
Structure Determination and Analysis of the Ceramic Material La0.987Ti1.627Nb3.307O13 by Synchrotron and Neutron Powder Diffraction and DFT Calculations



Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Structure Determination and Analysis
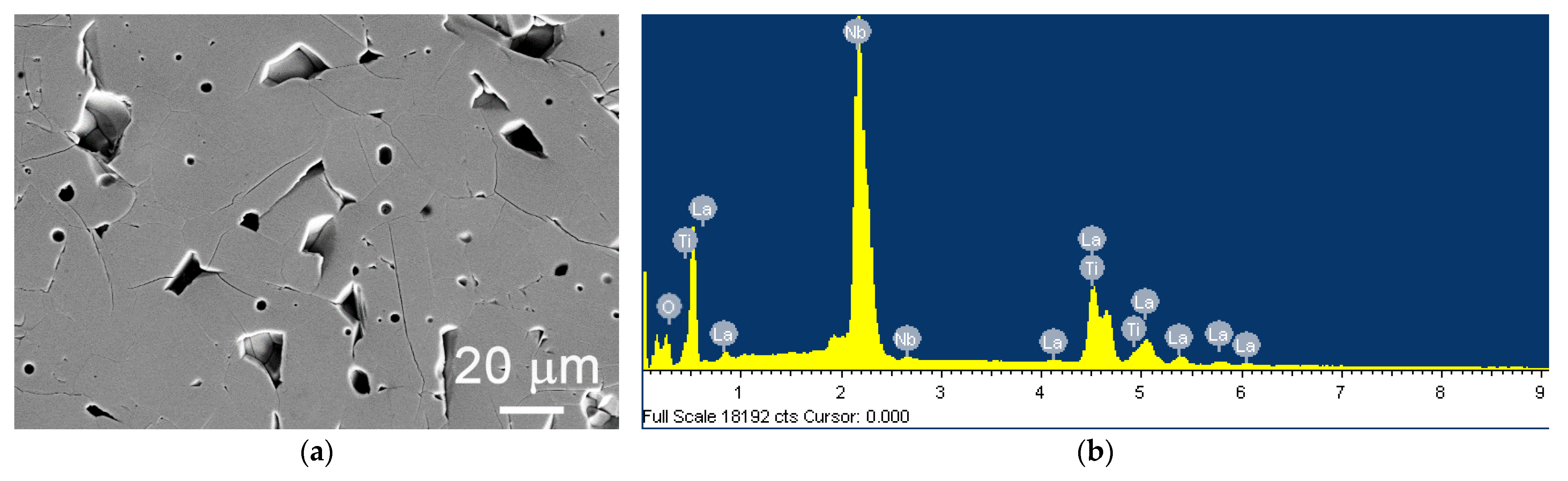
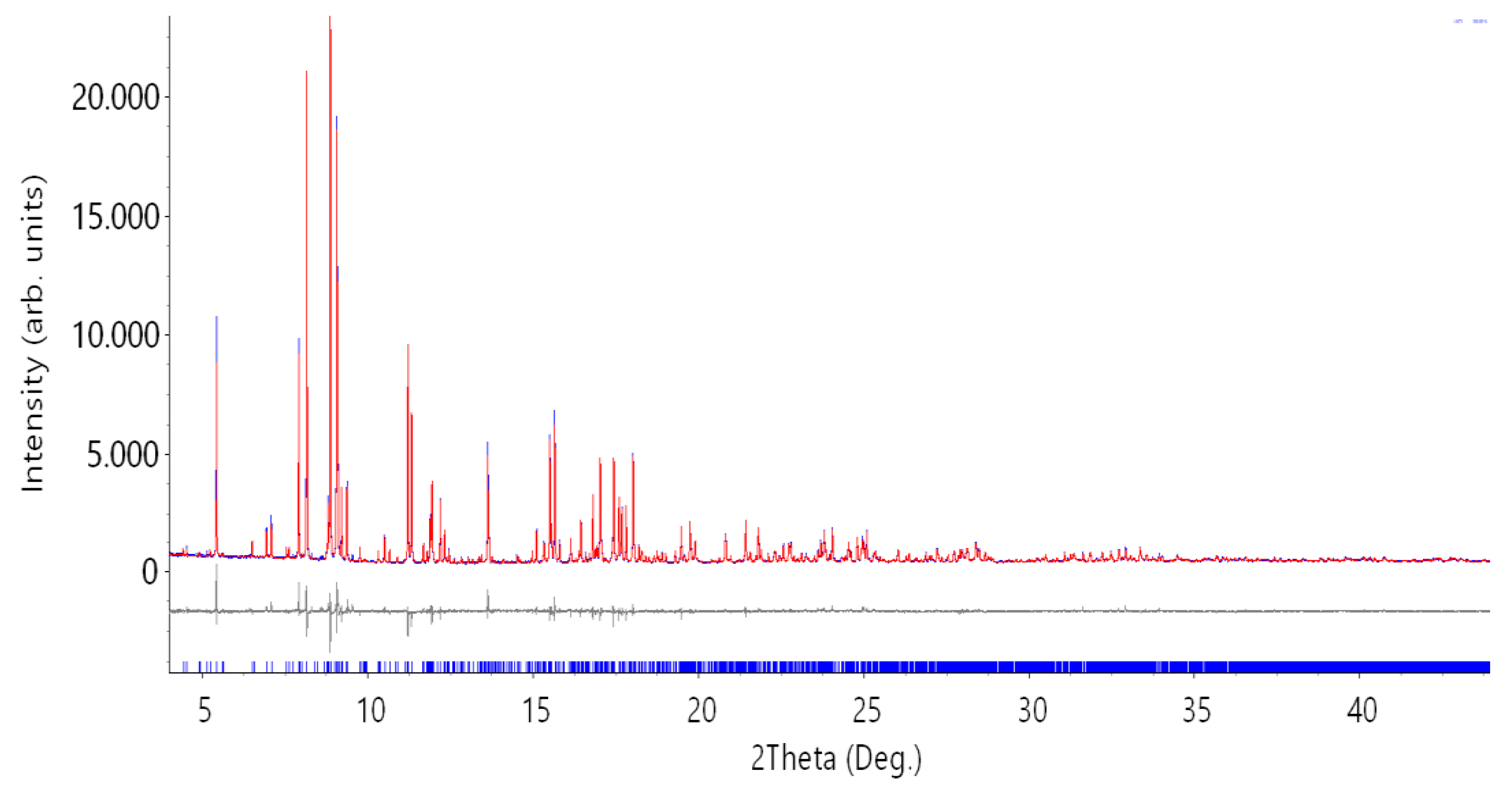
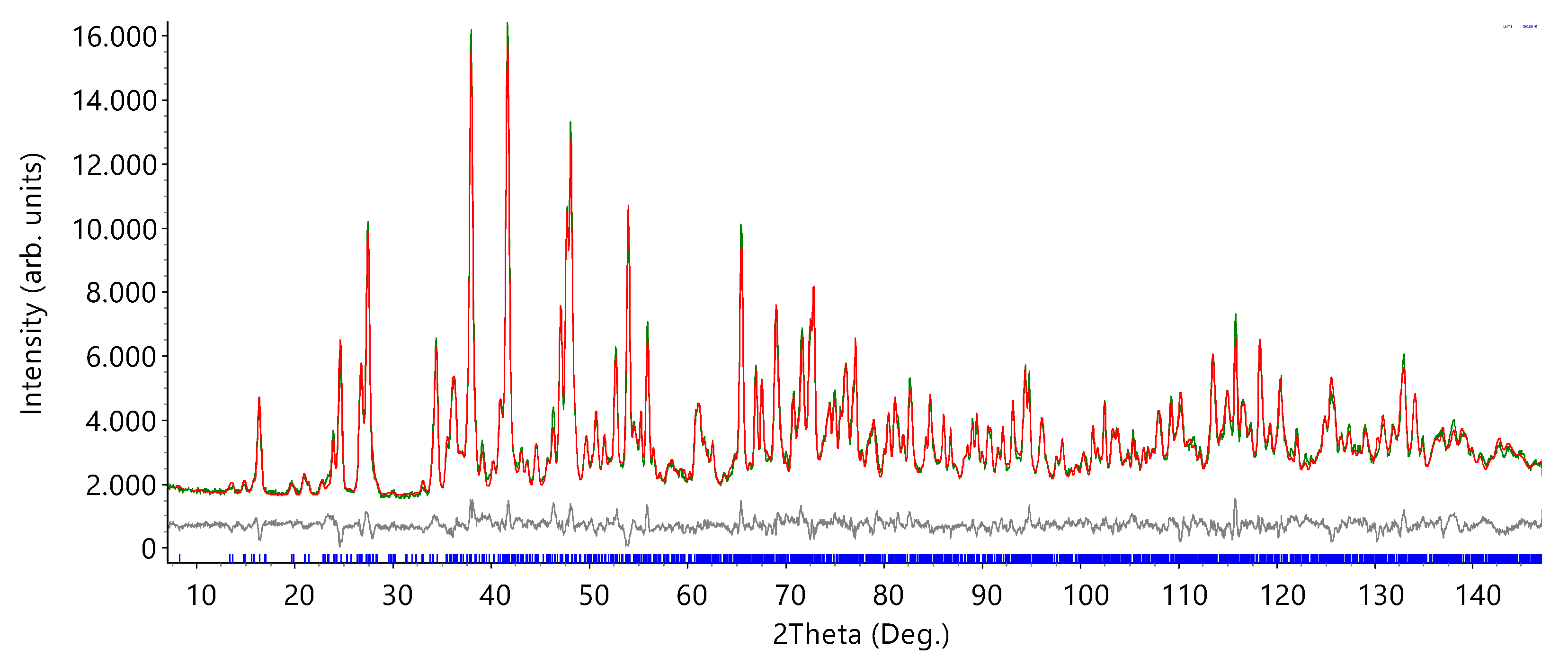
3.1.1. Structure Solution and Refinement
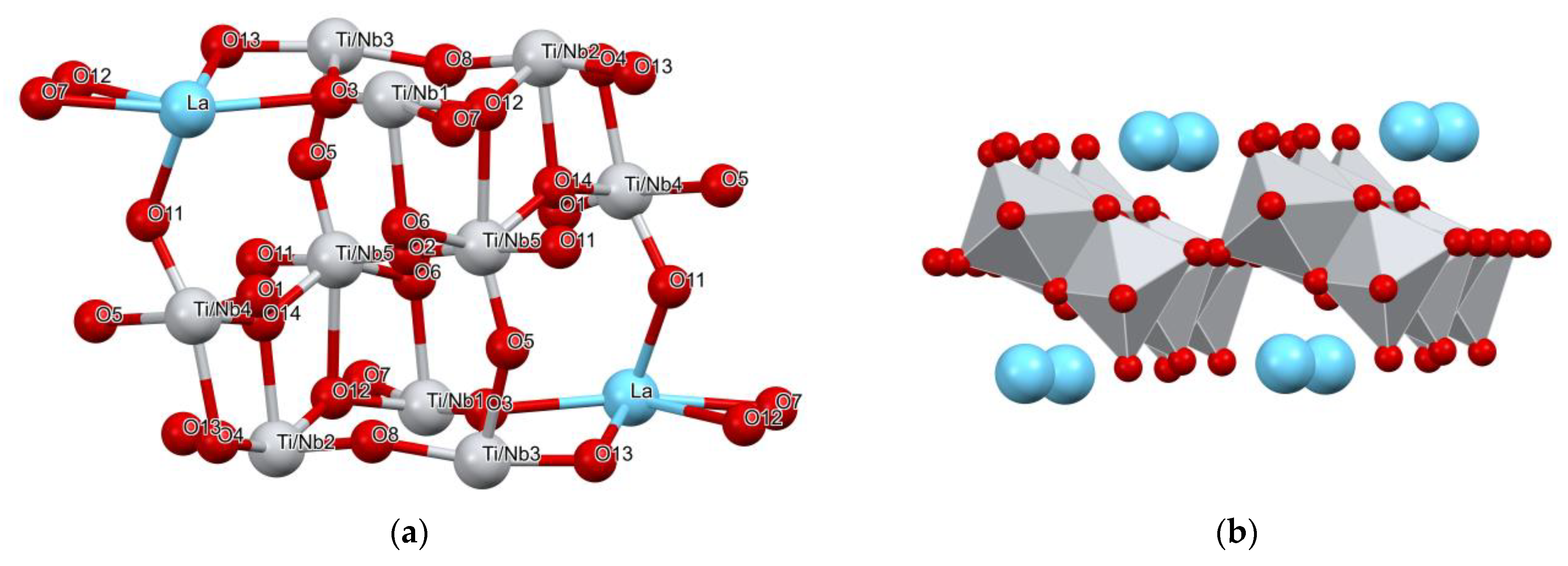
3.1.2. Structure Description
3.2. DFT Calculations
3.2.1. Optimization of Occupational Isomers
3.2.2. Thermodynamic Site Preferences
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Savchenko, E.P.; Godina, N.A.; Keler, É.K. Solid-Phase Reactions between Pentoxides of Niobium and Tantalum and Oxides of the Rare-Earth Elements. In Chemistry of High-Temperature Materials; Springer: Boston, MA, USA, 1969; pp. 108–113. [Google Scholar]
- MacChesney, J.B.; Sauer, H.A. The System La2O3—TiO5; Phase Equilibria and Electrical Properties. J. Am. Ceram. Soc. 1962, 45, 416–422. [Google Scholar] [CrossRef]
- Latie, L.; Villeneuve, G.; Conte, D.; le Flem, G. Ionic Conductivity of Oxides with General Formula LixLn1/3Nb1-XTixO3 (Ln = La, Nd). J. Solid State Chem. 1984, 51, 293–299. [Google Scholar] [CrossRef]
- Nadiri, A.; le Flem, G.; Delmas, C. Lithium Intercalation in Ln1/3NbO3 Perovskite-Type Phases (Ln = La, Nd). J. Solid State Chem. 1988, 73, 338–347. [Google Scholar] [CrossRef]
