


Anticancer Studies of Newly Synthesized Thiazole Derivatives: Synthesis, Characterization, Biological Activity, and Molecular Docking

 ,
, 
 ,
,  , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
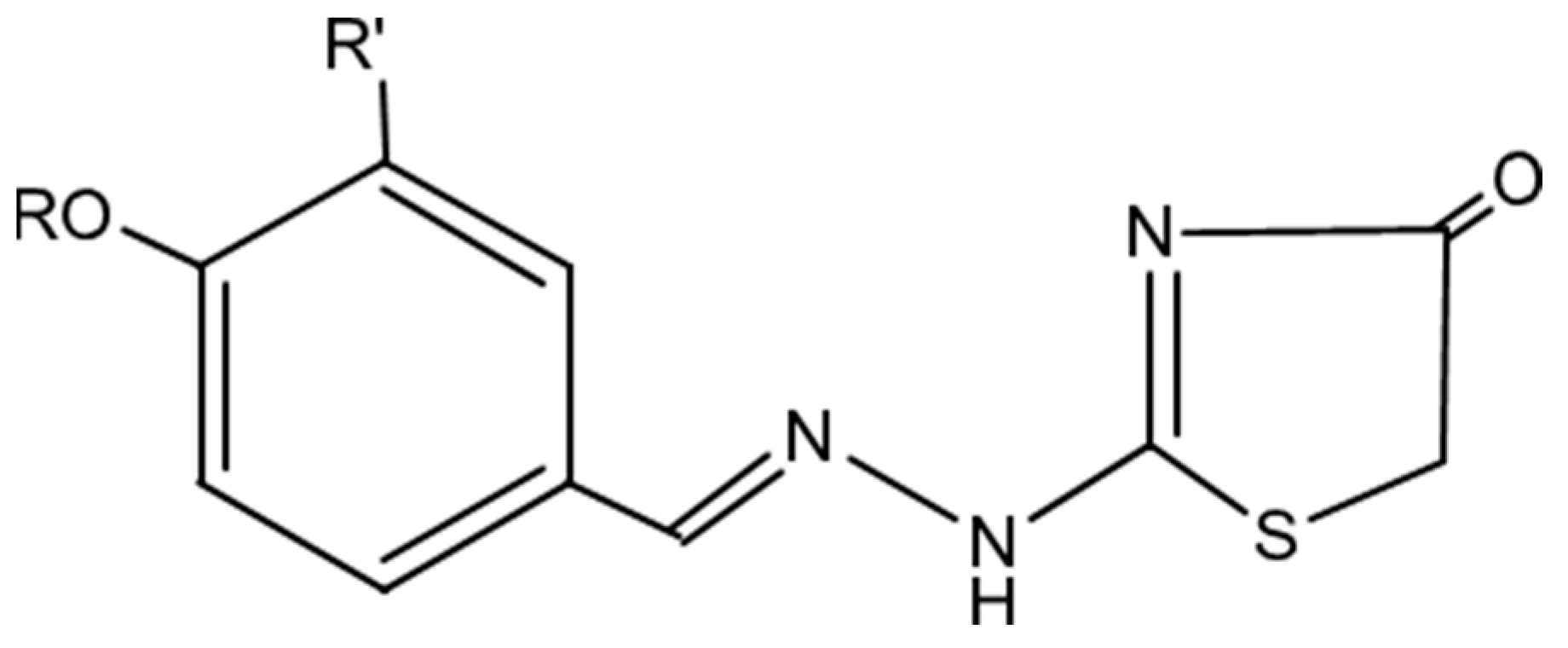
2.1. Chemistry
2.2. Evaluation of Biological Activity
2.2.1. In Vitro Cytotoxic Activity against Breast Cancer and Liver Cell Lines (MCF-7 and HepG2)
2.2.2. The VEGFR-2 Kinase Assay
2.2.3. Cell Cycle Assessment
2.2.4. Apoptosis Assay of Compound 4c by Using V-FITC/PI Annexin
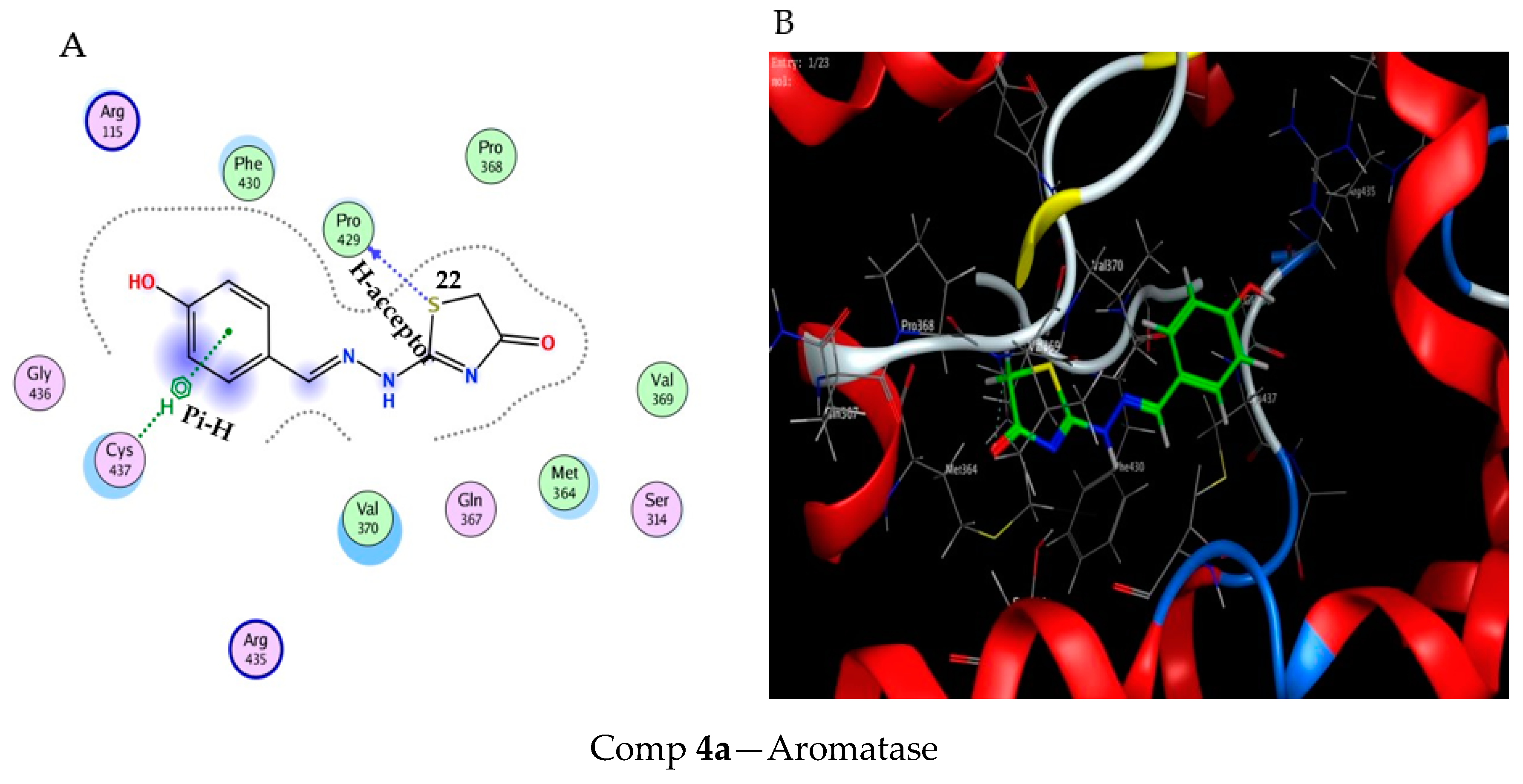
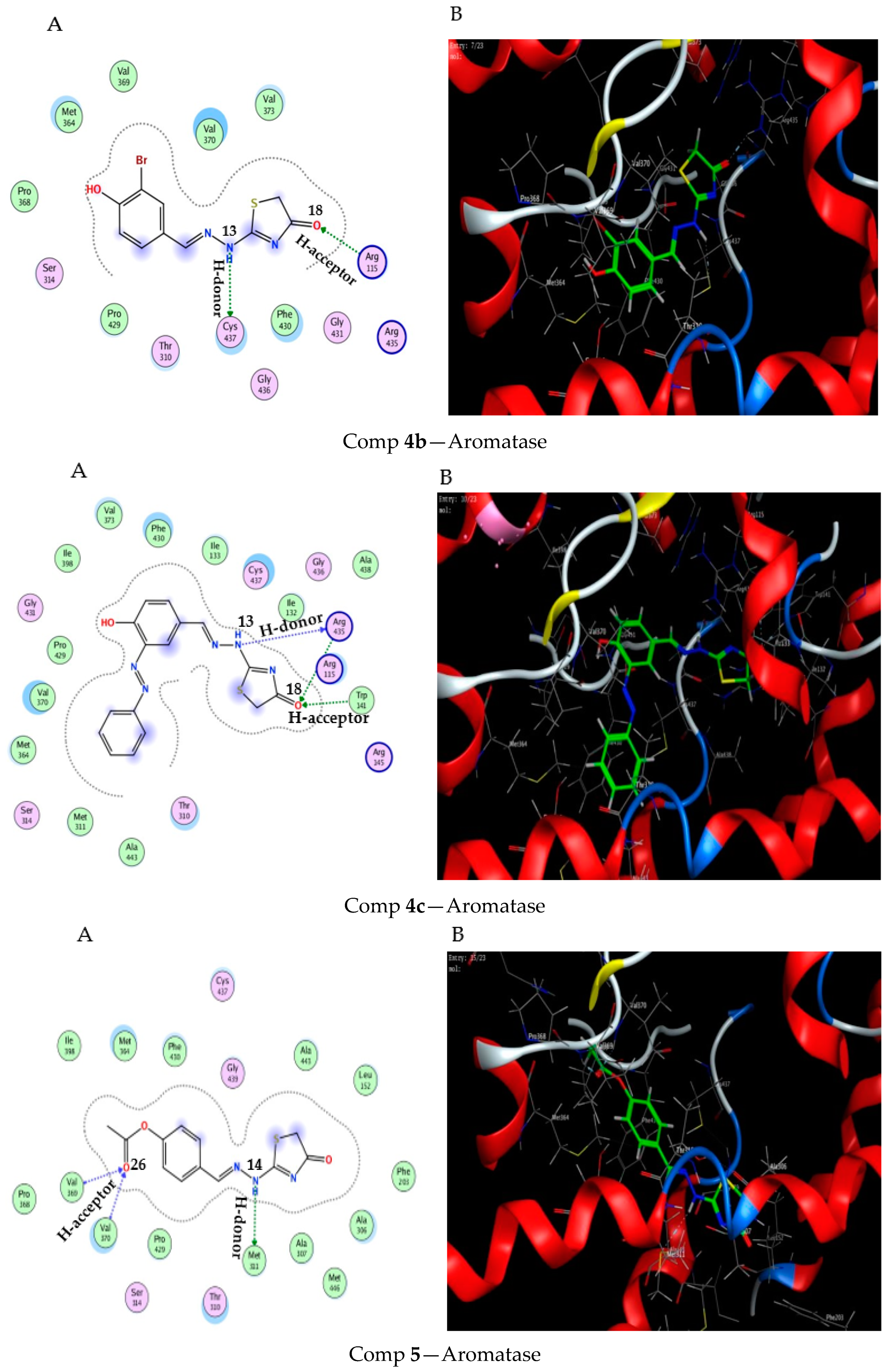
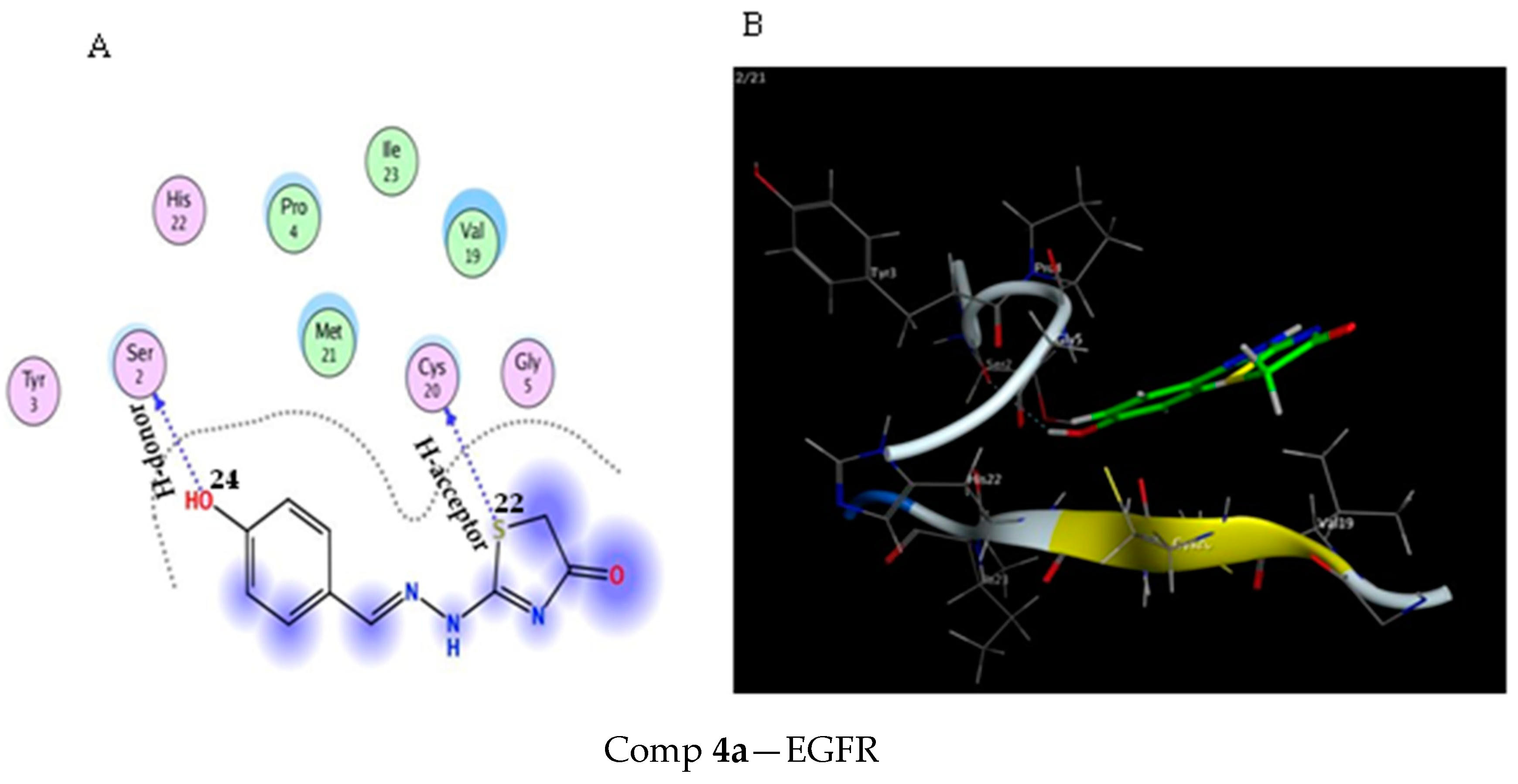
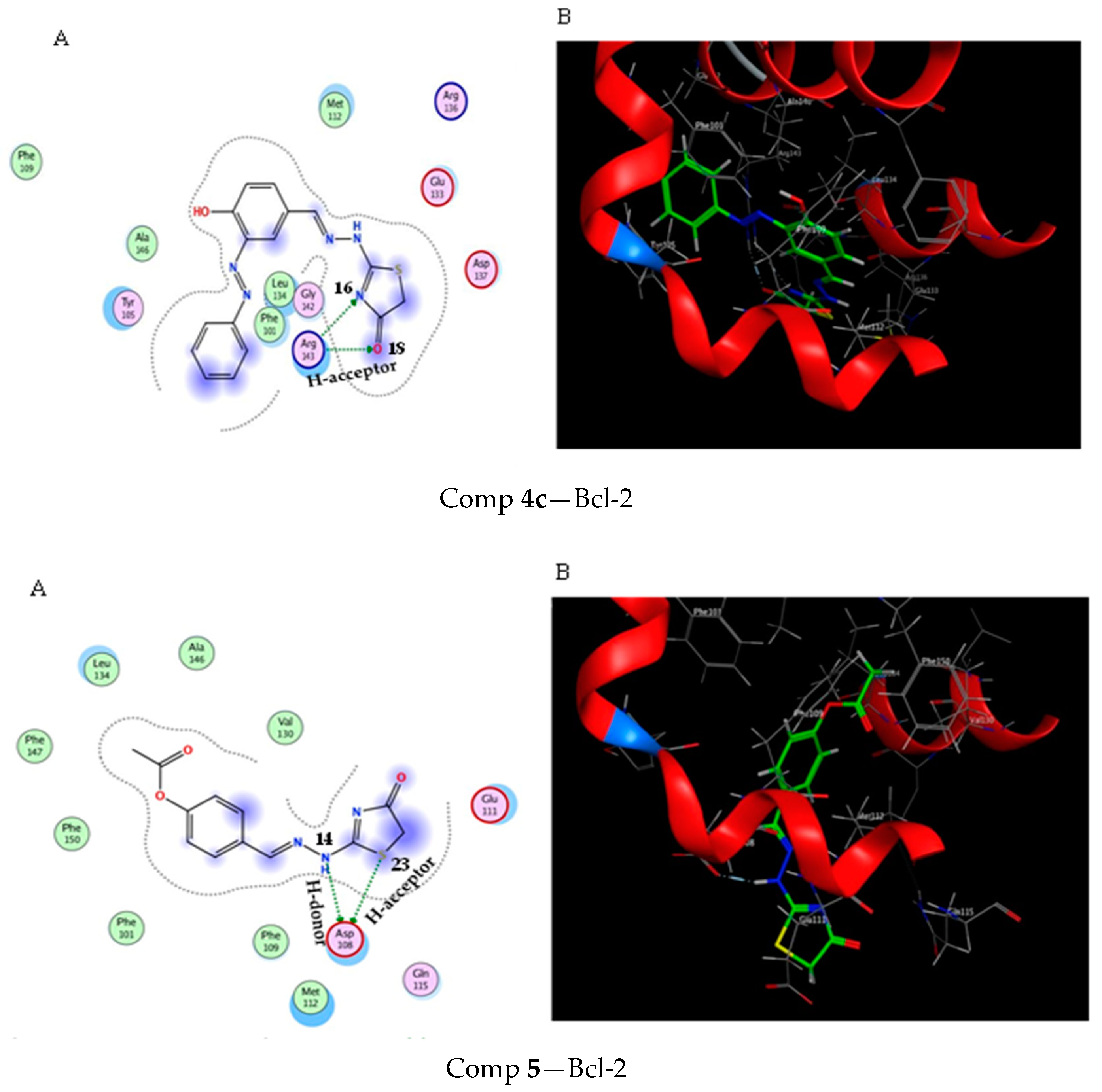
2.3. Molecular Docking Study
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthesis Guidelines for 2-Substituted Hydrazine Carbothioamides (2a–c)
2-(4-Hydroxybenzylidene)hydrazine Carbothioamide (2a)
2-(3-Bromo-4-hydroxybenzylidene)hydrazine-1-carbothioamide (2b)
4-Hydroxy-3-(phenyldiazenyl)benzylidene)hydrazine-1-carbothioamide (2c)
3.1.2. Synthesis of Thiazole-4 [5H]-One Derivatives (4a–c): General Technique
2-(2-(4-Hydroxybenzylidene)hydrazinyl)thiazole-4[5H]-one (4a)
2-(2-(3-Bromo-4-hydroxybenzylidene)hydrazinyl)thiazole-4[5H]-one (4b)
2-(2-(4-Hydroxy-3-phenyldiazenyl)benzylidene)hydrazinyl)thiazole-4[5H]-one (4c)
3.1.3. Synthesis 4-((2-(4-Oxo-4,5-Dihydrothiazol-2 Yl)Hydrazinylidene)methyl)phenyl Acetate (5)
3.1.4. Synthesis of 5-(4-Hydroxy-3-methoxybenzylidene)-2-(2-4-hydroxybenzylidene) Hydrazinyl) Thiazole-4[5H]-one (6)
3.2. Biological Assessment
3.2.1. The Ability of Cytotoxicity to Kill Cancer Cell Lines
3.2.2. Enzyme Assay for VEGFR-2
3.2.3. Study of the Cell Cycle of Chemical 4c
3.2.4. Apoptosis Assay with Annexin V FITC/PI
3.3. Molecular Docking Modeling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marwa, F.A.; Atiah, H.A. Design, synthesis, antiproliferative activity, and cell cycle analysis of new thiosemicarbazone derivatives targeting ribonucleotide reductase. Arab. J. Chem. 2021, 14, 102989. [Google Scholar]
- Mohammed, F.Z.; Rizzk, Y.W.; El Deen, I.M.; Mourad, A.A.; El Behery, M. Design, synthesis, cytotoxic screening and molecular docking studies of novel hybrid thiosemicarbazone derivatives as anticancer agents. Chem. Biodivers. 2021, 18, e2100580. [Google Scholar] [CrossRef]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef]
