Abstract
The adsorption equilibrium of methane (CH4) and carbon dioxide (CO2) on the metal–organic framework (MOF) UiO-66 is studied via molecular simulation. UiO-66 is a versatile MOF with vast potential for various adsorption processes, such as biogas upgrading, CO2 capture, and natural gas storage. The molecular simulations employ the grand canonical Monte Carlo (GCMC) method, covering a temperature range of 298–343 K and pressures up to 70 bar for CH4 and 30 bar for CO2. The accuracy of different forcefields in describing the adsorption equilibria is evaluated. Two modelling approaches are explored: (i) lumping each hydrogen atom in the MOF framework to the heavy atom it is bonded to (united atom approximation) and (ii) considering explicit hydrogen atoms. Additionally, the influence of electrical charges on CO2 adsorption is also evaluated. The findings indicate that the most effective forcefield to describe the adsorption equilibrium is a united atom forcefield based on the TraPPE parametrization. This approach also yields an accurate calculation of the isosteric heat of adsorption. In the case of CO2, it is observed that the use of electrical charges enhances the prediction of the heat of adsorption, especially in the low-coverage region.
1. Introduction
Metal–organic frameworks (MOFs) are highly ordered crystalline materials composed of metal ion cluster nodes connected by organic linkers. These frameworks offer versatility through customization by altering the metal-center/organic-linker combination and incorporating specific functionalities. MOFs have been studied for several applications, including gas storage, catalysis, sensing, and drug delivery. Among these applications, the study of adsorption-based separations stands out as one the most interesting fields of study [1,2,3,4,5,6,7,8].
UiO-66, like ZIF-8, ZIF-67, or HKUST-1, stands out as an interesting material within the vast array of MOFs due to its suitability for separation processes. Notably, UiO-66 boasts excellent adsorption properties, alongside remarkable thermal and mechanical stability. Additionally, UiO-66 demonstrates exceptional resistance to various solvents, such as water, dimethylformamide, benzene, and acetone. Even under high mechanical pressures of up to 10,000 kg/cm2, UiO-66 retains its structural integrity without compromise [9,10,11].
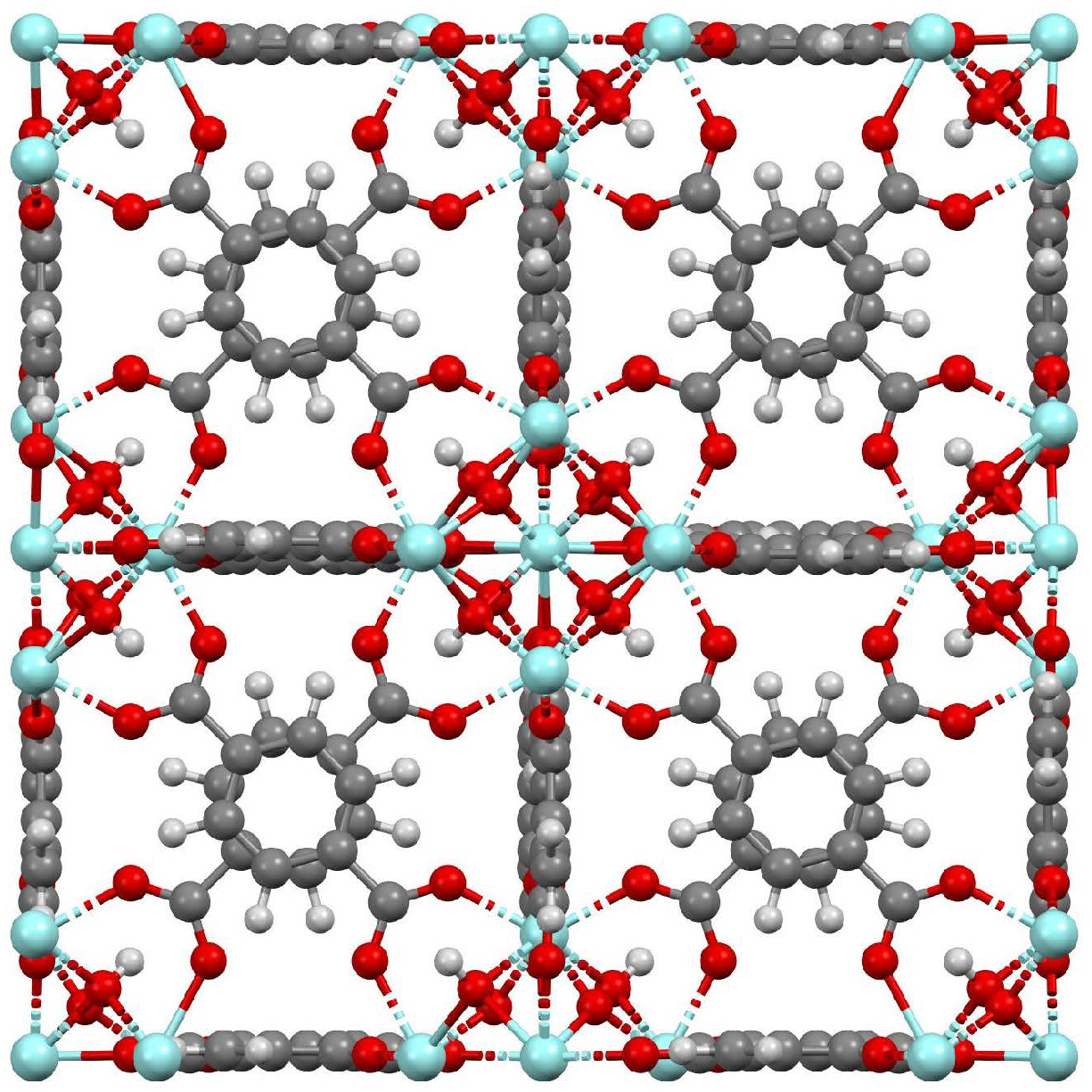
The UiO-66 framework is based on the linkage of zirconium oxide clusters Zr6O4(OH)4 through 1,4-benzene-dicarboxylate (BDC) ligands in a three-dimensional structure with tetrahedral and octahedral cages of approximately 8 Å and 11 Å, respectively. The windows accessing the cages have apertures of around 6 Å [12,13]. This molecular configuration promotes an exceptional stability due to the high degree of coordination of Zr−O metal centers to the organic linkers [10,11]. Several authors have reported UiO-66 surface areas of 1000–1400 m2/g and pore volumes of 0.4–0.8 cm3/g [11,13,14,15,16,17,18,19,20]. The UiO-66 framework structure is shown in Figure 1, which was generated from the CIF file reported by Yang et al. [21]. It shows an axial view of a simulation box containing 2 × 2 × 2 unit cells of UiO-66.
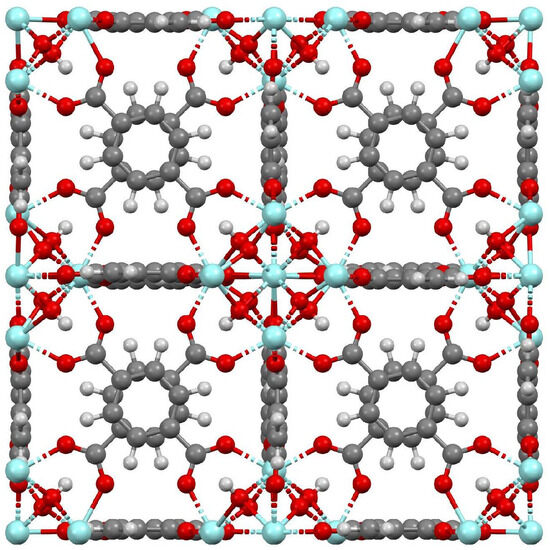
Figure 1.
UiO-66 framework structure. Color coding: gray—carbon; white—hydrogen; red—oxygen; light blue—zirconium.
UiO-66 has high potential as an adsorbent for applications in gas and liquid phase separations. It has been evaluated for the adsorption of hexane and xylene isomers [16,19,22,23], boron [24], arsenic [25], and diverse water contaminants [9]. UiO-66 has also been studied for gas storage [21] and, recently, it was studied for neon adsorption [26].