- Belous, A.; Pashkova, E.; Gavrilenko, O.; V’yunov, O.; Kovalenko, L. Solid Electrolytes Based on Lithium-Containing Lanthanum Metaniobates. J. Eur. Ceram. Soc. 2004, 24, 1301–1304. [Google Scholar] [CrossRef]
- Takahashi, J.; Kageyama, K.; Hayashi, T. Dielectric Properties of Double-Oxide Ceramics in the System Ln2O3-TiO2 (Ln = La, Nd and Sm). Jpn. J. Appl. Phys. 1991, 30, 2354–2358. [Google Scholar] [CrossRef]
- Škapin, S.D.; Kolar, D.; Suvorov, D. Phase Stability and Equilibria in the La2O3-TiO2 System. J. Eur. Ceram. Soc. 2000, 20, 1179–1185. [Google Scholar] [CrossRef]
- Škapin, S.; Kolar, D.; Suvorov, D. X-ray Diffraction and Microstructural Investigation of the Al2O3-La2O3-TiO2 System. J. Am. Ceram. Soc. 1993, 76, 2359–2362. [Google Scholar] [CrossRef]
- Suvorov, D.; Valant, M.; Škapin, S.; Kolar, D. Microwave Dielectric Properties of Ceramics with Compositions along the La2/3TiO3(STAB)-LaAlO3 Tie Line. J. Mater. Sci. 1998, 33, 85–89. [Google Scholar] [CrossRef]
- Škapin, S.D.; Škapin, A.S.; Suvorov, D.; Gaberšček, M. A Stabilization Mechanism for the Perovskite La2/3TiO3 Compound with Fe2O3: A Structural and Electrical Investigation. J. Eur. Ceram. Soc. 2008, 28, 2025–2032. [Google Scholar] [CrossRef]