- Loibl, S.; Poortmans, P.; Morrow, M.; Denkert, C.; Curigliano, G. Breast Cancer. Lancet. 2021, 397, 1750–1769. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Narayan, P.; Osgood, C.L.; Wedam, S.; Prowell, T.M.; Gao, J.J.; Shah, M.; Krol, D.; Wahby, S.; Royce, M.; et al. U.S. FDA Drug Approvals for Breast Cancer: A Decade in Review. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 1072–1086. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Peng, C.; Xie, X.; Peng, F. New Advances in Targeted Therapy of HER2-Negative Breast Cancer. Front. Oncol. 2022, 12, 828438. [Google Scholar] [CrossRef]
- Mukhopadhyay, K.D.; Liu, Z.; Bandyopadhyay, A.; Kirma, N.B.; Tekmal, R.R.; Wang, S.; Sun, L.Z. Aromatase expression increases the survival and malignancy of estrogen receptor positive breast cancer cells. PLoS ONE 2015, 10, e0121136. [Google Scholar] [CrossRef]
- Wu, S.; Ye, J.; Wang, Z.; Lin, S.X.; Lu, M.; Liang, Y.; Wu, C.L. Expression of aromatase in tumor related stroma is associated with human bladder cancer progression. Cancer Biol. Ther. 2018, 19, 175–180. [Google Scholar] [CrossRef]
- Chumsri, S.; Howes, T.; Bao, T.; Sabnis, G.; Brodie, A. Aromatase, aromatase inhibitors, and breast cancer. J. Steroid Biochem. Mol. Biol. 2011, 125, 13–22. [Google Scholar] [CrossRef]
- Sasaki, T.; Hiroki, K.; Yamashita, Y. The role of epidermal growth factor receptor in cancer metastasis and microenvironment. Biomed. Res. Int. 2013, 2013, 546318. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Jonathan, C.; David, A.Q.; Troy, M.R.; Felix, Y.F.; Alan, A. Transcription-associated cyclin-dependent kinases as targets and biomarkers for cancer therapy. Cancer Discov. 2020, 10, 351–370. [Google Scholar]
- Johnson, L.N.; De Moliner, E.; Brown, N.R.; Song, H.; Barford, D.; Endicott, J.A.; Noble, M.E. Structural studies with inhibitors of the cell cycle regulatory kinase cyclin-dependent protein kinase 2. Pharmacol. Ther. 2002, 93, 113–124. [Google Scholar] [CrossRef]
- Kontopidis, G.; McInnes, C.; Pandalaneni, S.R.; McNae, I.; Gibson, D.; Mezna, M.; Fischer, P.M. Differential binding of inhibitors to active and inactive CDK2 provides insights for drug design. Chem. Biol. 2006, 13, 201–211. [Google Scholar] [CrossRef]
- Campbell, K.J.; Tait, S.W.G. Targeting BCL-2 regulated apoptosis in cancer. Open Biol. 2018, 8, 180002. [Google Scholar] [CrossRef]
- Murphy, G.J.; Holder, J.C. PPAR-γ agonists: Therapeutic role in diabetes, inflammation and cancer. Trends Pharmacol. Sci. 2000, 21, 469–474. [Google Scholar] [CrossRef]
- Dayam, R.; Sanchez, T.; Clement, O.; Shoemaker, R.; Sei, S.; Neamati, N. β-Diketo acid pharmacophore hypothesis. 1. Discovery of a novel class of HIV-1 integrase inhibitors. J. Med. Chem. 2005, 48, 111–120. [Google Scholar] [CrossRef]