The potential application of UiO-66 in carbon dioxide (CO2)/methane (CH4) separation has been thoroughly studied. Cavka et al. [15] studied the adsorption of CH4 and CO2 on UiO-66, between 298 K and 343 K, and up to 80 and 30 bar, respectively. Yang et al. [18] studied the CO2/CH4 co-adsorption, concluding that UiO-66 can be a good candidate for pressure swing adsorption (PSA) applications, as it presents a good selectivity and working capacity; also, UiO-66 is easily regenerable.
UiO-66 functionalized materials have also been studied [12,27]. Xian et al. [28] prepared a series of polyethylenimine (PEI)-impregnated UiO-66 samples, showing that this approach can significantly increase the CO2 adsorption capacity and CO2/CH4 selectivity. Jasuja and Walton [14] synthesized a dimethyl-functionalized UiO-66, enhancing the CO2/CH4 selectivity at high pressures.
Molecular simulation is a powerful and extensively utilized method for investigating gas adsorption equilibria on nanoporous materials, particularly in the realm of MOFs [29,30,31,32]. Classical forcefields, such as UFF, DREIDING, OPLS, and TraPPE, are commonly employed for these simulations. These forcefields have been previously employed with success to describe several MOFs, such as Cu-BTC, MOF-5, IRMOFs, and MIL materials, as evidenced by previous studies [26,29,33,34,35,36,37].
Regarding UiO-66, Yang et al. [18] used grand canonical Monte Carlo (GCMC) simulations to calculate the pure and mixed CO2/CH4 adsorption equilibrium. Additionally, molecular dynamics were employed to study the co-diffusion of mixtures containing both species. The study revealed that CO2 preferentially occupies the UiO-66 tetrahedral cages, while CH4 is predominantly confined to the octahedral cages. Remarkably, CO2, being a slower-diffusing molecule, exhibited the intriguing effect of enhancing the mobility of the faster CH4 within UiO-66. The same group also used GCMC to study UiO-66 for CH4 storage. Other authors have used molecular simulation to study the adsorption of diverse species on UiO-66, including NH3, H2S, and CO2, [38] xylene isomers, [22,23] and water [39,40].
There is compelling evidence that UiO-66 is a very promising candidate for the adsorptive separation of CO2/CH4. Therefore, in this work, we employ GCMC molecular simulation to predict the CO2 and CH4 adsorption equilibria on UiO-66 at three different temperatures: 298 K, 313 K, and 343 K, covering the pressure ranges of 0–30 bar and 0–70 bar, respectively. We evaluate the influence of using various forcefields in our calculations by comparing the theoretical results with the experimental data published by Cavka et al. [15]. In this work, we use the TraPPE parametrization in the description of the crystalline structure as opposed to the approach taken by Yang et al. [18], in which the authors used the DREIDING and UFF forcefields to describe the MOF framework.
2. Methods
Molecular Models and Solid Lattice
In our GCMC molecular simulations, we adopt a rigid model for the UiO-66 framework. This involves nullifying the bond, angle, torsion, and improper torsion terms, while omitting the van der Waals and Coulombic interactions between the framework atoms. This approach is employed to optimize computational efficiency and save valuable computational time. The UiO-66 crystallographic structure is that obtained by Yang and co-workers through X-ray diffraction and further refinements resulting from geometrical optimization based on density-functional theory (DFT) [18].
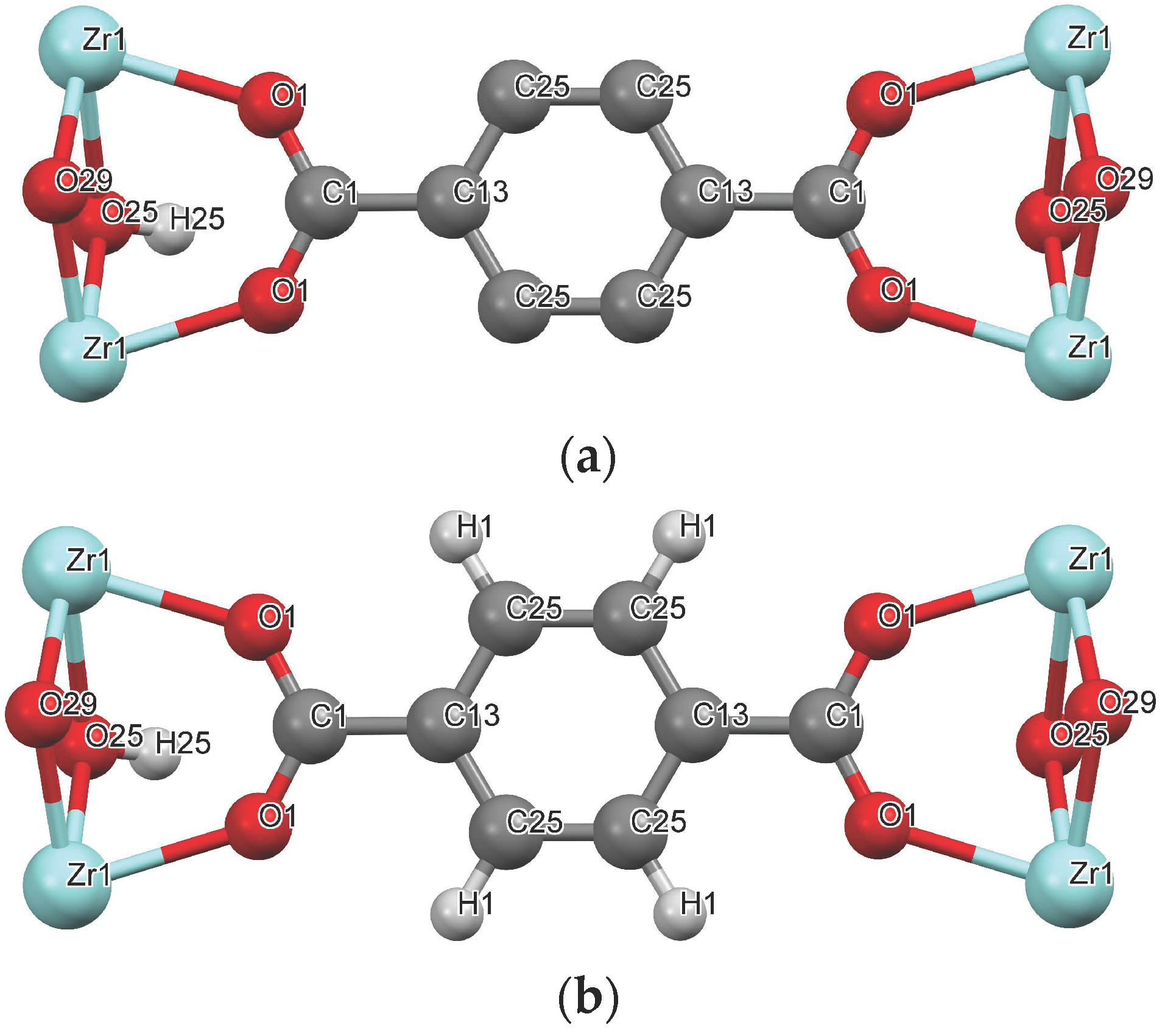
For the parameterization of the solid–fluid dispersive interactions, the Lennard-Jones (LJ) parameters of the organic linker were taken from those reported for similar organic molecules. Two different modelling approaches are studied. One approach is to unite each carbon of the framework and its bonded hydrogen(s) into a single interaction site; this is the united-atom approximation, here designated as UiO-66-UA (Figure 2a). Another approach is to treat each hydrogen and carbon atom as distinct interaction sites (UiO-66-EH), as shown in Figure 2b. Furthermore, the influence of considering electrical charges was evaluated for the case of CO2 adsorption.
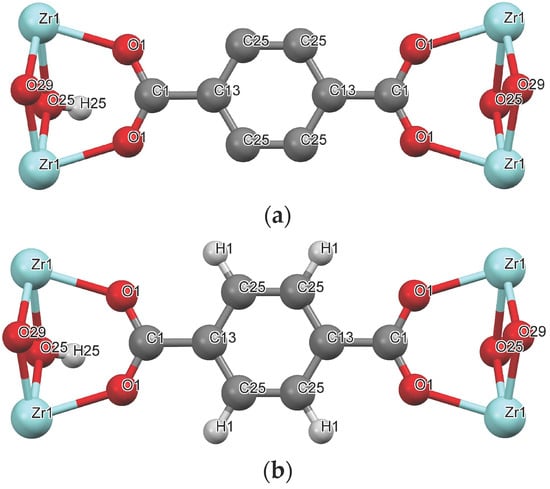
Figure 2.