- Inaguma, Y.; Katsumata, T.; Itoh, M.; Morii, Y. Crystal Structure of a Lithium Ion-Conducting Perovskite La2/3-XLi3xTiO3 (x = 0.05). J. Solid State Chem. 2002, 166, 67–72. [Google Scholar] [CrossRef]
- Golobič, A.; Meden, A.; Spreitzer, M.; Škapin, S.D. Phase Equilibria and Microwave Dielectric Properties in the Ternary La2O3–TiO2–Nb2O5 System. J. Eur. Ceram. Soc. 2021, 41, 7035–7043. [Google Scholar] [CrossRef]
- Yoshioka, H. Structure and Electrical Properties of A-Site-Deficient Perovskite Compounds in the La2/3TiO3-La1/3NbO3 System. J. Am. Ceram. Soc. 2002, 85, 1339–1342. [Google Scholar] [CrossRef]
- Ali, R.; Yashima, M.; Tanaka, M.; Yoshioka, H.; Mori, T.; Sasaki, S. High-Temperature Synchrotron X-Ray Powder Diffraction Study of the Orthorhombic-Tetragonal Phase Transition in La0.63(Ti0.92, Nb0.08)O3. J. Solid State Chem. 2002, 164. [Google Scholar] [CrossRef]
- Yashima, M.; Mori, M.; Kamiyama, T.; Oikawa, K.I.; Hoshikawa, A.; Torii, S.; Saitoh, K.; Tsuda, K. High-Temperature Phase Transition in Lanthanum Titanate Perovskite La0.64(Ti0.92,Nb0.08)O3. Chem. Phys. Lett. 2003, 375, 240–246. [Google Scholar] [CrossRef]
- Zhang, J.; Zuo, R. A Novel Self-Composite Property-Tunable LaTiNbO6 Microwave Dielectric Ceramic. Mater. Res. Bull. 2016, 83, 568–572. [Google Scholar] [CrossRef]
- Bian, J.J.; Li, Y.Z.; Yuan, L.L. Structural Stability and Microwave Dielectric Properties of (1 − x)Ln1/3NbO3-XLn2/3TiO3 (Ln: La, Nd; 0 ≤ x ≤ 0.8). Mater. Chem. Phys. 2009, 116, 102–106. [Google Scholar] [CrossRef]