- Kikkawa, R.; Hatanaka, I.; Yasuda, H.; Kobayashi, N.; Shigeta, Y.; Terashima, H.; Tsuboshima, M. Effect of a new aldose reductase inhibitor, (E)-3-carboxymethyl-5-[(2E)-methyl-3-phenylpropenylidene] rhodamine (ONO-2235) on peripheral nerve disorders in streptozotocin-diabetic rats. Diabetologia 1983, 24, 290–292. [Google Scholar] [CrossRef] [PubMed]
- da Rocha Junior, L.F.; Rêgo, M.J.B.; Cavalcanti, M.B.; Pereira, M.C.; Pitta, M.G.D.R.; de Oliveira, P.S.; Gonçalves, S.M.; Duarte, A.L.; de Lima Mdo, C.; Pitta Ida, R.; et al. Synthesis of a novel thiazolidinedione and evaluation of its modulatory effect on IFN-γ, IL-6, IL-17A, and IL-22 production in PBMCs from rheumatoid arthritis patients. Biomed. Res. Int. 2013, 2013, 926060. [Google Scholar] [CrossRef]
- Chhabria, M.T.; Patel, S.; Modi, P.; Brahmkshatriya, P.S. Thiazole: A review on chemistry, synthesis and therapeutic importance of its derivatives. Curr. Top. Med. Chem. 2016, 16, 2841–2862. [Google Scholar] [CrossRef] [PubMed]
- Zervosen, A.; Lu, W.P.; Chen, Z.; White, R.E.; Demuth, T.P.J.; Frere, J.M. Interactions between penicillin-binding proteins (PBPs) and two novel classes of PBP inhibitors, arylalkylidene rhodanines and arylalkylidene iminothiazolidin-4-ones. Antimicrob. Agents Chemother. 2004, 48, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Bozdağ-Dündar, O.; Ozgen, O.; Menteşe, A.; Altanlar, N.; Atli, O.; Kendi, E.; Ertan, R. Synthesis and antimicrobial activity of some new thiazolyl thiazolidine-2,4-dione derivatives. Bioorg Med. Chem. 2007, 15, 6012–6017. [Google Scholar] [CrossRef] [PubMed]
- Ravi, S.; Chiruvella, K.K.; Rajesh, K.; Prabhu, V.; Raghavan, S.C. 5-Isopropylidene-3-ethyl rhodanine induce growth inhibition followed by apoptosis in leukemia cells. Eur. J. Med. Chem. 2010, 45, 2748–2752. [Google Scholar] [CrossRef] [PubMed]
- Aziz, H.A.; El-Saghier, A.M.M.; Badr, M.; Abuo-Rahma, G.E.D.A.; Shoman, M.E. Thiazolidine-2,4-dione-linked ciprofloxacin derivatives with broad-spectrum antibacterial, MRSA and topoisomerase inhibitory activities. Mol. Divers. 2022, 26, 1743–1759. [Google Scholar] [CrossRef] [PubMed]
- Alegaon, S.G.; Alagawadi, K.R. New thiazolidinedione-5-acetic acid amide derivatives: Synthesis, characterization and investigation of antimicrobial and cytotoxic properties. Med. Chem. Res. 2012, 21, 816–824. [Google Scholar] [CrossRef]
- Mohamed, S.F.; Flefel, E.M.; Amr, A.E.-G.; Abd El-Shafy, D.N. Anti-HSV-1 activity and mechanism of action of some new synthesized substituted pyrimidine, thiopyrimidine and thiazolopyrimidine derivatives. Eur. J. Med. Chem. 2010, 45, 1494–1501. [Google Scholar] [CrossRef] [PubMed]
