

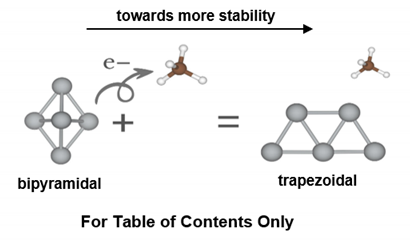
The Structural and Electronic Properties of the Ag5 Atomic Quantum Cluster Interacting with CO2, CH4, and H2O Molecules



Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
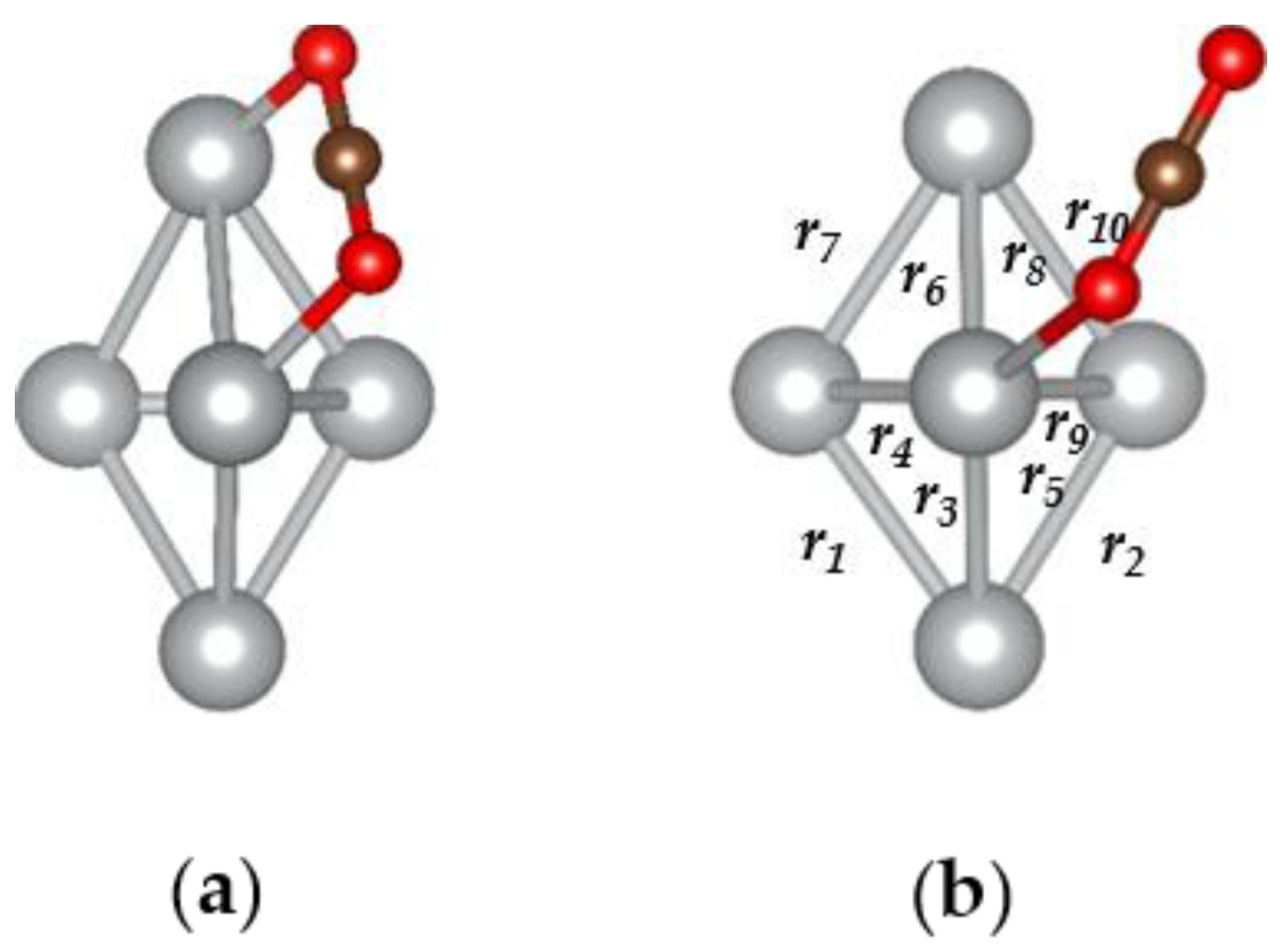
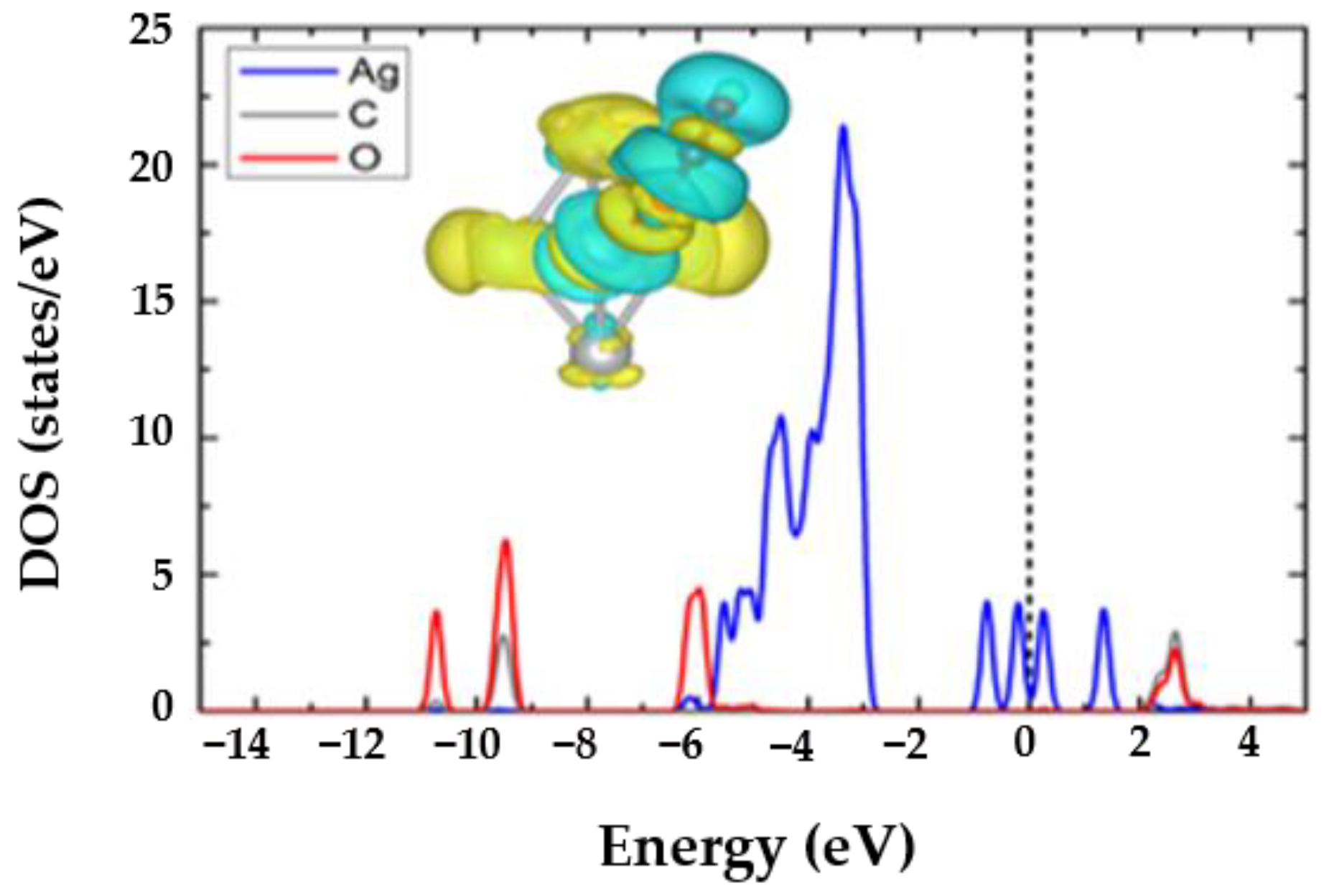
3.1. The CO2@Ag5 Structure
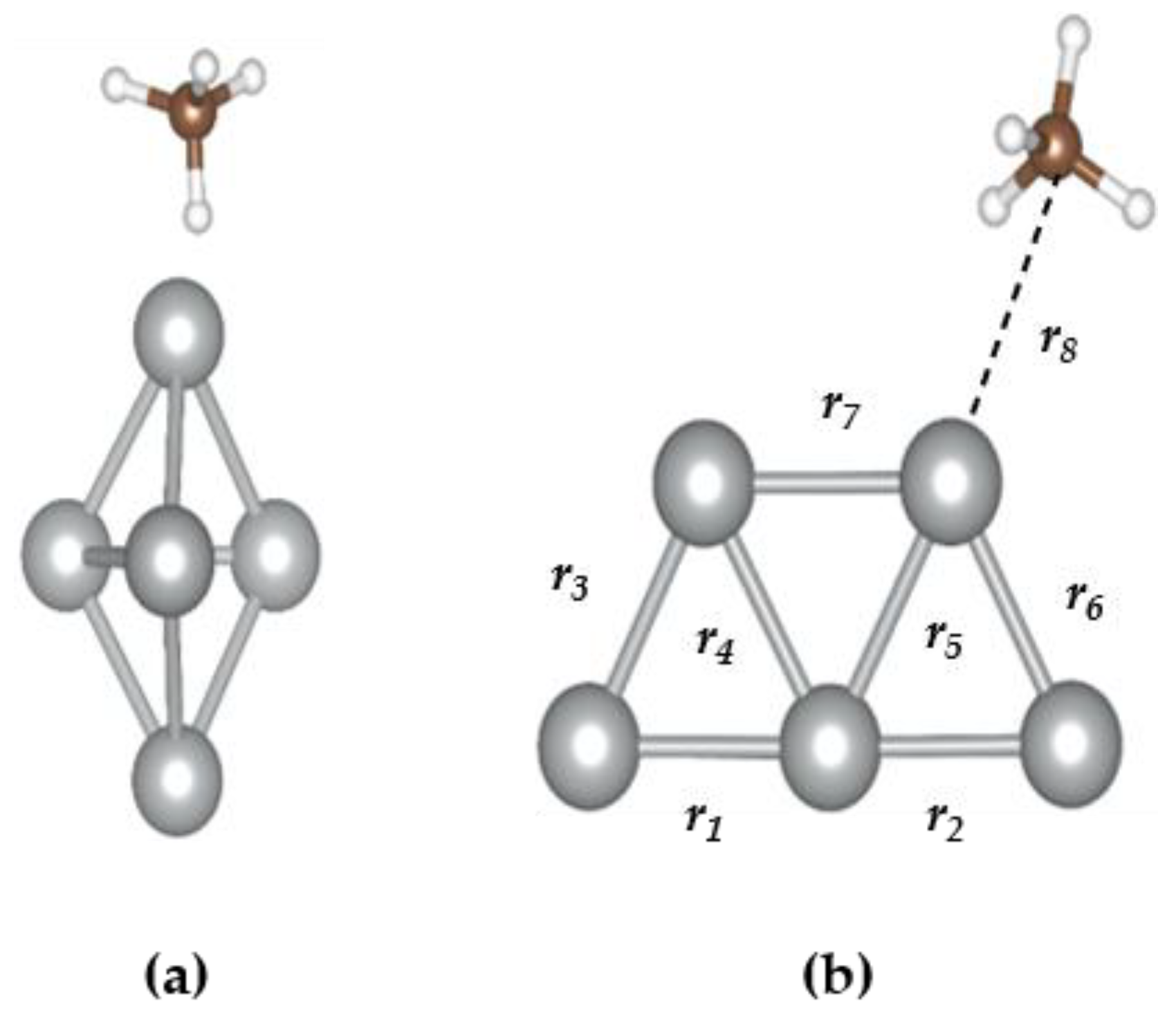
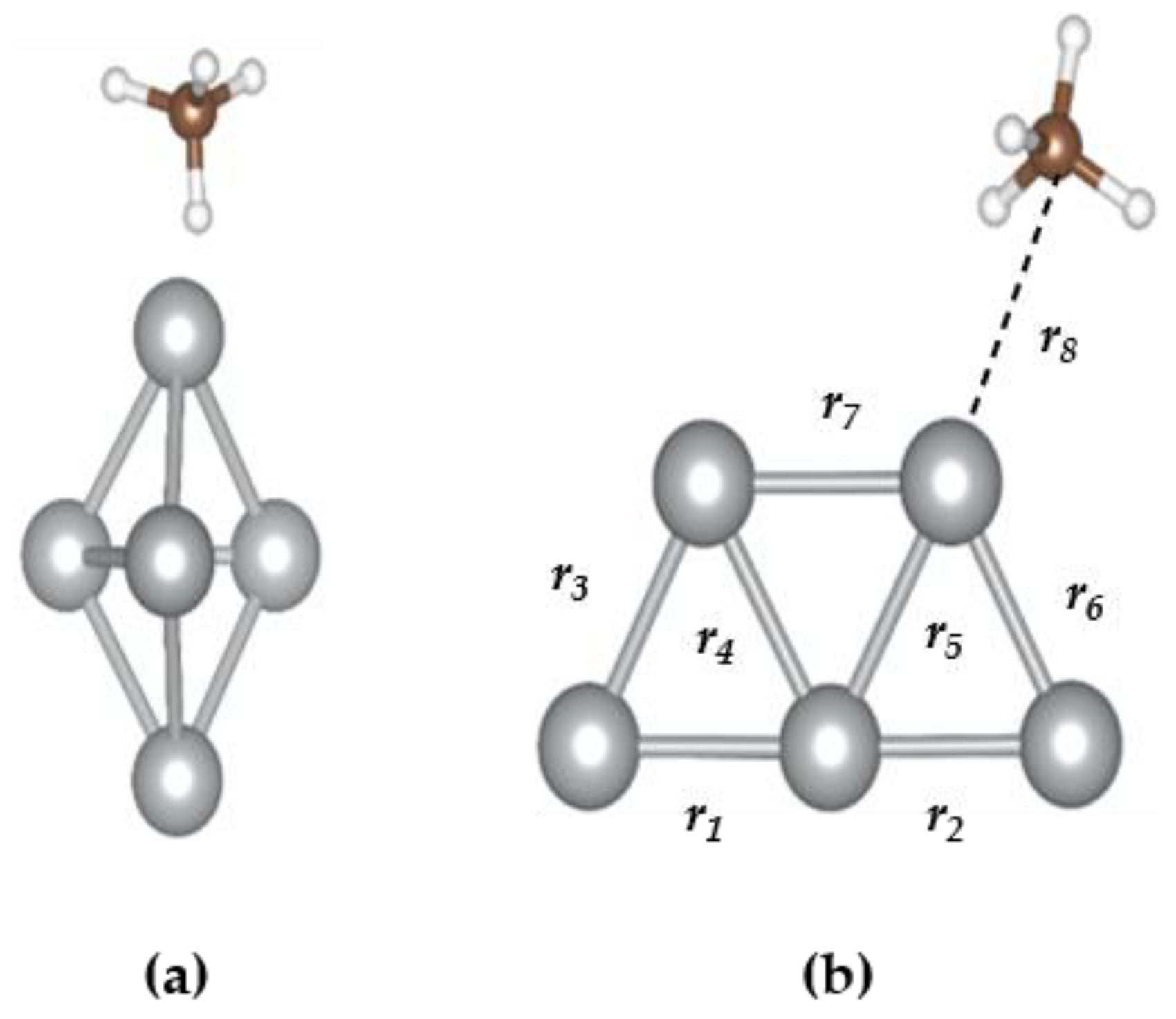
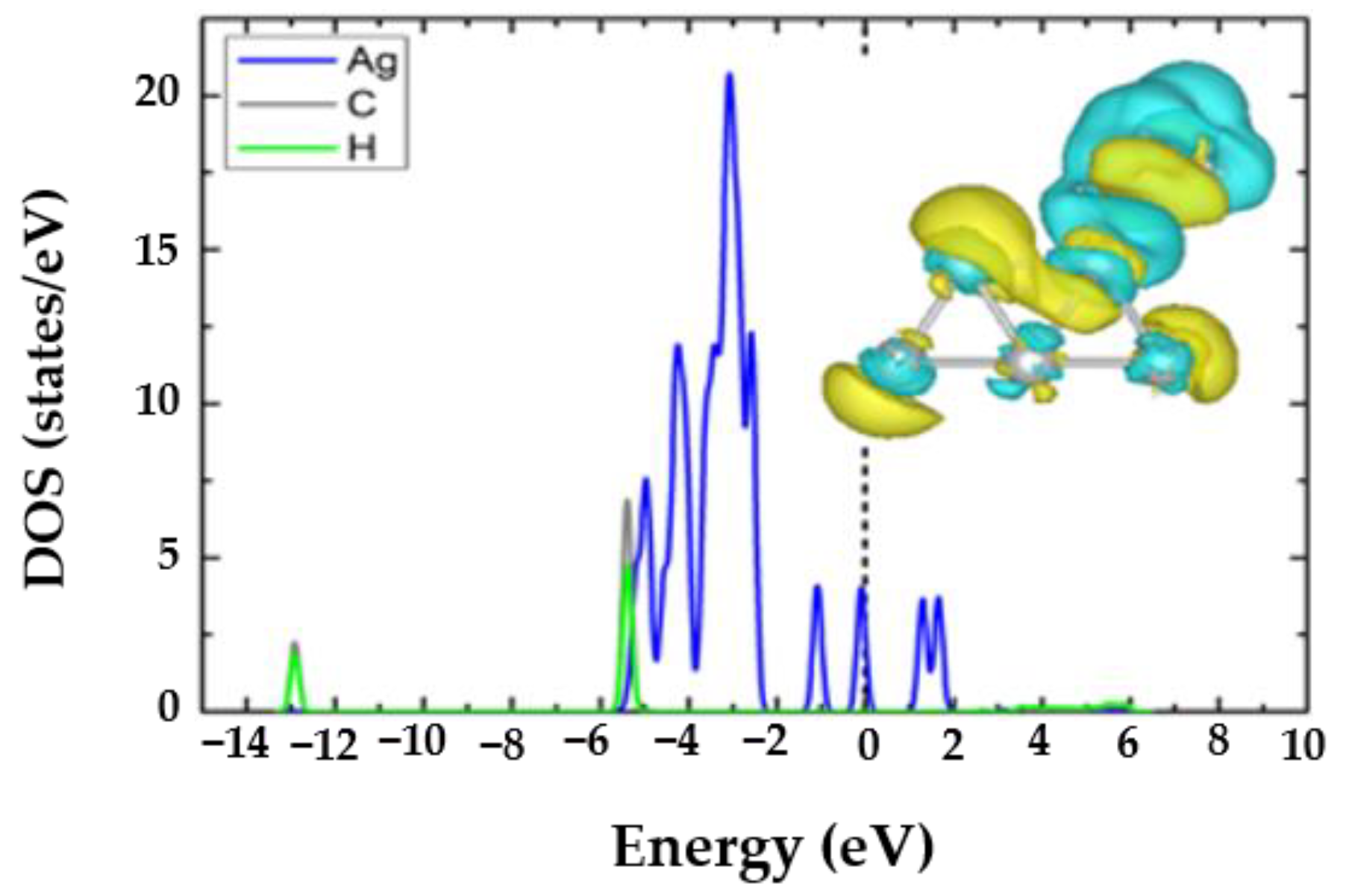
3.2. The CH4@Ag5 Structure
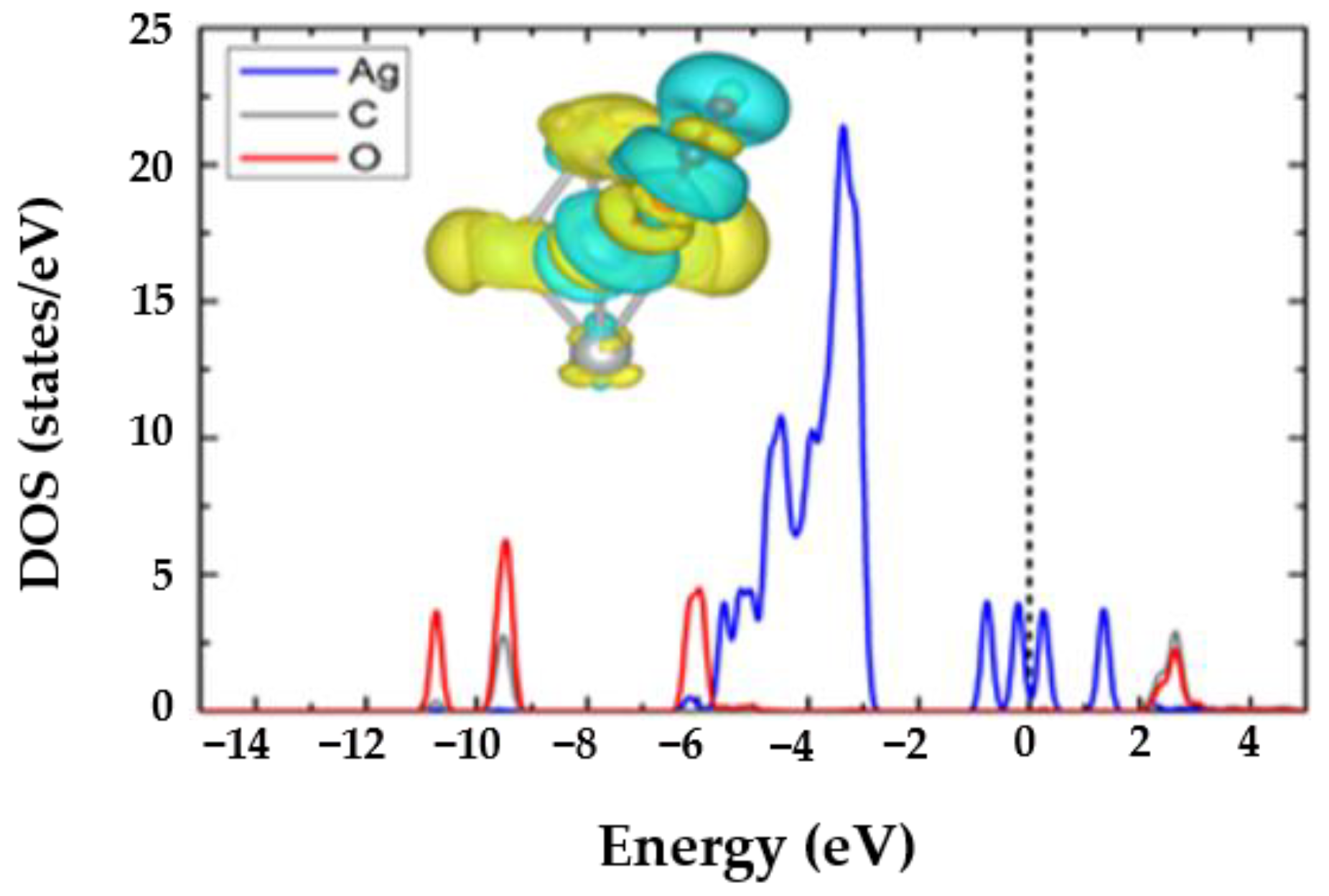
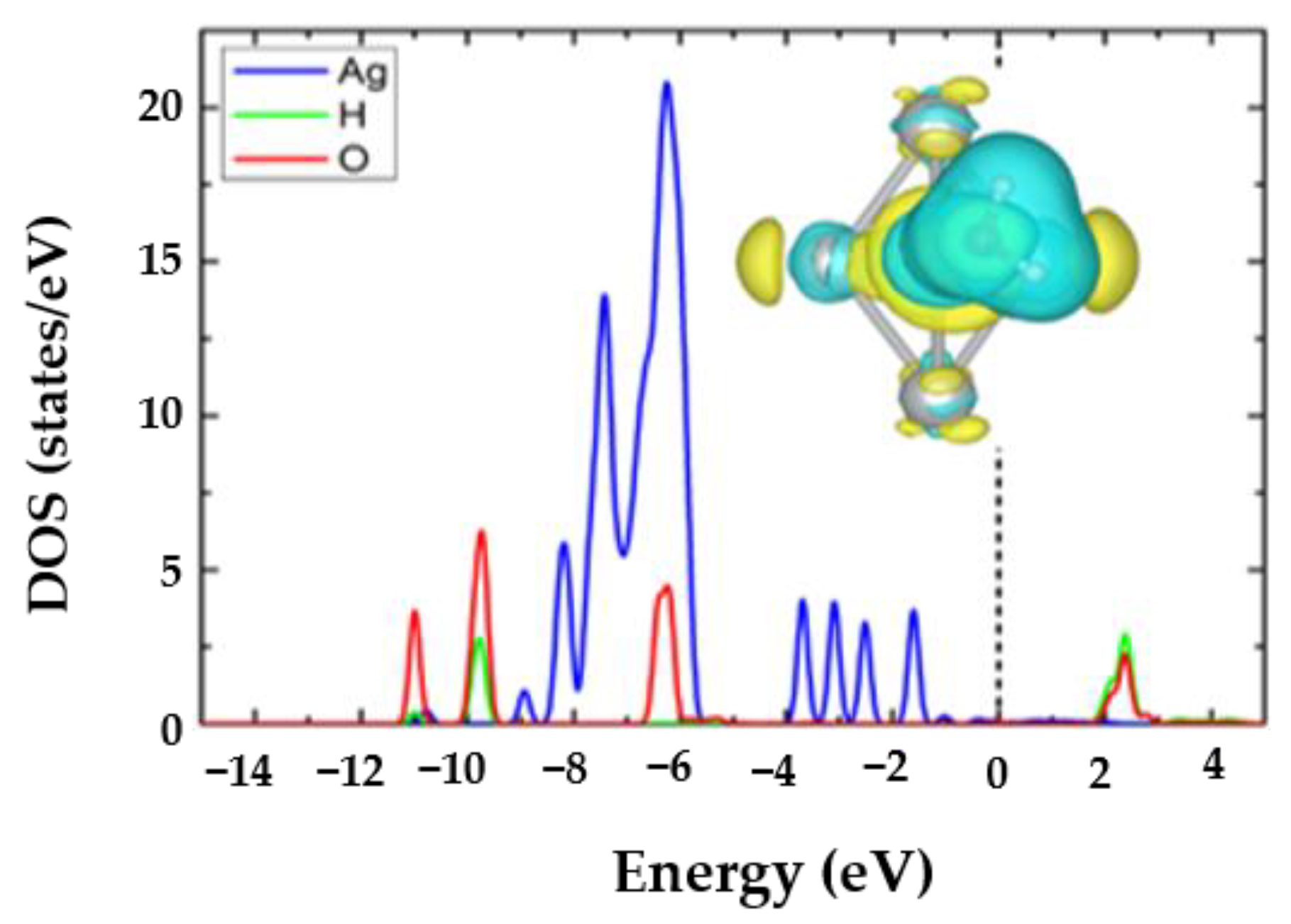
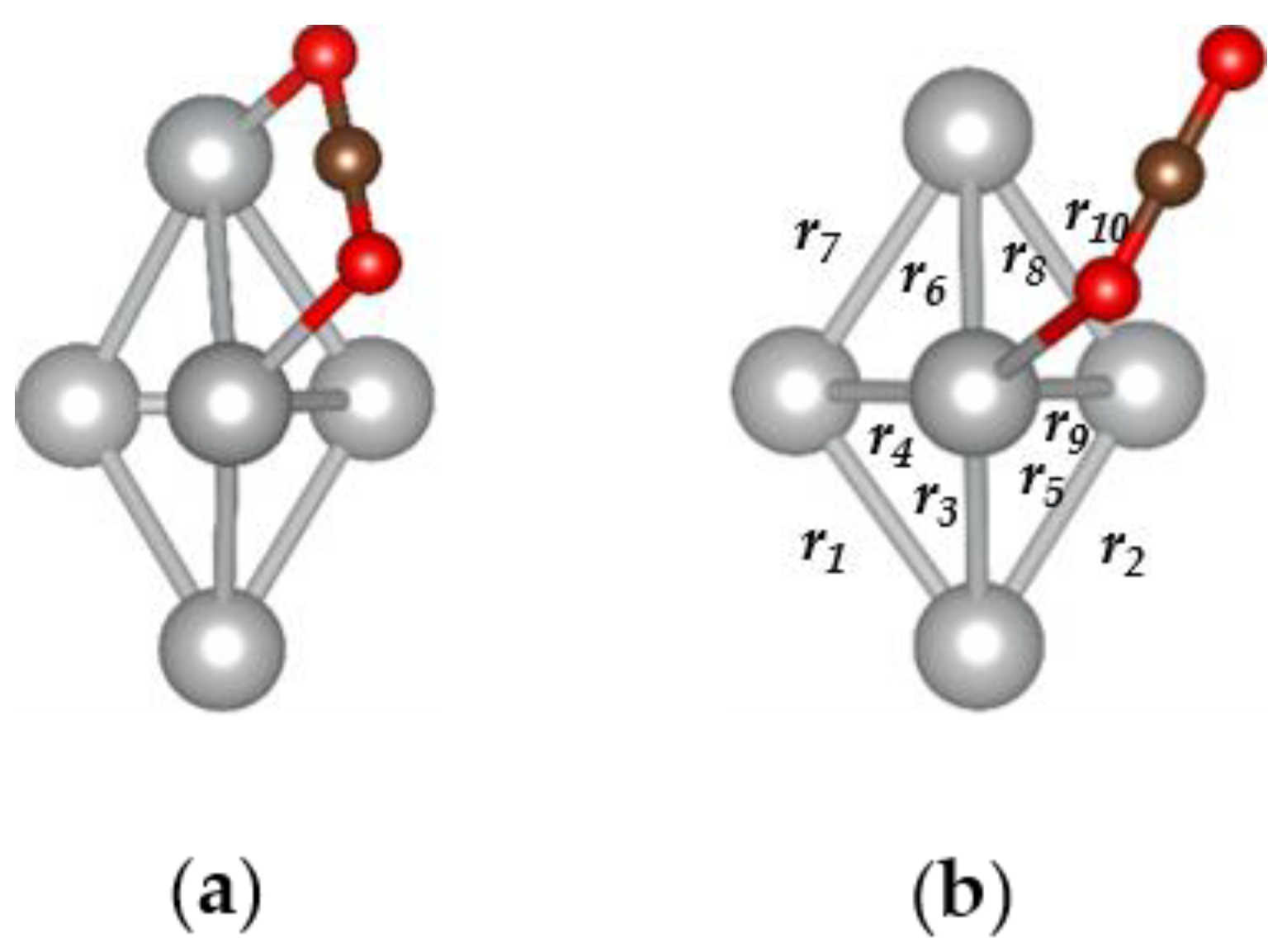
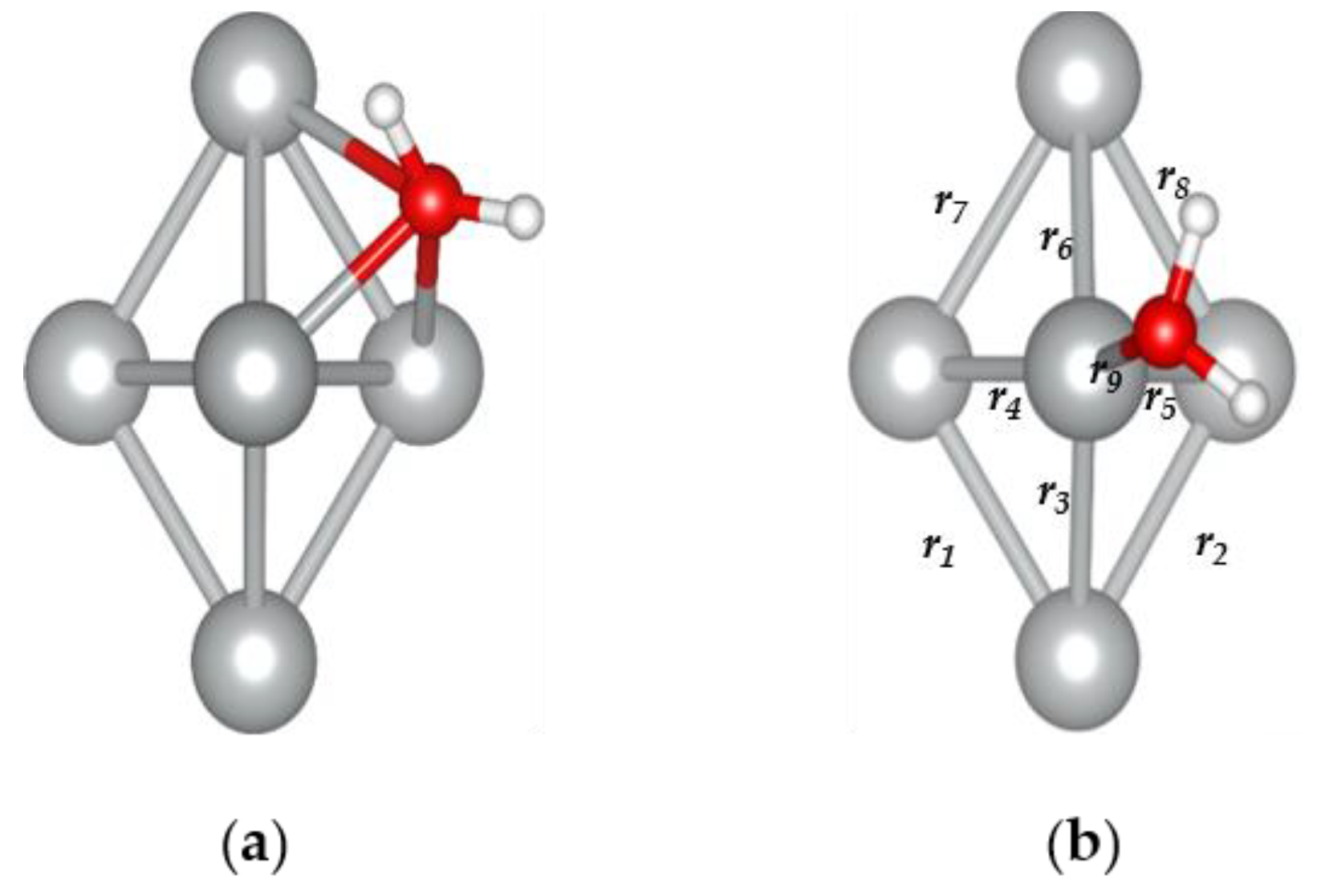
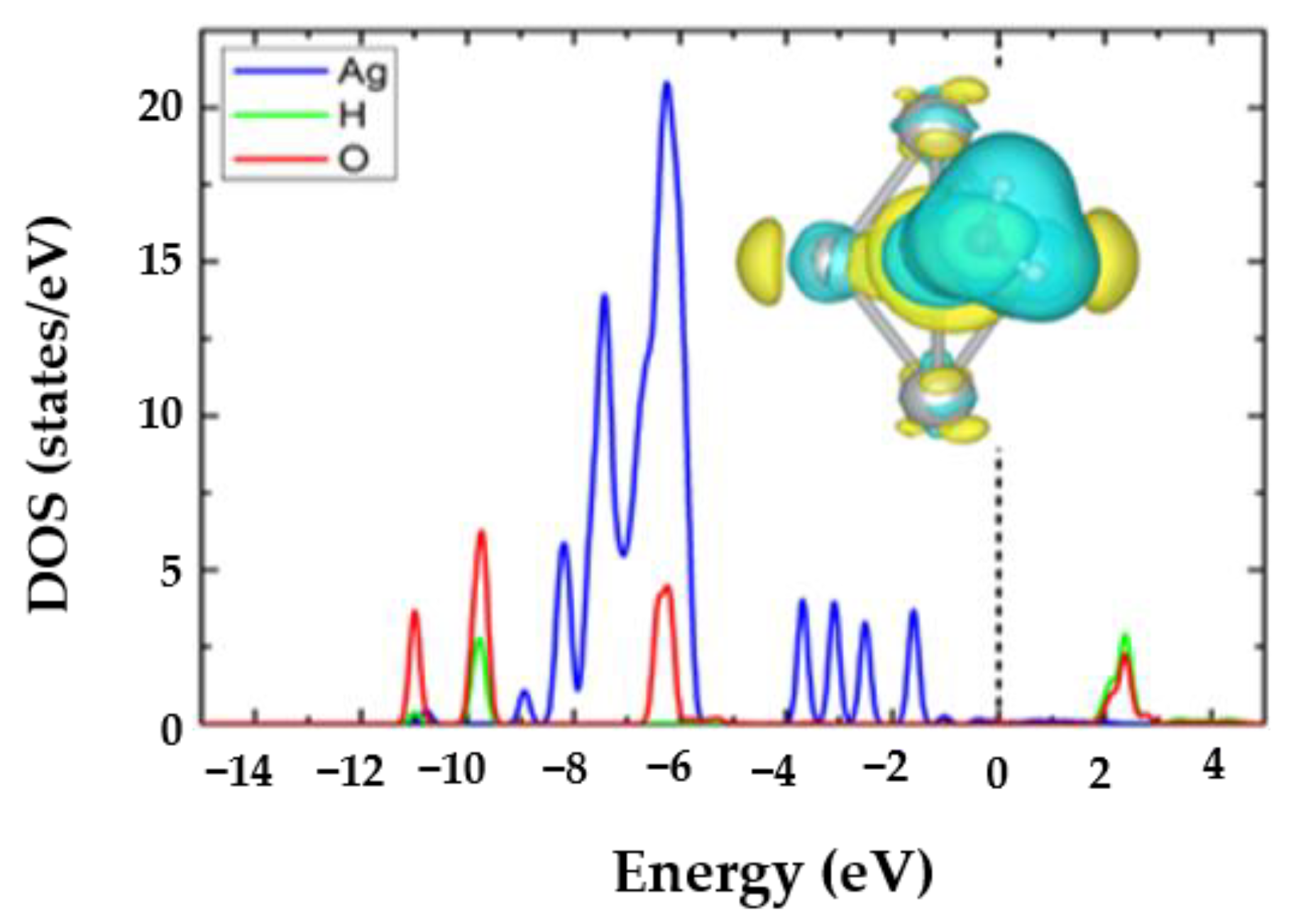
3.3. The H2O@Ag5 Structure
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferrando, R.; Jellinek, J.; Johnston, R.L. Nanoalloys: From theory to applications of alloy clusters and nanoparticles. Chem. Rev. 2008, 108, 845–910. [Google Scholar] [CrossRef] [PubMed]