- Golobič, A.; Škapin, S.D.; Suvorov, D.; Meden, A. Solving Structural Problems of Ceramic Materials. Croat. Chem. Acta 2004, 77, 435–446. [Google Scholar]
- Strahov, V.I.; Meljnikova, O.V.; Dib, M.; Pivovarova, A.P.; Karpinskaja, O.V. LaNb3O9-TiO2-Politermiceskoe Secenie v Sisteme La2O3-TiO2-Nb2O5. Zh. Neorg. Khim. 1996, 41, 1373–1375. [Google Scholar]
- Schouwink, P.; Didelot, E.; Lee, Y.S.; Mazet, T.; Černý, R. Structural and Magnetocaloric Properties of Novel Gadolinium Borohydrides. J. Alloys Compd. 2016, 664, 378–384. [Google Scholar] [CrossRef]
- Werner, P.E.; Eriksson, L.; Westdahl, M. TREOR, a Semi-Exhaustive Trial-and-Error Powder Indexing Program for All Symmetries. J. Appl. Crystallogr. 1985, 18, 367–370. [Google Scholar] [CrossRef]
- Visser, J.W. Fully Automatic Program for Finding the Unit Cell from Powder Data. J. Appl. Crystallogr. 1969, 2, 367–370. [Google Scholar] [CrossRef]
- Favre-Nicolin, V.; Černý, R. FOX, “Free Objects for Crystallography”: A Modular Approach to Ab Initio Structure Determination from Powder Diffraction. J. Appl. Crystallogr. 2002, 35, 734–743. [Google Scholar] [CrossRef]
- Coelho, A.A. TOPAS and TOPAS-Academic: An Optimization Program Integrating Computer Algebra and Crystallographic Objects Written in C++. J. Appl. Crystallogr. 2018, 51, 210–218. [Google Scholar] [CrossRef]