- Barsoum, F.F.; Girgis, A.S. Facile synthesis of bis (4,5-dihydro-1H-pyrazole-1-carboxamides) and their thio-analogues of potential PGE2 inhibitory properties. Eur. J. Med. Chem. 2009, 44, 2172–2177. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, M.; Jain, P. Synthetic and biological studies of pyrazolines and related heterocyclic compounds. Arab. J. Chem. 2014, 7, 553–596. [Google Scholar] [CrossRef]
- Hassan, A.S.; Moustafa, G.O.; Awad, H.M.; Nossier, E.S.; Mady, M.F. Design, synthesis, anticancer evaluation, enzymatic assays, and a molecular modeling study of novel pyrazole–indole hybrids. ACS Omega 2021, 6, 12361–12374. [Google Scholar] [CrossRef] [PubMed]
- Sharshira, E.M.; Hamada, N.M. Synthesis and in vitro antimicrobial activity of some pyrazolyl-1-carboxamide derivatives. Molecules 2011, 16, 7736–7745. [Google Scholar] [CrossRef] [PubMed]
- Hamadi, N.B.; Haouas, A.; Methamem, M.; Msaddek, M. Synthesis of new pyrazolines and their biological evaluation as antimicrobial agents. J. Chem. Res. 2012, 36, 563–565. [Google Scholar] [CrossRef]
- Havrylyuk, D.; Zimenkovsky, B.; Vasylenko, O.; Zaprutko, L.; Gzella, A.; Lesyk, R. Synthesis of novel thiazolone-based compounds containing pyrazoline moiety and evaluation of their anticancer activity. Eur. J. Med. Chem. 2009, 44, 1396–1404. [Google Scholar] [CrossRef] [PubMed]
- Sim, M.M.; Ng, S.B.; Buss, A.D.; Crasta, S.C.; Goh, K.L.; Lee, S.K. Benzylidene rhodanines as novel inhibitors of UDP-N-acetylmuramate/L-alanine ligase. Bioorg Med. Chem. Lett. 2002, 12, 697–699. [Google Scholar] [CrossRef] [PubMed]
- Turan-Zitouni, G.; Chevallet, P.; Kilic, F.S.; Erol, K. Synthesis of some thiazolyl-pyrazoline derivatives and preliminary investigation of their hypotensive activity. Eur. J. Med. Chem. 2000, 35, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Haroun, M.; Tratrat, C.; Kositzi, K.; Tsolaki, E.; Petrou, A.; Aldhubiab, B.; Attimarad, M.; Harsha, S.; Geronikaki, A.; Venugopala, K.N.; et al. New benzothiazole-based thiazolidinones as potent antimicrobial agents. Design, synthesis and biological evaluation. Curr. Top. Med. Chem. 2018, 18, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Sadek, B.; Al-Tabakha, M.M.; Fahelelbom, K.M.S. Antimicrobial prospect of newly synthesized 1, 3-thiazole derivatives. Molecules 2011, 16, 9386–9396. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Ramasamy, K.; Mani, V.; Mishra, R.K.; Majeed, A.B.A.; Clercq, E.D.; Narasimhan, B. Synthesis, antimicrobial, anticancer, antiviral evaluation and QSAR studies of 4-(1-aryl-2-oxo-1,2-dihydro-indol-3-ylideneamino)-N-substituted benzene sulfonamides. Arab. J. Chem. 2014, 7, 396–408. [Google Scholar] [CrossRef]
