko, L.L.; Trukhanov, A.V.; Kazakevich, I.S.; Trukhanov, S.V.; Prylutskyy, Y.I.; Ritter, U. Magnetic Anisotropy of the Graphite Nanoplatelet–Epoxy and MWCNT–Epoxy Composites with Aligned Barium Ferrite Filler. J. Mater. Sci. 2017, 52, 5345–5358. [Google Scholar] [CrossRef]
- Yakovenko, O.S.; Matzui, L.Y.; Vovchenko, L.L.; Lozitsky, O.V.; Prokopov, O.I.; Lazarenko, O.A.; Zhuravkov, A.V.; Oliynyk, V.V.; Launets, V.L.; Trukhanov, S. V Electrophysical Properties of Epoxy-Based Composites with Graphite Nanoplatelets and Magnetically Aligned Magnetite. Mol. Cryst. Liq. Cryst. 2018, 661, 68–80. [Google Scholar] [CrossRef]
- Trukhanov, S.V.; Kozlenko, D.P.; Trukhanov, A. V High Hydrostatic Pressure Effect on Magnetic State of Anion-Deficient La0. 70Sr0. 30MnOx Perovskite Manganites. J. Magn. Magn. Mater. 2008, 320, e88–e91. [Google Scholar] [CrossRef]
- Kozlovskiy, A.; Egizbek, K.; Zdorovets, M.V.; Ibragimova, M.; Shumskaya, A.; Rogachev, A.A.; Ignatovich, Z.V.; Kadyrzhanov, K. Evaluation of the Efficiency of Detection and Capture of Manganese in Aqueous Solutions of FeCeOx Nanocomposites Doped with Nb2O5. Sensors 2020, 20, 4851. [Google Scholar] [CrossRef]
- Trukhanov, S.V.; Fedotova, V.V.; Trukhanov, A.V.; Szymczak, H.; Botez, C.E. Cation Ordering and Magnetic Properties of Neodymium-Barium Manganites. Technol. Phys. 2008, 53, 49–54. [Google Scholar] [CrossRef]
- Kozlovskiy, A.; Kenzhina, I.; Alyamova, Z.A.; Zdorovets, M. Optical and Structural Properties of AlN Ceramics Irradiated with Heavy Ions. Opt. Mater. 2019, 91, 130–137. [Google Scholar] [CrossRef]
- APEX4 and SAINT, Bruker AXS LLC: Madison, WI, USA, 2021.
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of Silver and Molybdenum Microfocus X-Ray Sources for Single-Crystal Structure Determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09 Package; Gaussian Inc.: Pittsburgh, PA, USA, 2009. [Google Scholar]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP Density Functional Methods for a Large Set of Organic Molecules. J. Chem. Theory Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef]
- Ahsin, A.; Ayub, K. Superalkali-Based Alkalides Li3O@[12-Crown-4]M (Where M= Li, Na, and K) with Remarkable Static and Dynamic NLO Properties; A DFT Study. Mater. Sci. Semicond. Process. 2022, 138, 106254. [Google Scholar] [CrossRef]
- Ahsin, A.; Shah, A.B.; Ayub, K. Germanium-Based Superatom Clusters as Excess Electron Compounds with Significant Static and Dynamic NLO Response; a DFT Study. RSC Adv. 2022, 12, 365–377. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.; Millam, J.; Eppinnett, K.; Hovell, W.L.; Gilliland, R. GaussView, Version 3.09; Semichem Inc.: Shawnee Mission, KS, USA, 2003. [Google Scholar]
- Mostafa, A.E.; Eissa, M.S.; Elsonbaty, A.; Attala, K.; Abdel Salam, R.A.; Hadad, G.M.; Abdelshakour, M.A. Computer-Aided Design of an Eco-Friendly Imprinted Polymer Decorated Sensors Augmented by Self-Validated Ensemble Modeling Designs for the Quantitation of Drotaverine Hydrochloride in Dosage Form and Human Plasma. J. AOAC Int. 2023, qsad049. [Google Scholar] [CrossRef]
- Abkari, A.; Chaabane, I.; Guidara, K. DFT (B3LYP/LanL2DZ and B3LYP/6311G+ (d, p)) Comparative Vibrational Spectroscopic Analysis of Organic–Inorganic Compound Bis (4-Acetylanilinium) Tetrachlorocuprate (II). Phys. E Low-Dimensional Syst. Nanostructures 2016, 81, 136–144. [Google Scholar] [CrossRef]
- Allouche, A. Software News and Updates Gabedit—A Graphical User Interface for Computational Chemistry Softwares. J. Comput. Chem. 2012, 32, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful New Tools for Exploring 3D Structures of Biological Macromolecules for Basic and Applied Research and Education in Fundamental Biology, Biomedicine, Biotechnology, Bioengineering and Energy Sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Sherman, W.; Beard, H.S.; Farid, R. Use of an Induced Fit Receptor Structure in Virtual Screening. Chem. Biol. Drug Des. 2006, 67, 83–84. [Google Scholar] [CrossRef]
- Bakhite, E.A.; Marae, I.S.; Gad, M.A.; Mohamed, S.K.; Mague, J.T.; Abuelhassan, S. Pyridine Derivatives as Insecticides. Part 3. Synthesis, Crystal Structure, and Toxicological Evaluation of Some New Partially Hydrogenated Isoquinolines against Aphis Gossypii (Glover, 1887). J. Agric. Food Chem. 2022, 70, 9637–9644. [Google Scholar] [CrossRef]
- Wang, L.; Wang, W.Y.; Fang, X.Y.; Zhu, C.L.; Qiu, Y.Q. Intramolecular Photo-Induced Electron Transfer in Nonlinear Optical Chromophores: Fullerene (C60) Derivatives. Org. Electron. 2016, 33, 290–299. [Google Scholar] [CrossRef]
- Kleinman, D.A. Nonlinear Dielectric Polarization in Optical Media. Phys. Rev. 1962, 126, 1977. [Google Scholar] [CrossRef]
- Kelley, A.M. Frequency-Dependent First Hyperpolarizabilities from Linear Absorption Spectra. J. Opt. Soc. Am. B 2002, 19, 1890. [Google Scholar] [CrossRef]
- Ahsin, A.; Ali, A.; Ayub, K. Transition Metals Based Metalides TM-Janus-TM (Where TM=Sc–Zn and Janus=F6C6H6); A Theoretical Study of Nonconventional Metalides with Excellent Static and Dynamic Nonlinear Optical Properties. Mater. Sci. Semicond. Process. 2023, 162, 107506. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).