- Dovesi, R.; Orlando, R.; Civalleri, B.; Roetti, C.; Saunders, V.R.; Zicovich-Wilson, C.M. CRYSTAL: A Computational Tool for the Ab Initio Study of the Electronic Properties of Crystals. Z. Krist. 2005, 220, 571–573. [Google Scholar] [CrossRef]
- Bredow, T.; Heitjans, P.; Wilkening, M. Electric Field Gradient Calculations for LixTiS2 and Comparison with 7Li NMR Results. Phys. Rev. B Condens. Matter. Mater. Phys. 2004, 70, 115111–115119. [Google Scholar] [CrossRef]
- Corà, F. The Performance of Hybrid Density Functionals in Solid State Chemistry: The Case of BaTiO3. Mol. Phys. 2005, 103, 2483–2496. [Google Scholar] [CrossRef]
- Scaranto, J.; Giorgianni, S. A Quantum-Mechanical Study of CO Adsorbed on TiO2: A Comparison of the Lewis Acidity of the Rutile (1 1 0) and the Anatase (1 0 1) Surfaces. J. Mol. Struct. THEOCHEM 2008, 858, 72–76. [Google Scholar] [CrossRef]
- Towler, M.D.; Allan, N.L.; Harrison, N.M.; Saunders, V.R.; MacKrodt, W.C.; Aprà, E. Ab Initio Study of MnO and NiO. Phys. Rev. B 1994, 50, 5041–5054. [Google Scholar] [CrossRef]
- Ferrari, A.M.; Pisani, C. An Ab Initio Periodic Study of NiO Supported at the Pd(100) Surface. Part 1: The Perfect Epitaxial Monolayer. J. Phys. Chem. B 2006, 110, 7909–7917. [Google Scholar] [CrossRef]
- CRYSTAL. Available online: https://www.crystal.unito.it/basis_sets.html (accessed on 23 February 2023).