UiO-66 framework representation for the (a) UA and (b) ED models. Color coding: gray—carbon; white—hydrogen; red—oxygen; light blue—zirconium.
The TraPPE forcefield is employed in the simulations. In TraPPE, non-bonded interactions are described by pairwise-additive LJ 12-6 potentials and Coulombic interactions of partial charges with unlike LJ interactions computed through Lorentz–Berthelot combining rules.
Methane is modelled as a single chargeless LJ interaction site (Table 1). Carbon dioxide is represented as a three-site molecule. The intrinsic quadrupole moment is described by a base charge model. The CO bond length is 1.16 Å, and the bond angle O-C-O is 180°. As stated above, the non-bonded CO2 interactions are described by the pairwise-additive LJ 12-6 potentials and Coulombic interactions of partial charges given in Table 1.
The GCMC calculations for CO2 adsorption on UiO-66 were performed using three different forcefields, namely, the UA and chargeless MOF framework, the UA and partial charges placed on the framework atoms (UAq), and the explicit hydrogen forcefield with electrostatic charges (EHq). The use of these three approaches gives an understanding of the relevance of the hydrogen atoms and the electric charges of the MOF on the adsorption of CO2 by UiO-66. The electric charges employed in UAq and EHq are those reported by Yang et al. [18]. These partial charges were extracted from density-functional theory (DFT) calculations and the Mulliken charge partitioning method, as reported in [41]; such calculations were based on the PW91 GGA functional combined with the double numerical basis set containing a polarization function (DNP). The LJ interactions were truncated at Å, and analytical tail corrections were applied. The Coulombic electrostatic interactions were computed via the Ewald summation method utilizing a constant number of 5 inverse space vectors), an electrostatic cutoff equal to , and a damping factor equal to , where is the shortest length of the simulation box.

Table 1.
Lennard-Jones parameters and partial charges for the TraPPE forcefield.
Table 1.
Lennard-Jones parameters and partial charges for the TraPPE forcefield.
| Label | σ (Å) | ε/kB (K) | q (e) | Ref. |
|---|---|---|---|---|
| Methane | ||||
| CH4 | 3.73 | 148 | - | [42] |
| Carbon dioxide | ||||
| C_CO2 | 2.80 | 27.0 | +0.70 | [43] |
| O_CO2 | 3.05 | 79.0 | −0.35 | [43] |
The LJ parameterization of UiO-66 using the UA forcefield (UiO-66-UA) considers seven pseudo-atoms: Zr, C25, C13, C1, O25, O1, and O29, as illustrated in Figure 2a and indicated in Table 2. Hydrogen H25 is lumped with the pseudo-atom C25, whereas the vdW contribution of the polar H connected to hydroxyl O25 is neglected. The LJ parameters for the aromatic carbons are taken from the TraPPE-UA parameterization of toluene; the [CH]aro and the [C]aro-CHy correspond to the C25 and the C13, respectively. The C1 parameters assigned correspond to those of a carbon atom belonging to a carboxylic acid/ester group. The parameters for the oxygens O1 and O29 were taken from the TraPPE-UA description of ethers, where the LJ values correspond to a CHx-[O]-CHy. The values for oxygen O25 and the hydroxyl H were taken from the TraPPE-UA parameterization of alcohols, CHx-[O]-H and CHx-O-[H]. The Zr parameterization was taken from the UFF forcefield.
Table 2.
The Lennard-Jones parameters of the UiO-66 framework for the UA forcefield and partial charges for the UAq forcefield. The electric charges are those reported by Yang et al. [18]. The lumped charge for C25 is (−0.121) + (+0.127) = +0.006 (see Table 3).
The UiO-66-EH forcefield uses nine atoms, i.e., the seven atoms used in the UiO-66-UA forcefield, plus H1 and H25 (Figure 2b; Table 3). The LJ parameters for atoms Zr, C13, C1, O1, and O29 are the same as those for UiO-66-UA. The parameters of the atoms C25 and H1 are obtained through the TraPPE-EH parameterization of benzene, correspondent to a X(aro)-[C]-(aro)-X(aro) and [H]-C(aro). Due to information scarcity, the O25 and H25 parametrization employed corresponds to the TraPPE-UA of an alcohol.
Table 3.
The Lennard-Jones parameters of the UiO-66 framework for the EH forcefield and charges for the EHq forcefield.
3. Results and Discussion
3.1. Single-Component Adsorption Equilibria
In this study, we performed GCMC molecular simulations with different forcefields to investigate the adsorption of CO2 and CH4 on UiO-66 at three different temperatures: 298 K, 313 K, and 343 K. The pressure ranges examined were 0–30 bar for CO2 and 0–70 bar for CH4. The adsorption equilibria results can be analyzed through various interpretations, e.g., excess and absolute quantities. Here, we focus on reporting the adsorbed quantities in terms of the absolute amount adsorbed, as it is a quantity that is directly obtained from the GCMC calculations. The absolute amount adsorbed obtained from molecular simulations is calculated as
where is the ensemble average number of adsorbate molecules in the simulation box, is the adsorbent mass in the simulation box, and is Avogadro’s constant.
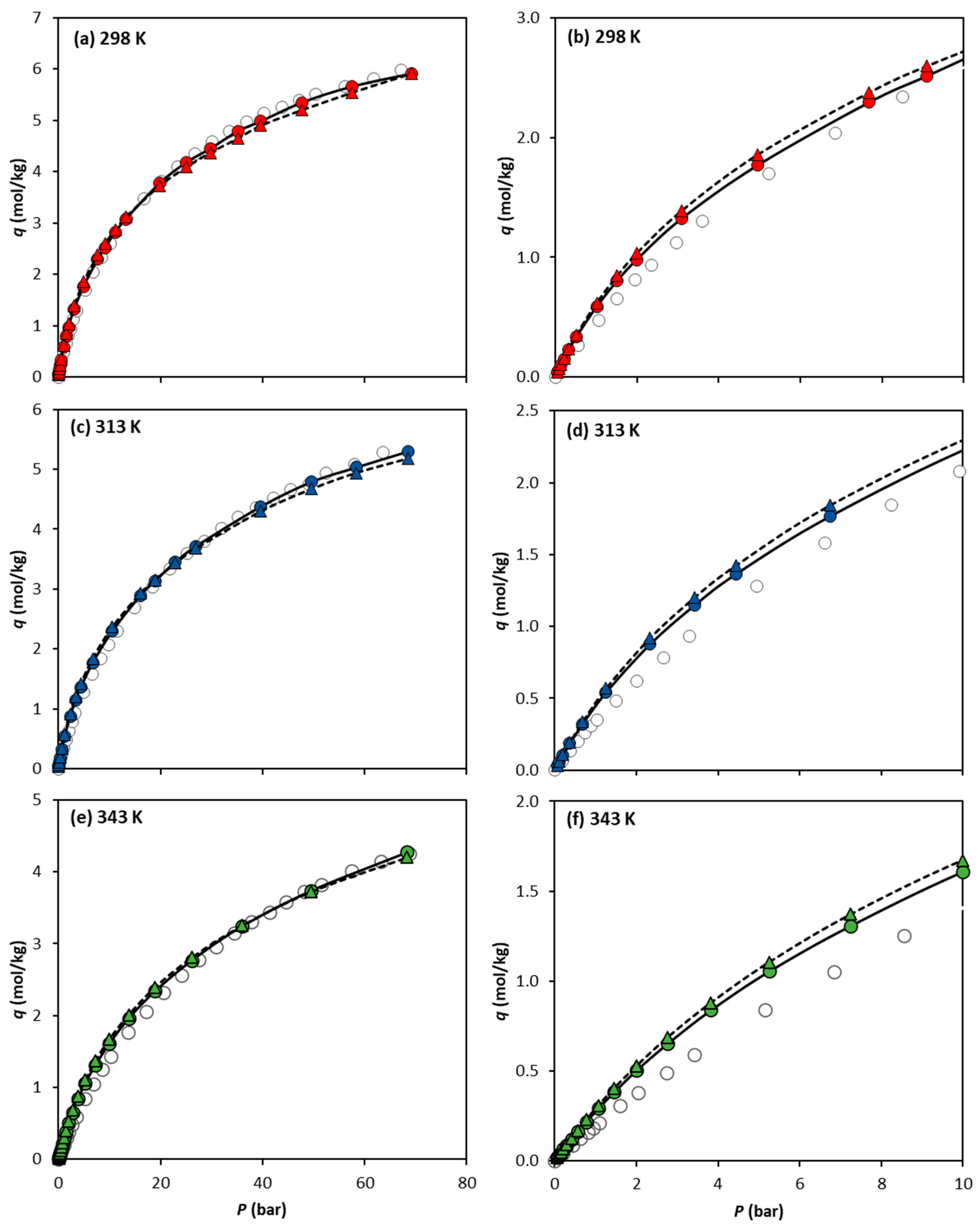
Figure 3 compares the absolute experimental adsorption isotherms of CH4 reported by Cavka et al. [14] at 298 K, 313 K, and 343 K, with the simulation results obtained using the UiO-66-UA and UiO-66-EH forcefields. Surprisingly, both forcefields demonstrate excellent agreement with the experimental CH4 adsorption data, despite covering a wide range of temperatures and pressures. It is also seen that the UiO-66-UA forcefield gives slightly better predictions, although the differences between both forcefields are not very significant.
