1. Introduction
The 229-thorium isotope has an exceptionally low-lying metastable nuclear excited state (
229mTh) [
1]. Current best estimates assign its value as
[
2,
3,
4], being comparable to electronic transition energies and notably within the range of laser spectroscopy [
1,
5,
6]. Taking advantage of this unique property, a number of precision spectroscopy experiments have been proposed, which could help detect certain types of dark matter or physics beyond the standard model [
6,
7].
Several experimental challenges prohibit an immediate measurement, most notably because of the fact that the isomeric energy is not precisely known [
8,
9,
10,
11]. Doping
Th into a solid-state host crystal would allow one to probe a mesoscopic number of nuclei (
to
) and lock the nuclei into the crystal lattice for observation [
6,
12]. Photons absorbed and emitted during excitation and de-excitation must pass the host material unperturbed; hence its band gap needs to exceed the isomer energy. Because of additional desirable features, a suitable host crystal is the calcium fluoride (CaF
2) compound [
13,
14].
Localized electronic states around the dopants may be energetically located in the band gap of pristine CaF
2, reducing the material’s optical transmission range [
13]. Even so, previous estimates for the energetically favored structural arrangement indicate transparency at the isomer energy [
13].
However, the extreme scarcity (only ∼
being globally available for research) and the need for further energy measurements prompted other proposals to populate the Th nuclei to their first excited state [
6]. Apart from direct laser excitation, there are two experimentally viable sources of
229mTh:
-decay of the 233-uranium isotope and
decay of 229-actinium, which both are to decay within the host crystal after doping [
6,
15,
16]. Thus, optical properties of doped CaF
2 crystals with these dopants and their favorable charge compensation schemes after decay also need to be considered.
Apart from photon detection upon de-excitation, another way to verify that the
Th nuclei are in the excited state is by measuring the interaction of the crystal electric field with the nuclear quadrupole moment (nuclear quadrupole resonance spectroscopy) [
17]. Importantly, the resonance frequency depends only on the known quadrupole moment of the nucleus and the electric field gradient (EFG) at the nucleus position [
18]. In order to set up experimental devices and provide reference samples, more accessible elements that have similar properties with respect to their nuclear quadrupole moments are neptunium and uranium [
19].
Finally, impurities of oxygen and cerium, the latter originating from the chemical process to prepare
Th for doping, cannot be completely ruled out [
20], which may give rise to undesirable optical features of the doped system.
2. Materials and Methods
All simulations are performed with the Vienna Ab-initio Simulation Package (VASP) version 6.1.0 [
21,
22,
23,
24,
25], implementing density functional theory within the projector augmented wave method [
26]. We use the generalized gradient approximation (GGA) of the exchange correlation functional as devised by Perdew et al. [
27] (PBE) for structural optimization and for the calculation of electric field gradients (EFGs)
. The single-shot variant of the
approximation [
28] (
) with PBE orbitals is used for band-gap calculations and the absorption coefficient is estimated via solution of the Bethe–Salpeter Equation (BSE) [
29]. Selected results are moreover calculated with the hybrid functional HSE06 [
30] in order to provide a reference for PBE calculations when a
comparison is not computationally feasible. Geometry optimization is performed with a conjugate gradient algorithm and a convergence criterion of
as the maximum force acting on an ion.
Initial positions (before relaxation) for interstitial ions are along the lattice vectors of the conventional cubic cell of pristine CaF
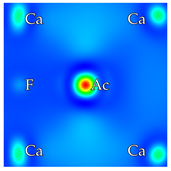
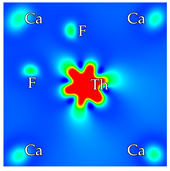
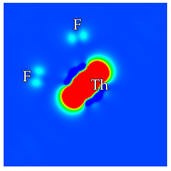
2, at the regular Ca-F distance from the dopant. Calcium vacancies are considered at nearest-neighbor positions. In total, there are 6 and 12 possibilities for interstitial fluorines and calcium vacancies, respectively. In cases where a fluorine is removed from the crystal, only the nearest neighbor F anions to the dopant are considered, of which there are 8. Reducing these numbers further depends on the symmetry of each scheme separately. Based on previous analysis and the local chemical environment, it is reasonable to assume that the dopant will be situated on a Ca site (substitutional doping) [
13]. The number of investigated charge compensation schemes depends on the amount of surplus fluorine and the extra charges introduced by the dopant. The following is a complete list of compensation schemes considered in this work (in Kröger–Vink notation [
31]) along with the number of symmetrically unique initial positions: no compensation (1),
(1), 2
(2), 3
(2),
(1), 2
(4),
+
(3),
+ 2
(7),
+ 3
(8),
+
(3), 2
+ 1
(13) and
(1).
Atoms that are ejected from the crystal due to substitutional doping or the creation of vacancies, as well as residuary ions from the compound that initially contained the dopant, must be accounted for in order to compare total energies between the compensation schemes, which are calculated as . Here “inside” refers to all atoms considered in the VASP calculation, and “environment” refers to the energetically favored compounds, created by the beforementioned remnants.