- Palekar, V.S.; Damle, A.J.; Shukla, S.R. Synthesis and antibacterial activity of some novel bis-1,2,4-triazolo [3,4-b]-1,3,4-thiadiazoles and bis-4-thiazolidinone derivatives from terephthalic dihydrazide. Eur. J. Med. Chem. 2009, 44, 5112–5116. [Google Scholar] [CrossRef] [PubMed]
- Hamama, W.S.; Ismail, M.A.; Shaaban, S.; Zoorob, H.H. Progress in the chemistry of 4-thiazolidinones. J. Heterocyclic Chem. 2008, 45, 939–956. [Google Scholar] [CrossRef]
- Kaminskyy, D.; Bednarczyk-Cwynar, B.; Vasylenko, O.; Kazakova, O.; Zimenkovsky, B.; Zaprutko, L.; Lesyk, R. Synthesis of new potential anticancer agents based on 4-thiazolidinone and oleanane scaffolds. Med. Chem. Res. 2012, 21, 3568–3580. [Google Scholar] [CrossRef]
- Kaur Manjal, S.; Kaur, R.; Bhatia, R.; Kumar, K.; Singh, V.; Shankar, R.; Kaur, R.; Rawal, R.K. Synthetic and medicinal perspective of thiazolidinones: A review. Bioorg Chem. 2017, 75, 406–423. [Google Scholar] [CrossRef] [PubMed]
- Gouda, M.A.; Abu-Hashem, A.A.; Hussein, H.A.R.; Aly, A.S. Recent Progress on Fused Thiadiazines: A Literature Review. Polycycl. Aromat. Compd. 2022, 42, 2861–2893. [Google Scholar] [CrossRef]
- Jilloju, P.C.; Persoons, L.; Kurapati, S.K.; Schols, D.; De Jonghe, S.; Daelemans, D.; Vedula, R.R. Discovery of (±)-3-(1 H-pyrazol-1-yl)-6,7-dihydro-5 H-[1,2,4] triazolo [3,4-b][1,3,4] thiadiazine derivatives with promising in vitro anticoronavirus and antitumoral activity. Mol. Divers. 2022, 26, 1357–1371. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, F.Z.; Rizzk, Y.W.; El-Deen, I.M.; Gad, E.M.; El Behery, M.; Mahdy, A.R. Discovery of 2-Amino-4H-1,3,4-thiadiazine-5 (6H)-one Derivatives and Their In Vitro Antitumor Investigation. ChemistrySelect 2022, 7, e202104333. [Google Scholar] [CrossRef]
- Kamat, V.; Santosh, R.; Poojary, B.; Nayak, S.P.; Kumar, B.K.; Sankaranarayanan, M.; Vootla, S.K. Pyridine-and thiazole-based hydrazides with promising anti-inflammatory and antimicrobial activities along with their in silico studies. ACS Omega 2020, 5, 25228–25239. [Google Scholar] [CrossRef] [PubMed]
- Morigi, R.; Locatelli, A.; Leoni, A.; Rambaldi, M. Recent patents on thiazole derivatives endowed with antitumor activity. Recent. Pat. Anticancer. Drug Discov. 2015, 10, 280–297. [Google Scholar] [CrossRef] [PubMed]
- Ghorab, M.M.; Al-Said, M.S. Antitumor activity of novel pyridine, thiophene and thiazole derivatives. Arch. Pharm. Res. 2012, 35, 965–973. [Google Scholar] [CrossRef]
- Kasralikar, H.M.; Jadhavar, S.C.; Bhansali, S.G.; Patwari, S.B.; Bhusare, S.R. Design, synthesis and docking studies of novel 1,2,3-triazolyl phenylthiazole analogs as potent anti-HIV-1 NNRT inhibitors. Med. Chem. 2017, 7, 268–275. [Google Scholar]