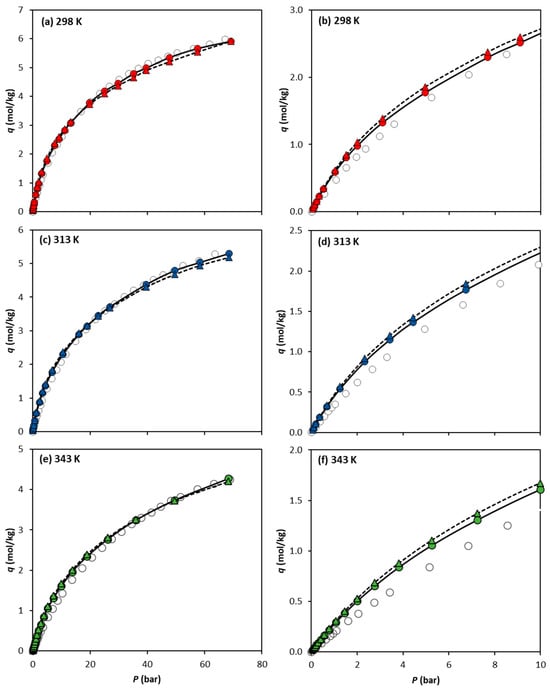
Figure 3.
Comparison of CH4 absolute adsorption equilibrium isotherms on UiO-66 at (a) 298 K, (c) 313 K, and (e) 343 K, obtained experimentally by Cavka et al. (open circles) and via molecular simulation using the forcefields UiO-66-UA (filled circles) and UiO-66-EH (filled triangles). The isotherm lower-pressure region up to 10 bar is detailed in (b) 298 K, (d) 313 K, and (f) 343 K. Lines are drawn as a guide for the eye.
Looking at the lower-pressure region (0–10 bar) represented in Figure 3b,d,f for 298 K, 313 K and 343 K, respectively, it can be observed that, in this region, the predictions using the UiO-66-UA or UiO-66-EH forcefields result in minimal differences in the adsorption data calculated. As the temperature increases, larger differences between the simulation data and experimental results are observed.
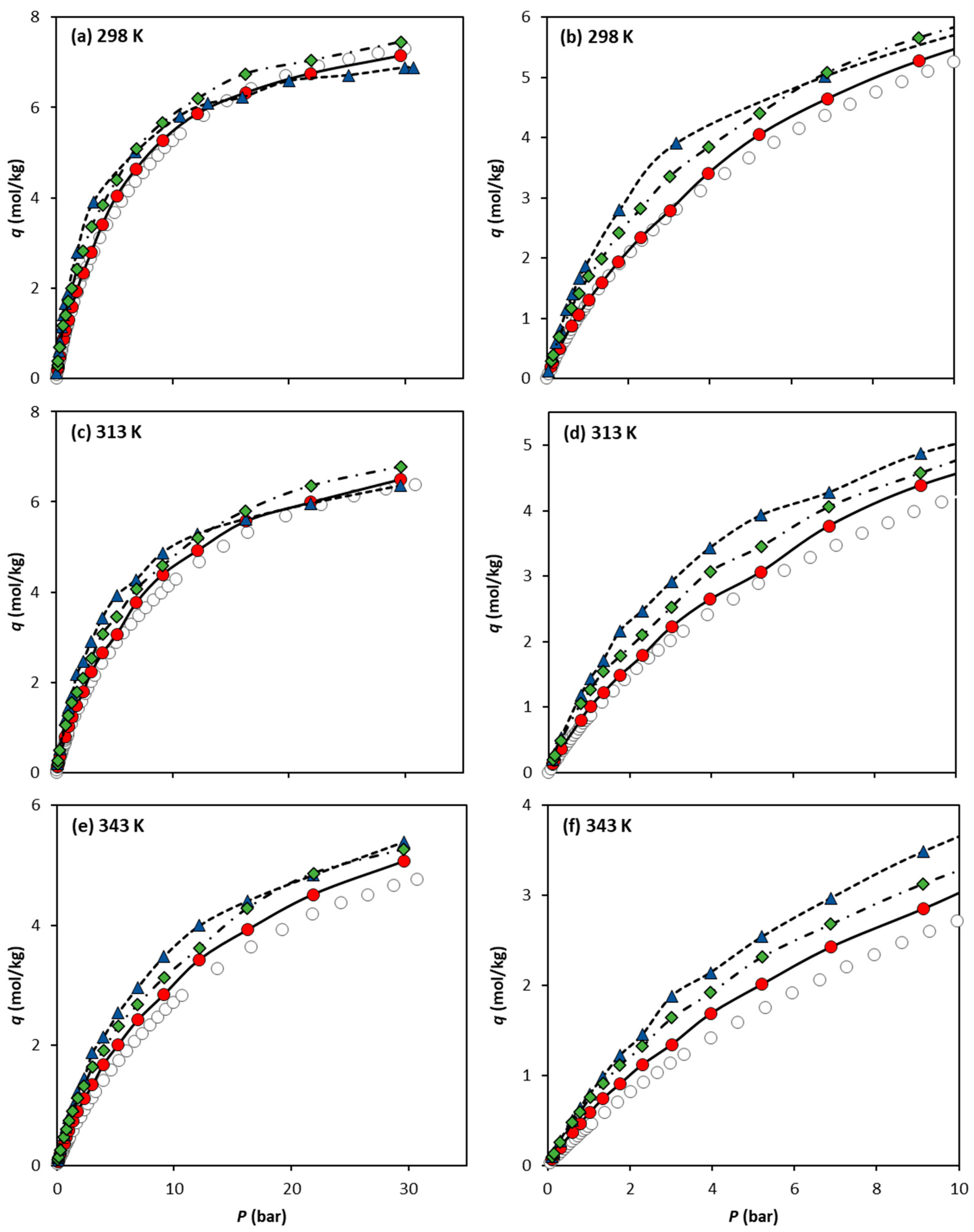
Figure 4 compares the CO2 adsorption equilibrium data measured experimentally by Cavka et al. [15] and our molecular simulation data obtained using the three different forcefields. For the sake of clarity, each experimental adsorption equilibrium isotherm is compared to the predicted GCMC data in a single graph (Figure 4a–c for 298 K, 313 K, and 343 K, respectively). The results obtained show that the UiO-66-UA forcefield, which neglects the electric charges (UiO-66-UA) in the solid, is the more accurate forcefield for the adsorption equilibrium predictions within the pressure and temperature ranges studied. It is widely acknowledged that simulated adsorption data are highly sensitive to the charge parametrization used. In cases where the partial charges are not well-tuned for a specific solid–fluid system, their impact on the results can be more detrimental than if they were entirely neglected, as seems to be the case here.