- Katz, E.; Willner, I. Integrated nanoparticle-biomolecule hybrid systems: Synthesis, properties, and applications. Angew. Chem.-Int. Ed. 2004, 43, 6042–6108. [Google Scholar] [CrossRef] [PubMed]
- Cobley, C.M.; Chen, J.; Cho, E.C.; Wang, L.V.; Xia, Y. Gold nanostructures: A class of multifunctional materials for biomedical applications. Chem. Soc. Rev. 2011, 40, 44–56. [Google Scholar] [CrossRef] [PubMed]
- González, B.S.; López-Quintela, M.A. New strategies and synthetic routes to synthesize fluorescent atomic quantum clusters. RSC Smart Mater. 2014, 2014, 25–50. [Google Scholar]
- Porto, V.; Borrajo, E.; Buceta, D.; Carneiro, C.; Huseyinova, S.; Domínguez, B.; Borgman, K.J.; Lakadamyali, M.; Garcia-Parajo, M.F.; Neissa, J.; et al. Silver Atomic Quantum Clusters of Three Atoms for Cancer Therapy: Targeting Chromatin Compaction to Increase the Therapeutic Index of Chemotherapy. Adv. Mater. 2018, 30, 1801317. [Google Scholar] [CrossRef] [PubMed]
- Tyo, E.C.; Vajda, S. Catalysis by clusters with precise numbers of atoms. Nat. Nanotechnol. 2015, 10, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Socaciu, L.D.; Hagen, J.; Le Roux, J.; Popolan, D.; Bernhardt, T.M.; Wöste, L.; Vajda, Š. Strongly cluster size dependent reaction behavior of CO with O2 on free silver cluster anions. J. Chem. Phys. 2004, 120, 2078–2081. [Google Scholar] [CrossRef]
- Lang, S.M.; Bernhardt, T.M.; Chernyy, V.; Bakker, J.M.; Barnett, R.N.; Landman, U. Selective C−H Bond Cleavage in Methane by Small Gold Clusters. Angew. Chem.-Int. Ed. 2017, 56, 13406–13410. [Google Scholar] [CrossRef]
- Park, J.; Kwon, S.G.; Jun, S.W.; Kim, B.H.; Hyeon, T. Large-scale synthesis of ultra-small-sized silver nanoparticles. ChemPhysChem 2012, 13, 2540–2543. [Google Scholar] [CrossRef]
- Fernando, A.; Weerawardene, K.L.D.M.; Karimova, N.V.; Aikens, C.M. Quantum Mechanical Studies of Large Metal, Metal Oxide, and Metal Chalcogenide Nanoparticles and Clusters. Chem. Rev. 2015, 115, 6112–6216. [Google Scholar] [CrossRef]