- Dall’Olio, S.; Dovesi, R.; Resta, R. Spontaneous Polarization as a Berry Phase of the Hartree-Fock Wave Function: The Case of KNbO3. Phys. Rev. B Condens. Matter. Mater. Phys. 1997, 56, 10105–10114. [Google Scholar] [CrossRef]
- Nb_986-31(631d)G_dallolio_1996. Available online: https://www.crystal.unito.it/Basis_Sets/niobium.html#Nb_986-31%28631d%29G_dallolio_1996 (accessed on 23 February 2023).
- Yang, J.; Dolg, M. Valence Basis Sets for Lanthanide 4f-in-Core Pseudopotentials Adapted for Crystal Orbital Ab Initio Calculations. Theor. Chem. Acc. 2005, 113, 212–224. [Google Scholar] [CrossRef]
- Towler, M. CRYSTAL Resources Page. Available online: https://vallico.net/mike_towler/crystal.html (accessed on 23 February 2023).
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Stokes, H.T.; Hatch, D.M. FINDSYM: Program for Identifying the Space-Group Symmetry of a Crystal. J. Appl. Crystallogr. 2005, 38, 237–238. [Google Scholar] [CrossRef]
- Fauquier, D.; Gasperin, M. Synthèse de Monocristaux et Étude Structurale de La Nb TiO6. Bull. Soc. franç. Min. Cristallogr. 1970, 93, 258–259. [Google Scholar] [CrossRef]
- Fischer, P.; Frey, G.; Koch, M.; Konnecke, M.; Pomjakushin, V.; Schefer, J.; Thut, R.; Schlumpf, N.; Burge, R.; Greuter, U.; et al. High-Resolution Powder Diffractometer HRPT for Thermal Neutrons at SINQ. Physica B Condens. Matter 2000, 276–278, 146–147. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Synchrotron Data | Neutron Data |
|---|---|---|
| Space group | ||
| a [Å] | 7.3321 (3) | 7.331 (2) |
| b [Å] | 7.4214 (4) | 7.418 (3) |
| c [Å] | 10.6730 (5) | 10.672 (4) |
| α [°] | 84.153 (4) | 84.14 (2) |
| β [°] | 80.159 (5) | 80.16 (3) |
| γ [°] | 60.367 (5) | 60.37 (3) |
| V [Å3] | 497.28 (5) 1 | 496.9 (4) 1 |
| Temperature | Ambient 2 | Ambient 2 |
| Wavelength [Å] | 0.49998 | 1.494 |
| Used 2θmin, 2θmax [°] | 3.999, 44.001 | 7.000, 147.000 |
| Used dmax, dmin [Å] | 7.165, 0.667 | 12.236, 0.779 |
| No. of profile points | 13335 | 2801 |
| No. of reflections | 3615 | 2220 |
| No. of profile parameters | 25 3 | 24 3 |
| No. of structural parameters | 68 | 68 |
| No. of restraints (occ.) | 5 | 5 |
| Rexp [%] | 2.271 | 1.717 |
| Rp [%] | 3.980 | 3.686 |
| Rwp [%] | 4.969 | 4.663 |
| Label | x | y | z | Occ. | Biso |
|---|---|---|---|---|---|
| La | 0.7995 (4) | 0.7550 (2) | 0.78768 (10) | 0.987 | 0.26 (2) |
| Ti1 | 0.2122 (6) | 0.7407 (4) | 0.1734 (2) | 0.458 | 0.107 (12) |