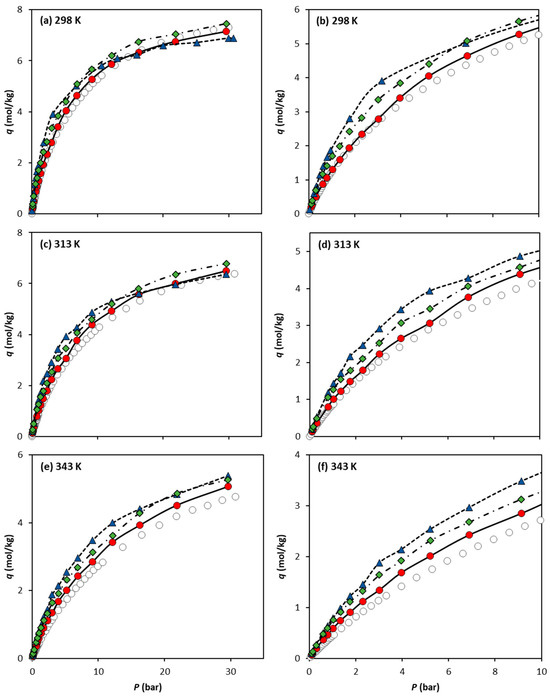
Figure 4.
Comparison of CO2 absolute adsorption equilibrium isotherms on UiO-66 at (a) 298 K, (c) 313 K, and (e) 343 K, obtained experimentally by Cavka et al. (open circles) and via molecular simulation using the forcefields UiO-66-UA (filled circles), UiO-66-EHq (filled triangles), and UiO-66-UAq (filled diamonds). The isotherm lower-pressure region up to 10 bar is detailed in (b) 298 K, (d) 313 K, and (f) 343 K. Lines are drawn as a guide for the eye.
A GCMC simulation of methane condensation at its normal boiling point, , within the porous structure of the MOF gives a maximum methane loading of ca. 0.0118 mol/g. By multiplying this value with the molar volume of methane at its normal boiling point, converts it into an equivalent pore volume. The obtained value of 0.45 cm3/g is in agreement with the theoretical pore volume of pristine UiO-66, but larger than the experimental pore volume of 0.36 cm3/g reported by Cavka et al. [15] for their sample, as measured from argon adsorption at 87 K (). This suggests that, based solely on gravimetric considerations, one might expect the simulated saturation adsorption capacity to surpass the experimental one by a factor of approximately 0.45/0.36. However, this discrepancy is not observed in our comparative analysis of the methane and carbon dioxide data.
It is important to note that UiO-66 is known to be susceptible to defects. Several authors [49,50,51] have argued that most, if not all, UiO-66 samples contain a significant number of missing linkers and/or missing cluster defects. Furthermore, research has shown that the presence of such defects can significantly impact the adsorption isotherms [40,52]. Given the likelihood that the sample of Cavka et al. [15] may also contain defects, it is somewhat surprising that the simulations, assuming a perfect crystal, align so closely with the experimental measurements.
The GCMC data obtained for both adsorbates show that explicitly considering the hydrogen atoms in the MOF modelling does not increase the accuracy of the simulated adsorption equilibria. This is observed more clearly in the calculations using the forcefield UiO-66-EHq for CO2 (e.g., 298 K, Figure 4a,b), where the isotherm does not have the same trend as the experimental data; i.e., the calculations overestimate the experimental values at lower pressures and, at a higher pressure, the obtained simulations underestimate them. The simulation results indicate that the explicit description of the UiO-66 electrical charges does not benefit the prediction of CO2 adsorption equilibrium. This indicates that the electrostatic parameterization obtained by Yang and co-workers [18,21] through quantum mechanics calculations is not suitable for being combined with the TraPPE forcefield.
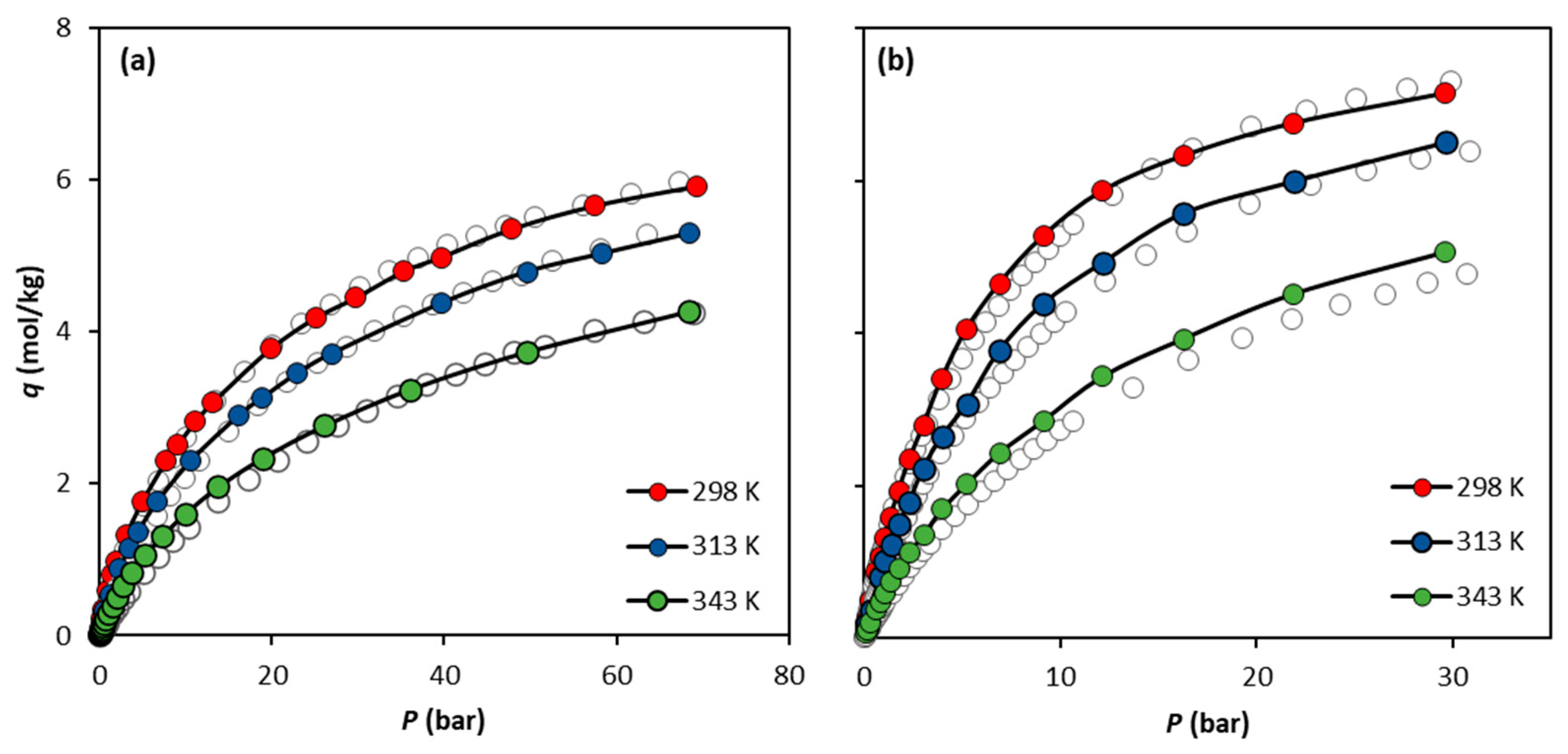
The calculations show that, similarly to CH4, the best forcefield to replicate the CO2 experimental data is the UiO-66-UA. Figure 5 presents an overall comparison between the experimental data and the simulation predictions for both CO2 and CH4 using the UiO-66-UA forcefield for the three temperatures studied. The data in Figure 5b show that, with the increase in temperature, the CO2 simulated data tend to slightly overestimate the experimental results.
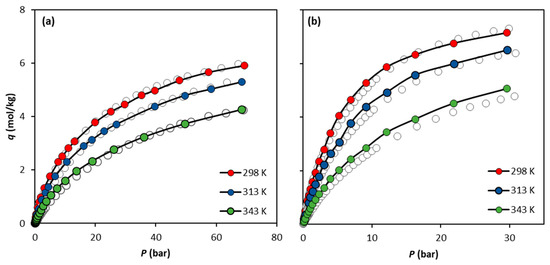
Figure 5.
Comparison of the adsorption equilibrium isotherms of (a) CH4 and (b) CO2 on UiO-66 at 298 K, 313 K, and 343 K obtained experimentally by Cavka et al. (open circles) and via molecular simulation using the forcefield UiO-66-UA (filled circles). Solid lines are drawn as a guide for the eye.