- Zhao, Y.X.; Li, X.N.; Yuan, Z.; Liu, Q.Y.; Shi, Q.; He, S.G. Methane activation by gold-doped titanium oxide cluster anions with closed-shell electronic structures. Chem. Sci. 2016, 7, 4730–4735. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chen, W. Sub-nanometre sized metal clusters: From synthetic challenges to the unique property discoveries. Chem. Soc. Rev. 2012, 41, 3594–3623. [Google Scholar] [CrossRef] [PubMed]
- López-Caballero, P.; Ramallo-López, J.M.; Giovanetti, L.J.; Buceta, D.; Miret-Artés, S.; López-Quintela, M.A.; Requejo, F.G.; de Lara-Castells, M.P. Exploring the properties of Ag5-TiO2 interfaces: Stable surface polaron formation, UV-Vis optical response, and CO2 photoactivation. J. Mater. Chem. A Mater. 2020, 8, 6842–6853. [Google Scholar] [CrossRef]
- López-Caballero, P.; Miret-Artés, S.; Mitrushchenkov, A.O.; De Lara-Castells, M.P. Ag5-induced stabilization of multiple surface polarons on perfect and reduced TiO2 rutile (110). J. Chem. Phys. 2020, 153, 164702. [Google Scholar] [CrossRef] [PubMed]