- Amr, A.; Sabrry, N.M.; Abdalla, M.M.; Abdel-Wahab, B.F. Synthesis, antiarrhythmic and anticoagulant activities of novel thiazolo derivatives from methyl 2-(thiazol-2-ylcarbamoyl) acetate. Eur. J. Med. Chem. 2009, 44, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Maghraby, M.T.; Abou-Ghadir, O.M.F.; Abdel-Moty, S.G.; Ali, A.Y.; Salem, O.I.A. Novel class of benzimidazole-thiazole hybrids: The privileged scaffolds of potent anti-inflammatory activity with dual inhibition of cyclooxygenase and 15-lipoxygenase enzymes. Bioorg Med. Chem. 2020, 28, 115403. [Google Scholar] [CrossRef] [PubMed]
- Ruberte, A.C.; Ramos-Inza, S.; Aydillo, C.; Talavera, I.; Encío, I.; Plano, D.; Sanmartín, C. Novel N,N′-disubstituted acylselenoureas as potential antioxidant and cytotoxic agents. Antioxidants 2020, 9, 55. [Google Scholar] [CrossRef] [PubMed]
- Suvannang, N.; Nantasenamat, C.; Isarankura-Na-Ayudhya, C.; Prachayasittikul, V. Molecular Docking of Aromatase Inhibitors. Molecules 2011, 16, 3597–3617. [Google Scholar] [CrossRef]
- Mansourian, M.; Hassanzadeh, F.; Shahlaei, M. Exploring the interaction between epidermal growth factor receptor tyrosine kinase and some of the synthesized inhibitors using combination of in-silico and in-vitro cytotoxicity methods. Res. Pharm. Sci. 2018, 13, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Ikwu, F.A.; Isyaku, Y.; Obadawo, B.S.; Lawal, H.A.; Ajibowu, S.A. In silico design and molecular docking study of CDK2 inhibitors with potent cytotoxic activity against HCT116 colorectal cancer cell line. J. Genet. Eng. Biotechnol. 2020, 18, 51. [Google Scholar] [CrossRef] [PubMed]
- Ramos, J.; Muthukumaran, J.; Freire, F.; Paquete-Ferreira, J.; Otrelo-Cardoso, A.R.; Svergun, D.; Panjkovich, A.; Santos-Silva, T. Shedding Light on the Interaction of Human Anti-Apoptotic Bcl-2 Protein with Ligands through Biophysical and in Silico Studies. Int. J. Mol. Sci. 2019, 20, 860. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival: Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Abou-Seri, S.M.; Eldehna, W.M.; Ali, M.M.; Abou El Ella, D.A. 1-Piperazinylphthalazines as potential VEGFR-2 inhibitors and anticancer agents: Synthesis and in vitro biological evaluation. Eur. J. Med. Chem. 2016, 107, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Amin, N.H.; Elsaadi, M.T.; Zaki, S.S.; Abdel-Rahman, H.M. Design, synthesis and molecular modeling studies of 2-styrylquinazoline derivatives as EGFR inhibitors and apoptosis inducers. Bioorg. Chem. 2020, 105, 104358. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, K.; Fukuda, T.; Isogai, H.; Okumura, K.; Krstic-Demonacos, M.; Isogai, E. Antimicrobial peptide FF/CAP18 induces apoptotic cell death in HCT116 colon cancer cells via changes in the metabolic profile. Int. J. Oncol. 2015, 46, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Mourad, A.A.; Rizzk, Y.W.; Zaki, I.; Mohammed, F.Z.; El Behery, M. Synthesis and cytotoxicity screening of some synthesized hybrid nitrogen molecules as anticancer agents. J. Mol. Struct. 2021, 1242, 130722. [Google Scholar] [CrossRef]
- Abu Almaaty, A.H.; Elgrahy, N.A.; Fayad, E.; Abu Ali, O.A.; Mahdy, A.R.; Barakat, L.A.; El Behery, M. Design, synthesis and anticancer evaluation of substituted cinnamic acid bearing 2-quinolone hybrid derivatives. Molecules 2021, 26, 4724. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 Values (µM) of Compounds | MCF-7 | HepG2 |
|---|---|---|
| 4a | 12.7 ± 0.77 | 6.69 ± 0.41 |
| 4b | 31.5 ± 1.91 | 51.7 ± 3.13 |
| 4c | 2.57 ± 0.16 | 7.26 ± 0.44 |
| 5 | 28.0 ± 1.69 | 26.8 ± 1.62 |
| Staurosporine | 6.77 ± 0.41 | 8.4 ± 0.51 |
| Compounds | VEGFR-2 IC50 Values (µM) |
|---|---|
| 4c | 0.15 |
| Sorafenib | 0.059 |
| Score (kcal/ mol) | RMSD | Ligand | Receptor (Key Amino Acids) | Interaction | Distance/E (kcal/mol) | |
|---|---|---|---|---|---|---|
| Comp 4a | −6.51 | 1.13 | S 22 | PRO 429 | H-acceptor | 2.96 (1.6) |
| 6-ring | CYS 437 | pi-H | 3.60 (−0.6) | |||
| Comp 4b | −6.45 | 0.63 | NH 13 | CYS 437 | H-donor | 3.71 (−0.7) |
| O 18 | ARG 115 | H-acceptor | 2.96 (−2.6) | |||
| Comp 4c | −7.91 | 1.35 | NH 13 | ARG 435 | H-donor | 2.99 (−4.4) |
| O 18 | TRP 141 | H-acceptor | 3.37 (−1.5) | |||
| O 18 | ARG 435 | H-acceptor | 3.56 (−0.6) | |||
| Comp 5 | −7.63 | 1.33 | NH 14 | MET 311 | H-donor | 3.70 (−1.9) |
| O 26 | VAL 369 | H-acceptor | 3.00 (−3.4) | |||
| O 26 | VAL370 | H-acceptor | 3.04 (−1.2) |
| Score (kcal/mol) | RMSD | Ligand | Receptor (Key Amino Acids) | Interaction | Distance/E (kcal/mol) | |
|---|---|---|---|---|---|---|
| Comp 4a | −4.35 | 1.47 | S 22 | CYS 20 | H-acceptor | 4.05 (−0.3) |
| S 22 | CYS 20 | H-acceptor | 3.85 (−0.3) | |||
| OH 24 | SER 2 | H-donor | 3.02 (−0.8) | |||
| Comp 4b | −4.5 | 1.21 | O 18 | GLY 5 | H-acceptor | 3.01 (−2.7) |
| Comp 4c | −4.72 | 1.33 | S 22 | CYS 20 | H-acceptor pi-cation | 3.48 (−0.5) |
| 6-ring | ASN 1 | 4.00 (−1.5) | ||||
| Comp 5 | −4.56 | 0.92 | NH 14 | CYS 20 | H-donor | 3.00 (−3.1) |
| NH 14 | CYS 20 | H-donor | 3.95 (−2.7) | |||
| S 23 | CYS 20 | H-acceptor | 4.21(−0.4) | |||
| S 23 | CYS 20 | H-acceptor | 3.64 (−0.4) |
| Score (kcal/mol) | RMSD | Ligand | Receptor (Key Amino Acids) | Interaction | Distance/E (kcal/mol) | |
|---|---|---|---|---|---|---|