3.2. Adsorption Energetics
The isosteric heat of adsorption, , quantifies the strength of the interactions between the adsorbate molecules and the MOF solid lattice. In this work, the heat of adsorption is obtained directly from statistical thermodynamics through the GCMC simulations using the co-variance formulation of Nicholson and Parsonage [53],
where the triangular brackets denote an ensemble average, the number of adsorbate molecules in the simulation box, and the configurational energy. The calculations rely on numerous particle insertions and deletions in the grand canonical ensemble. As a consequence, performing long simulations becomes necessary to achieve accurate statistics for calculating averages, particularly when dealing with very low loading conditions where the number of molecules is significantly reduced [54].
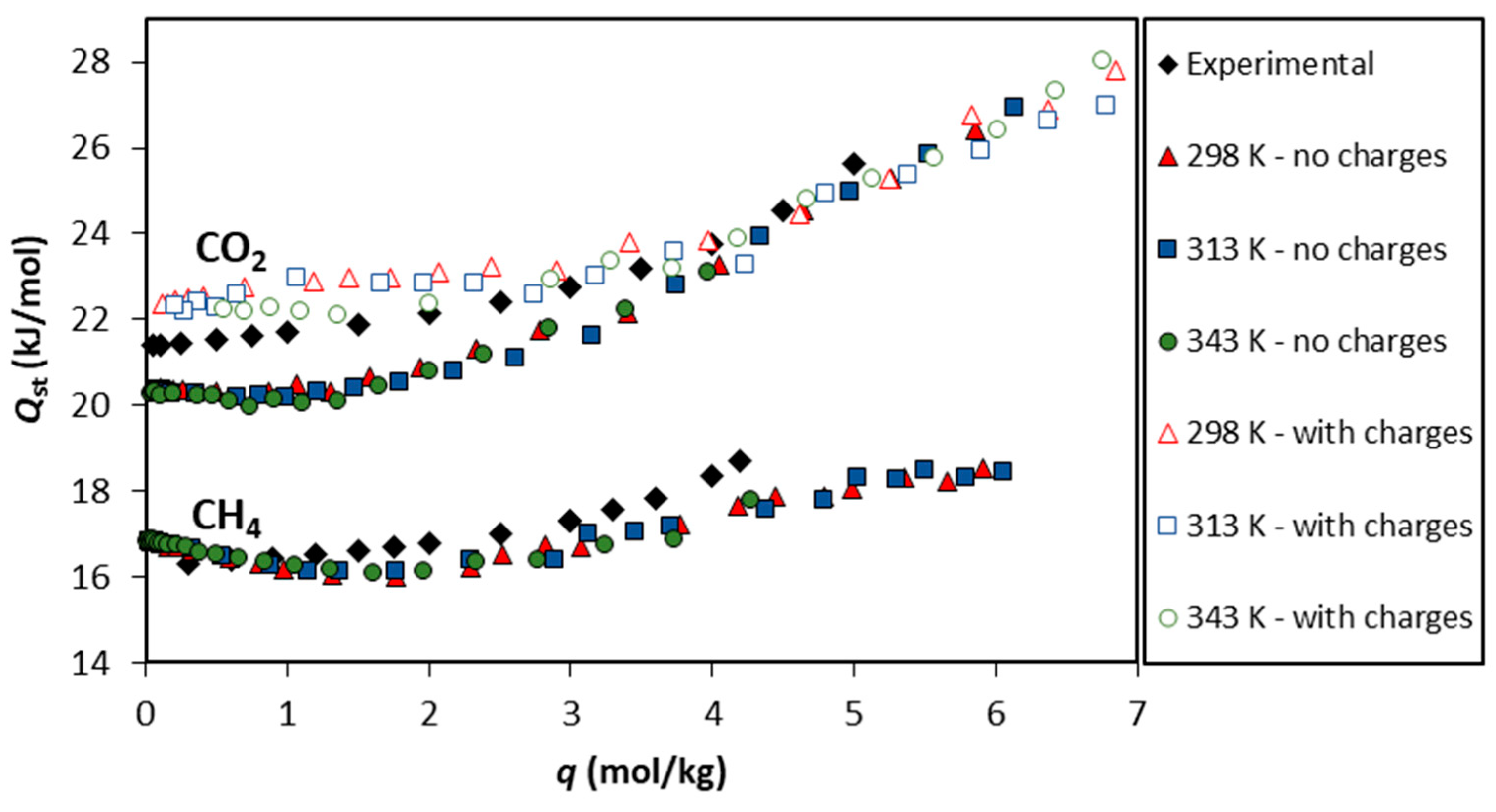
The isosteric heats calculated with the UiO-66-UA forcefield are presented in Figure 6 for both gases studied. Regarding the theoretical values for CH4, they range from 16 to 18 kJ/mol, slightly decreasing up to a loading of 2 mol/kg and then increasing the up to the maximum loading values studied (~6 mol/kg). The initial decrease in the isosteric heat of adsorption with loading suggests the influence of adsorbent heterogeneity [55].
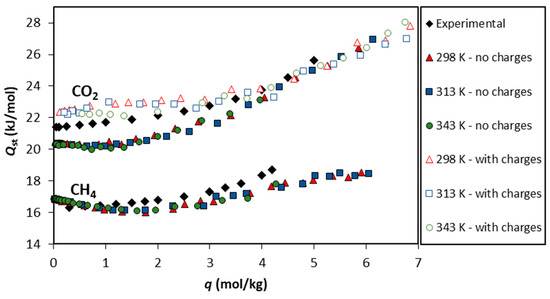
Figure 6.
The isosteric heat of adsorption () as a function of the CO2 and CH4 loadings. The closed colored symbols represent the values predicted via GCMC simulations using the UiO-66-UA forcefield at 298 K, 313 K, and 343 K. The open symbols correspond to the GCMC simulations using electrical charges at the same temperatures, while the black symbols denote the values calculated from the isosteric plot of the adsorption equilibrium experimental data [15].
The calculated from the experimental data reported by Cavka et al. [15] compares very well with the theoretical calculations, although the initial decrease in is not observed. Figure 6 also presents the results for as a function of the CO2 loading on UiO-66. The theoretical values obtained through simulations not considering the electrical charges are within 20 and 27 kJ/mol and show a plateau at low loadings (up to 2 mol/kg) and, for higher CO2 loadings, an increase is observed. The isosteric heat of adsorption calculated from the experimental data presents the same trend as the theoretical values with differences smaller than 2 kJ/mol. Additionally, calculations considering the electrical charges were also performed, and the obtained results show that this approach describes more accurately the heat of adsorption derived from the experimental data at low coverage, as depicted in Figure 6. The study of the heat of adsorption corroborates the success of the forcefields employed to replicate the experimental system.
4. Conclusions
In this work, we report the successful prediction of the CH4 and CO2 adsorption equilibrium on UiO-66 MOF using GCMC molecular simulations with parametrizations based on the TraPPE forcefield. We cover pressures up to 70 bar for methane and up to 30 bar for carbon dioxide; the temperature range analyzed was 298–343 K for both species.
We successfully employed two models: one considering explicit hydrogen atoms (EH) and the other lumping the MOF’s hydrogen atoms to the heavier atom it is bonded to (UA). Both approaches lead to excellent agreement with the experimental CH4 data over the wide temperature and pressure ranges studied. Regarding the adsorption of CO2, the utilization of electrical charges was also evaluated; the results showed that, using the parametrization employed, the most effective forcefield to describe CO2 adsorption is a chargeless UA forcefield.
The CH4 isosteric heat of adsorption obtained from the experimental data can be described with high accuracy using the UiO-66-UA forcefield. In the case of CO2, it is observed that the use of electrical charges enhances the prediction of the heat of adsorption, especially in the low-coverage region.