- Hagen, J.; Socaciu, L.D.; Le Roux, J.; Popolan, D.; Bernhardt, T.M.; Wöste, L.; Mitrić, R.; Noack, H.; Bonačić-Koutecký, V. Cooperative Effects in the Activation of Molecular Oxygen by Anionic Silver Clusters. J. Am. Chem. Soc. 2004, 126, 3442–3443. [Google Scholar] [CrossRef] [PubMed]
- Alotaibi, M.; Wu, Q.; Lambert, C. Computational Studies of Ag5 Atomic Quantum Clusters Deposited on Anatase and Rutile TiO2 Surfaces. Appl. Surf. Sci. 2022, 613, 156054. [Google Scholar] [CrossRef]
- Gogoi, D.; Namdeo, A.; Golder, A.K.; Peela, N.R. Ag-doped TiO2 photocatalysts with effective charge transfer for highly efficient hydrogen production through water splitting. Int. J. Hydrog. Energy 2020, 45, 2729–2744. [Google Scholar] [CrossRef]
- Preda, G.; Pacchioni, G. Formation of oxygen active species in Ag-modified CeO2 catalyst for soot oxidation: A DFT study. Catal. Today 2011, 177, 31–38. [Google Scholar] [CrossRef]
- Porto, V.; Buceta, D.; Domínguez, B.; Carneiro, C.; Borrajo, E.; Fraile, M.; Davila-Ferreira, N.; Arias, I.R.; Blanco, J.M.; Blanco, M.C.; et al. Silver Clusters of Five Atoms as Highly Selective Antitumoral Agents through Irreversible Oxidation of Thiols. Adv. Funct. Mater. 2022, 32, 2113028. [Google Scholar] [CrossRef]
- Arias, I.R.; Buceta, D.; Barone, G.; Giménez-López, M.C.; Lozano, H.; Lazzari, M.; López-Quintela, M.A. Ag5 nanoclusters with dual catalytic antiradical activities. J. Colloid Interface Sci. 2022, 628, 437–447. [Google Scholar] [CrossRef]
- Greeley, J.; Jaramillo, T.F.; Bonde, J.; Chorkendorff, I.; Nørskov, J.K. Computational high-throughput screening of electrocatalytic materials for hydrogen evolution. Nat. Mater. 2006, 5, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Chem. Chem. Phys. 2001, 155, 871–964. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, 1133–1138. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter. 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Kresse, D.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA: A three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Wu, Q.; Hou, S.; Buceta, D.; Ordo, H.J.L.; Arturo, L.M.; Lambert, C.J. Tuning the surface states of TiO2 using Cu5 atomic clusters. Appl. Surf. Sci. 2022, 594, 153455. [Google Scholar] [CrossRef]
- Yang, L.; Huang, H.; Luo, X.; He, H.; Gao, F.; Zhou, Y. Unpaired Electron-Induced Wide-Range Light Absorption within Zn (or Cu) MOFs Containing Electron-Withdrawing Ligands: A Theoretical and Experimental Study. J. Phys. Chem. A 2020, 124, 5314–5322. [Google Scholar] [CrossRef]
- Guo, S.; Li, Y.; Liu, L.; Zhang, X.; Zhang, S. Theoretical evidence for new adsorption sites of CO2 on the Ag electrode surface. arXiv 2020, arXiv:2007.00827v1. Available online: https://api.semanticscholar.org/CorpusID:220302831 (accessed on 12 September 2023).