| Nb1 | 0.2122 (6) | 0.7407 (4) | 0.1734 (2) | 0.542 | 0.107 (12) |
| Ti2 | 0.2911 (5) | 0.2594 (3) | 0.82544 (17) | 0.173 | 0.107 (12) |
| Nb2 | 0.2911 (5) | 0.2594 (3) | 0.82544 (17) | 0.827 | 0.107 (12) |
| Ti3 | 0.7093 (5) | 0.2402 (3) | 0.1775 (4) | 0.103 | 0.107 (12) |
| Nb3 | 0.7093 (5) | 0.2402 (3) | 0.1775 (4) | 0.897 | 0.107 (12) |
| Ti4 | 0.1090 (7) | 0.2489 (4) | 0.5639 (2) | 0.585 | 0.107 (12) |
| Nb4 | 0.1090 (7) | 0.2489 (4) | 0.5639 (2) | 0.349 | 0.107 (12) |
| Ti5 | 0.3692 (5) | 0.7842 (3) | 0.45570 (18) | 0.308 | 0.107 (12) |
| Nb5 | 0.3692 (5) | 0.7842 (3) | 0.45570 (18) | 0.692 | 0.107 (12) |
| O1 | 0.0 | 0.5 | 0.5 | 1.0 | 0.77 (2) |
| O2 | 0.5 | 0.5 | 0.5 | 1.0 | 0.77 (2) |
| O3 | 0.4030 (10) | 0.4478 (10) | 0.2108 (6) | 1.0 | 0.77 (2) |
| O4 | 0.0223 (13) | 0.3255 (7) | 0.7700 (4) | 1.0 | 0.77 (2) |
| O5 | 0.2933 (12) | 0.8649 (7) | 0.6191 (4) | 1.0 | 0.77 (2) |
| O6 | 0.1443 (17) | 0.7719 (8) | 0.3890 (4) | 1.0 | 0.77 (2) |
| O7 | 0.0117 (10) | 0.0390 (10) | 0.1744 (5) | 1.0 | 0.77 (2) |
| O8 | 0.2104 (10) | 0.5593 (10) | 0.7863 (6) | 1.0 | 0.77 (2) |
| O9 | 0.7311 (16) | 0.2901 (7) | 0.9985 (5) | 1.0 | 0.77 (2) |
| O10 | 0.2333 (16) | 0.2847 (8) | 0.9891 (4) | 1.0 | 0.77 (2) |
| O11 | 0.7829 (11) | 0.9077 (6) | 0.5820 (4) | 1.0 | 0.77 (2) |
| O12 | 0.5924 (11) | 0.1835 (7) | 0.7838 (4) | 1.0 | 0.77 (2) |
| O13 | 0.3885 (10) | 0.9608 (10) | 0.8278 (6) | 1.0 | 0.77 (2) |
| O14 | 0.3637 (16) | 0.2213 (8) | 0.6106 (4) | 1.0 | 0.77 (2) |
| Functional/Pseudopotentials | Opt? | Relative Energies of Isomers [kcal/mol] | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DFT | PP La | PP Nb | 11222 | 12122 | 12212 | 12221 | 21122 | 21212 | 21221 | 22112 | 22121 | 22211 | |
| B3LYP | SDD | HAY | Yes | 5.95 | 18.65 | −13.30 | −4.37 | 15.22 | −9.33 | −3.62 | −6.89 | 0.27 | −2.72 |
| B3LYP | HAY | HAY | Yes | 6.55 | 17.70 | −13.54 | −5.05 | 15.95 | −8.42 | −3.03 | −7.03 | 0.17 | −2.89 |
| B3LYP | SDD | None | Yes | 6.61 | 20.72 | −12.87 | −4.38 | 17.01 | −9.88 | −3.26 | −7.04 | 1.59 | −3.41 |
| B3LYP | HAY | None | Yes | 6.36 | 20.27 | −13.67 | −5.01 | 16.94 | −10.49 | −3.68 | −7.81 | 1.00 | −4.05 |
| B3LYP | None | None | Yes | 6.31 | 20.64 | −13.66 | −5.30 | 17.87 | −10.29 | −3.82 | −7.17 | 1.16 | −3.49 |
| PBE | SDD | HAY | Yes | 5.85 | 16.84 | −11.77 | −3.83 | 13.72 | −8.38 | −3.42 | −6.43 | 0.31 | −1.81 |
| BLYP | SDD | HAY | Yes | 6.90 | 17.30 | −12.02 | −4.35 | 14.84 | −8.79 | −3.55 | −6.60 | 0.09 | −2.79 |