Author Contributions
Conceptualization, R.P.P.L.R. and J.P.B.M.; methodology, J.P.B.M.; software, J.P.B.M.; formal analysis, J.M.M.M.; investigation, J.M.M.M.; writing—original draft preparation, R.P.P.L.R.; writing—review and editing, R.P.P.L.R. and J.P.B.M.; visualization, J.M.M.M. and R.P.P.L.R.; supervision, J.P.B.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by national funds from FCT/MCTES (Portugal) through Associate Laboratory for Green Chemistry–LAQV [UIDB/50006/2020; UIDP/50006/2020]. Rui Ribeiro also acknowledges funding through the Norma Transitória DL 57/2016 Program Contract.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Millward, A.R.; Yaghi, O.M. Metal-Organic Frameworks with Exceptionally High Capacity for Storage of Carbon Dioxide at Room Temperature. J. Am. Chem. Soc. 2005, 127, 17998–17999. [Google Scholar] [CrossRef] [PubMed]
- Rowsell, J.L.C.; Yaghi, O.M. Metal–Organic Frameworks: A New Class of Porous Materials. Microporous Mesoporous Mater. 2004, 73, 3–14. [Google Scholar] [CrossRef]
- Meek, S.T.; Greathouse, J.A.; Allendorf, M.D. Metal-Organic Frameworks: A Rapidly Growing Class of Versatile Nanoporous Materials. Adv. Mater. 2011, 23, 249–267. [Google Scholar] [CrossRef] [PubMed]
- Mueller, U.; Schubert, M.; Teich, F.; Puetter, H.; Schierle-Arndt, K.; Pastre, J. Metal-Organic Frameworks-Prospective Industrial Applications. J. Mater. Chem. 2006, 16, 626–636. [Google Scholar] [CrossRef]
- Schneemann, A.; Bon, V.; Schwedler, I.; Senkovska, I.; Kaskel, S.; Fischer, R.A. Flexible metal-organic frameworks. Chem. Soc. Rev. 2014, 43, 6062–6096. [Google Scholar] [CrossRef]
- Chen, X.; Li, M.; Lin, M.; Lu, C.; Kumar, A.; Pan, Y.; Liu, J.; Peng, Y. Current and promising applications of Hf(iv)-based MOFs in clinical cancer therapy. J. Mater. Chem. B 2023, 11, 5693–5714. [Google Scholar] [CrossRef]
- Xu, Z.; Wu, Z.; Huang, S.; Ye, K.; Jiang, Y.; Liu, J.; Liu, J.; Lu, X.; Li, B. A metal-organic framework-based immunomodulatory nanoplatform for anti-atherosclerosis treatment. J. Control. Release 2023, 354, 615–625. [Google Scholar] [CrossRef]
- Ma, D.; Li, Z.; Zhu, J.; Zhou, Y.; Chen, L.; Mai, X.; Liufu, M.; Wu, Y.; Li, Y. Inverse and highly selective separation of CO2/C2H2 on a thulium–organic framework. J. Mater. Chem. A 2020, 8, 11933–11937. [Google Scholar] [CrossRef]
- Ahmadijokani, F.; Molavi, H.; Rezakazemi, M.; Tajahmadi, S.; Bahi, A.; Ko, F.; Aminabhavi, T.M.; Li, J.-R.; Arjmand, M. UiO-66 metal–organic frameworks in water treatment: A critical review. Prog. Mater. Sci. 2022, 125, 100904. [Google Scholar] [CrossRef]
- Wu, H.; Yildirim, T.; Zhou, W. Exceptional Mechanical Stability of Highly Porous Zirconium Metal–Organic Framework UiO-66 and Its Important Implications. J. Phys. Chem. Lett. 2013, 4, 925–930. [Google Scholar] [CrossRef]
- Cavka, J.H.; Jakobsen, S.; Olsbye, U.; Guillou, N.; Lamberti, C.; Bordiga, S.; Lillerud, K.P. A New Zirconium Inorganic Building Brick Forming Metal Organic Frameworks with Exceptional Stability. J. Am. Chem. Soc. 2008, 130, 13850–13851. [Google Scholar] [CrossRef] [PubMed]
- Chavan, S.; Vitillo, J.G.; Uddin, M.J.; Bonino, F.; Lamberti, C.; Groppo, E.; Lillerud, K.-P.; Bordiga, S. Functionalization of UiO-66 Metal−Organic Framework and Highly Cross-Linked Polystyrene with Cr(CO)3: In Situ Formation, Stability, and Photoreactivity. Chem. Mater. 2010, 22, 4602–4611. [Google Scholar] [CrossRef]
- Winarta, J.; Shan, B.; McIntyre, S.M.; Ye, L.; Wang, C.; Liu, J.; Mu, B. A Decade of UiO-66 Research: A Historic Review of Dynamic Structure, Synthesis Mechanisms, and Characterization Techniques of an Archetypal Metal–Organic Framework. Cryst. Growth Des. 2020, 20, 1347–1362. [Google Scholar] [CrossRef]
- Jasuja, H.; Walton, K.S. Experimental Study of CO2, CH4, and Water Vapor Adsorption on a Dimethyl-Functionalized Ui0-66 Framework. J. Phys. Chem. C 2013, 117, 7062–7068. [Google Scholar] [CrossRef]
- Cavka, J.H.; Grande, C.A.; Mondino, G.; Blom, R. High Pressure Adsorption of CO2 and CH4 on Zr-MOFs. Ind. Eng. Chem. Res. 2014, 53, 15500–15507. [Google Scholar] [CrossRef]
- Bárcia, P.S.; Guimarães, D.; Mendes, P.A.P.; Silva, J.A.C.; Guillerm, V.; Chevreau, H.; Serre, C.; Rodrigues, A.E. Reverse Shape Selectivity in the Adsorption of Hexane and Xylene Isomers in MOF UiO-66. Microporous Mesoporous Mater. 2011, 139, 67–73. [Google Scholar] [CrossRef]
- Kandiah, M.; Nilsen, M.H.; Usseglio, S.; Jakobsen, S.; Olsbye, U.; Tilset, M.; Larabi, C.; Quadrelli, E.A.; Bonino, F.; Lillerud, K.P. Synthesis and Stability of Tagged UiO-66 Zr-MOFs. Chem. Mater. 2010, 22, 6632–6640. [Google Scholar] [CrossRef]
- Yang, Q.Y.; Wiersum, A.D.; Jobic, H.; Guillerm, V.; Serre, C.; Llewellyn, P.L.; Maurin, G. Understanding the Thermodynamic and Kinetic Behavior of the CO2/CH4 Gas Mixture within the Porous Zirconium Terephthalate UiO-66(Zr): A Joint Experimental and Modeling Approach. J. Phys. Chem. C 2011, 115, 13768–13774. [Google Scholar] [CrossRef]
- Moreira, M.A.; Santos, J.C.; Ferreira, A.F.P.; Loureiro, J.M.; Ragon, F.; Horcajada, P.; Shim, K.-E.; Hwang, Y.-K.; Lee, U.H.; Chang, J.-S.; et al. Reverse Shape Selectivity in the Liquid-Phase Adsorption of Xylene Isomers in Zirconium Terephthalate MOF UiO-66. Langmuir 2012, 28, 5715–5723. [Google Scholar] [CrossRef]
- Edubilli, S.; Gumma, S. A systematic evaluation of UiO-66 metal organic framework for CO2/N2 separation. Sep. Purif. Technol. 2019, 224, 85–94. [Google Scholar] [CrossRef]
- Yang, Q.; Guillerm, V.; Ragon, F.; Wiersum, A.D.; Llewellyn, P.L.; Zhong, C.; Devic, T.; Serre, C.; Maurin, G. CH4 storage and CO2 capture in highly porous zirconium oxide based metal-organic frameworks. Chem. Commun. 2012, 48, 9831–9833. [Google Scholar] [CrossRef] [PubMed]
- Lennox, M.J.; Düren, T. Understanding the Kinetic and Thermodynamic Origins of Xylene Separation in UiO-66(Zr) via Molecular Simulation. J. Phys. Chem. C 2016, 120, 18651–18658. [Google Scholar] [CrossRef]
- Granato, M.A.; Martins, V.D.; Ferreira, A.F.P.; Rodrigues, A.E. Adsorption of xylene isomers in MOF UiO-66 by molecular simulation. Microporous Mesoporous Mater. 2014, 190, 165–170. [Google Scholar] [CrossRef]
- Lyu, J.; Liu, H.; Zeng, Z.; Zhang, J.; Xiao, Z.; Bai, P.; Guo, X. Metal–Organic Framework UiO-66 as an Efficient Adsorbent for Boron Removal from Aqueous Solution. Ind. Eng. Chem. Res. 2017, 56, 2565–2572. [Google Scholar] [CrossRef]
- Wang, C.; Liu, X.; Chen, J.P.; Li, K. Superior removal of arsenic from water with zirconium metal-organic framework UiO-66. Sci. Rep. 2015, 5, 16613. [Google Scholar] [CrossRef] [PubMed]
- Barreto, J.; Xavier, M.D.G.; Ribeiro, R.P.P.L.; Martins, D.; Esteves, I.A.A.C.; Branco, M.; Tirolien, T.; Mota, J.P.B.; Bonfait, G. Neon Adsorption on HKUST-1 and UiO-66 Metal–Organic Frameworks over Wide Pressure and Temperature Ranges. J. Chem. Eng. Data 2019, 64, 5407–5414. [Google Scholar] [CrossRef]