| Comp 4a | −5.21 | 1.57 | O 18 | LEU 83 | H-acceptor | 3.09 (−3.3) |
| Comp 4b | −5.44 | 1.1 | S 22 | ASP 86 | H-acceptor | 4.02 (−1.5) |
| OH 24 | ASP 145 | H-donor | 2.80 (−4.3) | |||
| O 18 | LEU 83 | H-acceptor | 3.17 (−2.4) | |||
| Comp 4c | −6.67 | 0.93 | OH 24 | ASP 86 | H-donor | 3.08 (−2.6) |
| O 18 | LYS 33 | H-acceptor | 2.99 (−8.4) | |||
| Comp 5 | −6.64 | 1.4 | NH 14 | LEU 83 | H-donor | 3.16 (−2.3) |
| S 23 | ASP 86 | H-acceptor | 3.49 (−1.9) |
| Score (kcal/mol) | RMSD | Ligand | Receptor (Key Amino Acids) | Interaction | Distance/E (kcal/mol) | |
|---|---|---|---|---|---|---|
| Comp 4a | −4.6 | 1.17 | N 16 | ARG 143 | H-acceptor | 2.99 (−5.4) |
| O 18 | ARG 143 | H-acceptor | 3.30 (−0.9) | |||
| Comp 4b | −5.15 | 0.57 | NH 13 | GLU 133 | H-donor | 2.91 (−1.5) |
| S 22 | GLU 33 | H-acceptor | 3.59 (−1.1) | |||
| S 22 | ASP 37 | H-acceptor | 3.77 (−1.1) | |||
| O 18 | ARG 143 | H-acceptor | 3.14 (−1.2) | |||
| 6-ring | MET 112 | pi-H | 3.59 (−0.7) | |||
| Comp 4c | −5.53 | 1.07 | N 16 | ARG 143 | H-acceptor | 3.52 (−0.9) |
| O 18 | ARG 143 | H-acceptor | 2.98 (−3.4) | |||
| Comp 5 | −5.7 | 1.29 | NH 14 | ASP 108 | H-donor | 2.93 (−7.2) |
| S 23 | ASP 108 | H-acceptor | 4.03 (−1.6) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Salmi, F.A.; Alrohaimi, A.H.; Behery, M.E.; Megahed, W.; Abu Ali, O.A.; Elsaid, F.G.; Fayad, E.; Mohammed, F.Z.; Keshta, A.T. Anticancer Studies of Newly Synthesized Thiazole Derivatives: Synthesis, Characterization, Biological Activity, and Molecular Docking. Crystals 2023, 13, 1546. https://doi.org/10.3390/cryst13111546
Al-Salmi FA, Alrohaimi AH, Behery ME, Megahed W, Abu Ali OA, Elsaid FG, Fayad E, Mohammed FZ, Keshta AT. Anticancer Studies of Newly Synthesized Thiazole Derivatives: Synthesis, Characterization, Biological Activity, and Molecular Docking. Crystals. 2023; 13(11):1546. https://doi.org/10.3390/cryst13111546
Chicago/Turabian StyleAl-Salmi, Fawziah A., Abdulmohsen H. Alrohaimi, Mohammed El Behery, Walaa Megahed, Ola A. Abu Ali, Fahmy G. Elsaid, Eman Fayad, Faten Z. Mohammed, and Akaber T. Keshta. 2023. "Anticancer Studies of Newly Synthesized Thiazole Derivatives: Synthesis, Characterization, Biological Activity, and Molecular Docking" Crystals 13, no. 11: 1546. https://doi.org/10.3390/cryst13111546
APA StyleAl-Salmi, F. A., Alrohaimi, A. H., Behery, M. E., Megahed, W., Abu Ali, O. A., Elsaid, F. G., Fayad, E., Mohammed, F. Z., & Keshta, A. T. (2023). Anticancer Studies of Newly Synthesized Thiazole Derivatives: Synthesis, Characterization, Biological Activity, and Molecular Docking. Crystals, 13(11), 1546. https://doi.org/10.3390/cryst13111546