- Zhang, X.G.; Jin, X.; Wu, D.Y.; Tian, Z.Q. Selective Electrocatalytic Mechanism of CO2 Reduction Reaction to CO on Silver Electrodes: A Unique Reaction Intermediate. J. Phys. Chem. C 2018, 122, 25447–25455. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Loffreda, D.; Koper, M.T.M.; Sautet, P. Introducing structural sensitivity into adsorption-energy scaling relations by means of coordination numbers. Nat. Chem. 2015, 7, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.G.; Liu, Y.; Zhan, C.; Jin, X.; Chi, Q.; Wu, D.Y.; Zhao, Y.; Tian, Z.Q. Reaction Selectivity for Plasmon-Driven Carbon Dioxide Reduction on Silver Clusters: A Theoretical Prediction. J. Phys. Chem. C 2019, 123, 11101–11108. [Google Scholar] [CrossRef]
- Freund, H.-J.; Roberts, M.W. Surface chemistry of carbon dioxide. Surf. Sci. Rep. 1996, 25, 225–273. [Google Scholar] [CrossRef]
- Zhang, X.; Lim, E.; Kim, S.K.; Bowen, K.H. Photoelectron spectroscopic and computational study of (M-CO2)− anions, M = Cu, Ag, Au. J. Chem. Phys. 2015, 143, 174305. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. A Quantum Theory of Molecular Structure and Its Applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Verdinelli, V.; Germán, E.; Luna, C.R.; Marchetti, J.M.; Volpe, M.A.; Juan, A. Theoretical study of hydrogen adsorption on Ru-decorated (8,0) single-walled carbon nanotube. J. Phys. Chem. C 2014, 118, 27672–27680. [Google Scholar] [CrossRef]
- Du, J.; Jiang, G. A Systematic Theoretical Study of UC6: Structure, Bonding Nature, and Spectroscopy. Inorg. Chem. 2017, 56, 13794–13800. [Google Scholar] [CrossRef]
- Polynskaya, J.G.; Lebedev, A.V.; Knizhnik, A.A.; Sinitsa, A.S.; Smirnov, R.V.; Potapkin, B.V. Influence of charge state and active site structure of tetrahedral copper and silver clusters on the methane activation. Comput. Theor. Chem. 2019, 1147, 51–61. [Google Scholar] [CrossRef]
- Cheng, Z.; Fine, N.A.; Lo, C.S. Platinum nanoclusters exhibit enhanced catalytic activity for methane dehydrogenation. Top. Catal. 2012, 55, 345–352. [Google Scholar] [CrossRef]
- Nasresfahani, S.; Safaiee, R.; Sheikhi, M.H. A dispersion-corrected DFT insight into the structural, electronic and CH4 adsorption properties of small tin-oxide clusters. J. Alloys Compd. 2018, 757, 382–392. [Google Scholar] [CrossRef]
- Cuong, N.T.; Nguyen, H.M.T.; Pham-Ho, M.P.; Nguyen, M.T. Optical properties of the hydrated charged silver tetramer and silver hexamer encapsulated inside the sodalite cavity of an LTA-type zeolite. Phys. Chem. Chem. Phys. 2016, 18, 18128–18136. [Google Scholar] [CrossRef] [PubMed]
- Pang, R.; Zhang, X.G.; Zhou, J.Z.; Wu, D.Y.; Tian, Z.Q. SERS Chemical Enhancement of Water Molecules from Halide Ion Coadsorption and Photoinduced Charge Transfer on Silver Electrodes. J. Phys. Chem. C 2017, 121, 10445–10454. [Google Scholar] [CrossRef]
- Baetzold, R.C. Silver-Water Clusters: A Computation of Agn(H2O)m for n = 1–6; M = 1–8. J. Phys. Chem. C 2017, 121, 11811–11823. [Google Scholar] [CrossRef]
- Baetzold, R.C. Silver-water clusters: A theoretical description of Agn(H2O)m for n = 1–4; m = 1–4. J. Phys. Chem. C 2015, 119, 8299–8309. [Google Scholar] [CrossRef]
- Qin, C.; Whitten, J.L. Adsorption of O, H, OH, and H2O on Ag(100). J. Phys. Chem. B 2005, 109, 8852–8856. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-Atom Fragments for Describing Molecular Charge Densities. Theoret. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Guerra, C.F.; Handgraaf, J.W.; Baerends, E.J.; Bickelhaupt, F.M. Voronoi Deformation Density (VDD) Charges: Assessment of the Mulliken, Bader, Hirshfeld, Weinhold, and VDD Methods for Charge Analysis. J. Comput. Chem. 2004, 25, 189–210. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Bond Length (Å) |
|---|---|
| r1 | 2.82 |
| r2 | 2.80 |
| r3 | 2.76 |
| r4 | 2.67 |
| r5 | 2.72 |
| r6 | 2.81 |
| r7 | 2.81 |
| r8 | 2.80 |
| r9 (Ag-O) | 2.56 |
| r10 (Ag-C) | 3.25 |
| Symbol | Bond Length (Å) |
|---|---|
| r1 | 2.62 |
| r2 | 2.62 |
| r3 | 2.63 |
| r4 | 2.65 |
| r5 | 2.65 |
| r6 | 2.63 |
| r7 | 2.65 |
| r8 (Ag-C) | 3.60 |
| Symbol | Bond Length (Å) |
|---|---|
| r1 | 2.80 |
| r2 | 2.80 |
| r3 | 2.68 |
| r4 | 2.72 |
| r5 | 2.81 |
| r6 | 2.80 |
| r7 | 2.80 |
| r8 | 2.80 |
| r9 (Ag-O) | 2.36 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alotaibi, M.; Alotaibi, T.; Alshammari, M.; Ismael, A.K. The Structural and Electronic Properties of the Ag5 Atomic Quantum Cluster Interacting with CO2, CH4, and H2O Molecules. Crystals 2023, 13, 1691. https://doi.org/10.3390/cryst13121691
Alotaibi M, Alotaibi T, Alshammari M, Ismael AK. The Structural and Electronic Properties of the Ag5 Atomic Quantum Cluster Interacting with CO2, CH4, and H2O Molecules. Crystals. 2023; 13(12):1691. https://doi.org/10.3390/cryst13121691
Chicago/Turabian StyleAlotaibi, Moteb, Turki Alotaibi, Majed Alshammari, and Ali K. Ismael. 2023. "The Structural and Electronic Properties of the Ag5 Atomic Quantum Cluster Interacting with CO2, CH4, and H2O Molecules" Crystals 13, no. 12: 1691. https://doi.org/10.3390/cryst13121691
APA StyleAlotaibi, M., Alotaibi, T., Alshammari, M., & Ismael, A. K. (2023). The Structural and Electronic Properties of the Ag5 Atomic Quantum Cluster Interacting with CO2, CH4, and H2O Molecules. Crystals, 13(12), 1691. https://doi.org/10.3390/cryst13121691