- Garibay, S.J.; Cohen, S.M. Isoreticular synthesis and modification of frameworks with the UiO-66 topology. Chem. Commun. 2010, 46, 7700–7702. [Google Scholar] [CrossRef]
- Xian, S.; Wu, Y.; Wu, J.; Wang, X.; Xiao, J. Enhanced Dynamic CO2 Adsorption Capacity and CO2/CH4 Selectivity on Polyethylenimine-Impregnated UiO-66. Ind. Eng. Chem. Res. 2015, 54, 11151–11158. [Google Scholar] [CrossRef]
- Ribeiro, R.P.P.L.; Camacho, B.C.R.; Lyubchyk, A.; Esteves, I.A.A.C.; Cruz, F.J.A.L.; Mota, J.P.B. Experimental and Computational Study of Ethane and Ethylene Adsorption in the MIL-53(Al) Metal Organic Framework. Microporous Mesoporous Mater. 2016, 230, 154–165. [Google Scholar] [CrossRef]
- Ribeiro, R.P.P.L.; Mota, J.P.B. Surface Area and Porosity of Co3(ndc)3(dabco) Metal–Organic Framework and Its Methane Storage Capacity: A Combined Experimental and Simulation Study. J. Phys. Chem. C 2021, 125, 2411–2423. [Google Scholar] [CrossRef]
- Rogacka, J.; Seremak, A.; Luna-Triguero, A.; Formalik, F.; Matito-Martos, I.; Firlej, L.; Calero, S.; Kuchta, B. High-throughput screening of metal—Organic frameworks for CO2 and CH4 separation in the presence of water. Chem. Eng. J. 2021, 403, 126392. [Google Scholar] [CrossRef]
- Becker, T.M.; Heinen, J.; Dubbeldam, D.; Lin, L.-C.; Vlugt, T.J.H. Polarizable Force Fields for CO2 and CH4 Adsorption in M-MOF-74. J. Phys. Chem. C 2017, 121, 4659–4673. [Google Scholar] [CrossRef] [PubMed]
- Skoulidas, A.I. Molecular Dynamics Simulations of Gas Diffusion in Metal−Organic Frameworks: Argon in CuBTC. J. Am. Chem. Soc. 2004, 126, 1356–1357. [Google Scholar] [CrossRef] [PubMed]
- Boyd, P.G.; Moosavi, S.M.; Witman, M.; Smit, B. Force-Field Prediction of Materials Properties in Metal-Organic Frameworks. J. Phys. Chem. Lett. 2017, 8, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhong, C. Molecular Simulation of Carbon Dioxide/Methane/Hydrogen Mixture Adsorption in Metal−Organic Frameworks. J. Phys. Chem. B 2006, 110, 17776–17783. [Google Scholar] [CrossRef]
- Martín-Calvo, A.; García-Pérez, E.; Manuel Castillo, J.; Calero, S. Molecular simulations for adsorption and separation of natural gas in IRMOF-1 and Cu-BTC metal-organic frameworks. Phys. Chem. Chem. Phys. 2008, 10, 7085–7091. [Google Scholar] [CrossRef]
- Sladekova, K.; Campbell, C.; Grant, C.; Fletcher, A.J.; Gomes, J.R.B.; Jorge, M. The effect of atomic point charges on adsorption isotherms of CO2 and water in metal organic frameworks. Adsorption 2020, 26, 663–685. [Google Scholar] [CrossRef]
- Demir, H.; Walton, K.S.; Sholl, D.S. Computational Screening of Functionalized UiO-66 Materials for Selective Contaminant Removal from Air. J. Phys. Chem. C 2017, 121, 20396–20406. [Google Scholar] [CrossRef]
- Planchais, A.; Devautour-Vinot, S.; Salles, F.; Ragon, F.; Devic, T.; Serre, C.; Maurin, G. A Joint Experimental/Computational Exploration of the Dynamics of Confined Water/Zr-Based MOFs Systems. J. Phys. Chem. C 2014, 118, 14441–14448. [Google Scholar] [CrossRef]
- Ghosh, P.; Colón, Y.J.; Snurr, R.Q. Water adsorption in UiO-66: The importance of defects. Chem. Commun. 2014, 50, 11329–11331. [Google Scholar] [CrossRef]
- Ramsahye, N.A.; Maurin, G.; Bourrelly, S.; Llewellyn, P.; Loiseau, T.; Ferey, G. Charge distribution in metal organic framework materials: Transferability to a preliminary molecular simulation study of the CO2 adsorption in the MIL-53 (Al) system. Phys. Chem. Chem. Phys. 2007, 9, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.G.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 1. United-Atom Description of n-Alkanes. J. Phys. Chem. B 1998, 102, 2569–2577. [Google Scholar] [CrossRef]
- Potoff, J.J.; Siepmann, J.I. Vapor–liquid equilibria of mixtures containing alkanes, carbon dioxide, and nitrogen. AIChE J. 2001, 47, 1676–1682. [Google Scholar] [CrossRef]
- Wick, C.D.; Siepman, J.I.; Klotz, W.L.; Schure, M.R. Temperature effects on the retention of n-alkanes and arenes in helium-squalane gas-liquid chromatography. Experiment and molecular simulation. J. Chromatogr. A 2002, 954, 181–190. [Google Scholar] [CrossRef]
- Kamath, G.; Cao, F.; Potoff, J.J. An Improved Force Field for the Prediction of the Vapor−Liquid Equilibria for Carboxylic Acids. J. Phys. Chem. B 2004, 108, 14130–14136. [Google Scholar] [CrossRef]
- Stubbs, J.M.; Potoff, J.J.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 6. United-Atom Description for Ethers, Glycols, Ketones, and Aldehydes. J. Phys. Chem. B 2004, 108, 17596–17605. [Google Scholar] [CrossRef]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A., III; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Rai, N.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 10. Explicit-Hydrogen Description of Substituted Benzenes and Polycyclic Aromatic Compounds. J. Phys. Chem. B 2013, 117, 273–288. [Google Scholar] [CrossRef]
- Shearer, G.C.; Chavan, S.; Bordiga, S.; Svelle, S.; Olsbye, U.; Lillerud, K.P. Defect Engineering: Tuning the Porosity and Composition of the Metal–Organic Framework UiO-66 via Modulated Synthesis. Chem. Mater. 2016, 28, 3749–3761. [Google Scholar] [CrossRef]
- Thornton, A.W.; Babarao, R.; Jain, A.; Trousselet, F.; Coudert, F.X. Defects in metal–organic frameworks: A compromise between adsorption and stability? Dalton Trans. 2016, 45, 4352–4359. [Google Scholar] [CrossRef]
- Wu, H.; Chua, Y.S.; Krungleviciute, V.; Tyagi, M.; Chen, P.; Yildirim, T.; Zhou, W. Unusual and Highly Tunable Missing-Linker Defects in Zirconium Metal–Organic Framework UiO-66 and Their Important Effects on Gas Adsorption. J. Am. Chem. Soc. 2013, 135, 10525–10532. [Google Scholar] [CrossRef] [PubMed]
- Vandenbrande, S.; Verstraelen, T.; Gutiérrez-Sevillano, J.J.; Waroquier, M.; Van Speybroeck, V. Methane Adsorption in Zr-Based MOFs: Comparison and Critical Evaluation of Force Fields. J. Phys. Chem. C 2017, 121, 25309–25322. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, D.; Parsonage, N.G. Computer Simulation and the Statistical Mechanics of Adsorption; Academic Press: New York, NY, USA, 1982. [Google Scholar]
- Vlugt, T.J.H.; García-Pérez, E.; Dubbeldam, D.; Ban, S.; Calero, S. Computing the Heat of Adsorption using Molecular Simulations: The Effect of Strong Coulombic Interactions. J. Chem. Theory Comput. 2008, 4, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Babarao, R.; Hu, Z.; Jiang, J.; Chempath, S.; Sandler, S.I. Storage and Separation of CO2 and CH4 in Silicalite, C168 Schwarzite, and IRMOF-1: A Comparative Study from Monte Carlo Simulation. Langmuir 2007, 23, 659–666. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).