| B3LYP | SDD | HAY | No | 22.74 | 27.81 | −31.10 | −0.71 | 31.03 | −22.90 | 5.78 | −25.20 | 7.02 | −12.61 |
| Site Isomer | Titanium on Sites | Statistical Occupancy Index (exp.) | Preference Order (exp.) | Relative Energy [kcal/mol] | Preference Order (calc.) |
|---|---|---|---|---|---|
| 11222 | 1 and 2 | 0.079 | 7. | 5.95 | 8. |
| 12122 | 1 and 3 | 0.103 | 5. | 18.65 | 10. |
| 12212 | 1 and 4 | 0.268 | 1. | −13.30 | 1. |
| 12221 | 1 and 5 | 0.141 | 3. | −4.37 | 4. |
| 21122 | 2 and 3 | 0.018 | 10. | 15.22 | 9. |
| 21212 | 2 and 4 | 0.101 | 6. | −9.33 | 2. |
| 21221 | 2 and 5 | 0.053 | 9. | −3.62 | 5. |
| 22112 | 3 and 4 | 0.060 | 8. | −6.89 | 3. |
| 22121 | 3 and 5 | 0.032 | 9. | 0.27 | 7. |
| 22211 | 4 and 5 | 0.180 | 2. | −2.72 | 6. |
| 11222 | 1 and 2 | 0.079 | 7. | 5.95 | 8. |
| Ti on Site | Considered Isomers | Average Energy of Isomers | Preference Order (calc.) | Preference Order (exp.) |
|---|---|---|---|---|
| 1 | 11222, 12122, 12212, 12221 | 1.73 | 3. | 2. |
| 2 | 11222, 21122, 21212, 21221 | 2.06 | 4. | 4. |
| 3 | 12122, 21122, 22112, 22121 | 6,81 | 5. | 5. |
| 4 | 12212, 21212, 22112, 22211 | −8.06 | 1. | 1. |
| 5 | 12221, 21221, 22121, 22211 | −2.61 | 2. | 3. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stare, K.; Stare, J.; Škapin, S.D.; Spreitzer, M.; Meden, A. Structure Determination and Analysis of the Ceramic Material La0.987Ti1.627Nb3.307O13 by Synchrotron and Neutron Powder Diffraction and DFT Calculations. Crystals 2023, 13, 439. https://doi.org/10.3390/cryst13030439
Stare K, Stare J, Škapin SD, Spreitzer M, Meden A. Structure Determination and Analysis of the Ceramic Material La0.987Ti1.627Nb3.307O13 by Synchrotron and Neutron Powder Diffraction and DFT Calculations. Crystals. 2023; 13(3):439. https://doi.org/10.3390/cryst13030439
Chicago/Turabian StyleStare, Katarina, Jernej Stare, Srečo Davor Škapin, Matjaž Spreitzer, and Anton Meden. 2023. "Structure Determination and Analysis of the Ceramic Material La0.987Ti1.627Nb3.307O13 by Synchrotron and Neutron Powder Diffraction and DFT Calculations" Crystals 13, no. 3: 439. https://doi.org/10.3390/cryst13030439
APA StyleStare, K., Stare, J., Škapin, S. D., Spreitzer, M., & Meden, A. (2023). Structure Determination and Analysis of the Ceramic Material La0.987Ti1.627Nb3.307O13 by Synchrotron and Neutron Powder Diffraction and DFT Calculations. Crystals, 13(3), 439. https://doi.org/10.3390/cryst13030439