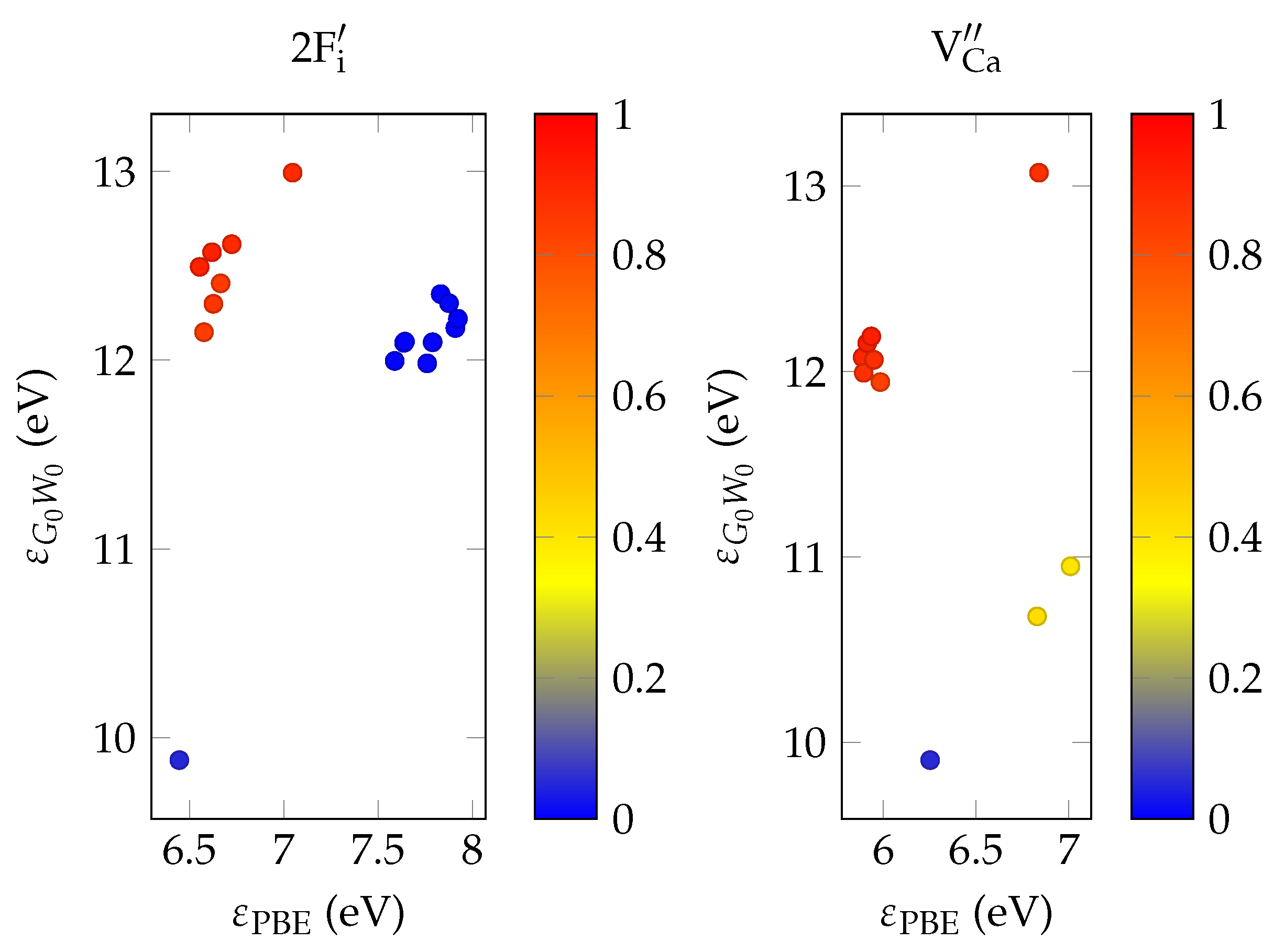
Concerning the predictive capabilities of the resulting total energies, noteworthy drawbacks stem from the idealized model of a static, infinitely extending bulk and not necessarily from methodical reasons. In particular, surface energies and kinetic effects are completely unaccounted for in this model. For instance, experimental evidence suggests there is a high mobility of fluorine anions in the crystal bulk during annealing whereas displacements of calcium ions may be hindered due to their increased mass and ionic radius [
32]. On the other hand, formation energies predicted by GGA-DFT are expected to deviate from the experiment by
[
33], smaller than most energy differences between compensation schemes observed in this work.
Initially, structures are relaxed at relatively high doping concentrations, with one impurity per 31 calcium atoms in a structure of the conventional unit cell of CaF2. For computational reasons, optical calculations are also performed with this system size. In order to determine the EFG, energetically favorable structures are again relaxed in a super structure (1 dopant per 107 Ca atoms).
The energy cutoff is converged at 675 with the criterion that the total energy deviates at most by /atom to a reference calculation. All doped systems are studied by using a -point sampling of the first Brillouin zone. Furthermore, we use Gaussian smearing with a smearing parameter of , which may be smaller in cases where partial occupancies would be observed by using this setting. In fully compensated systems and for pristine CaF2, spin polarization is not considered. Scalar relativistic effects are considered but corrections due to spin-orbit coupling are neglected.
Because a main concern of this work are optical properties, we use pseudopotentials of the GW type whenever possible. For calcium, we choose the Ca_sv_GW variant whereas for fluorine, the F_GW_new type is used. Cerium also has a Ce_GW type available; for all other heavy elements, we choose the standard Ac, U, Th and Np versions.
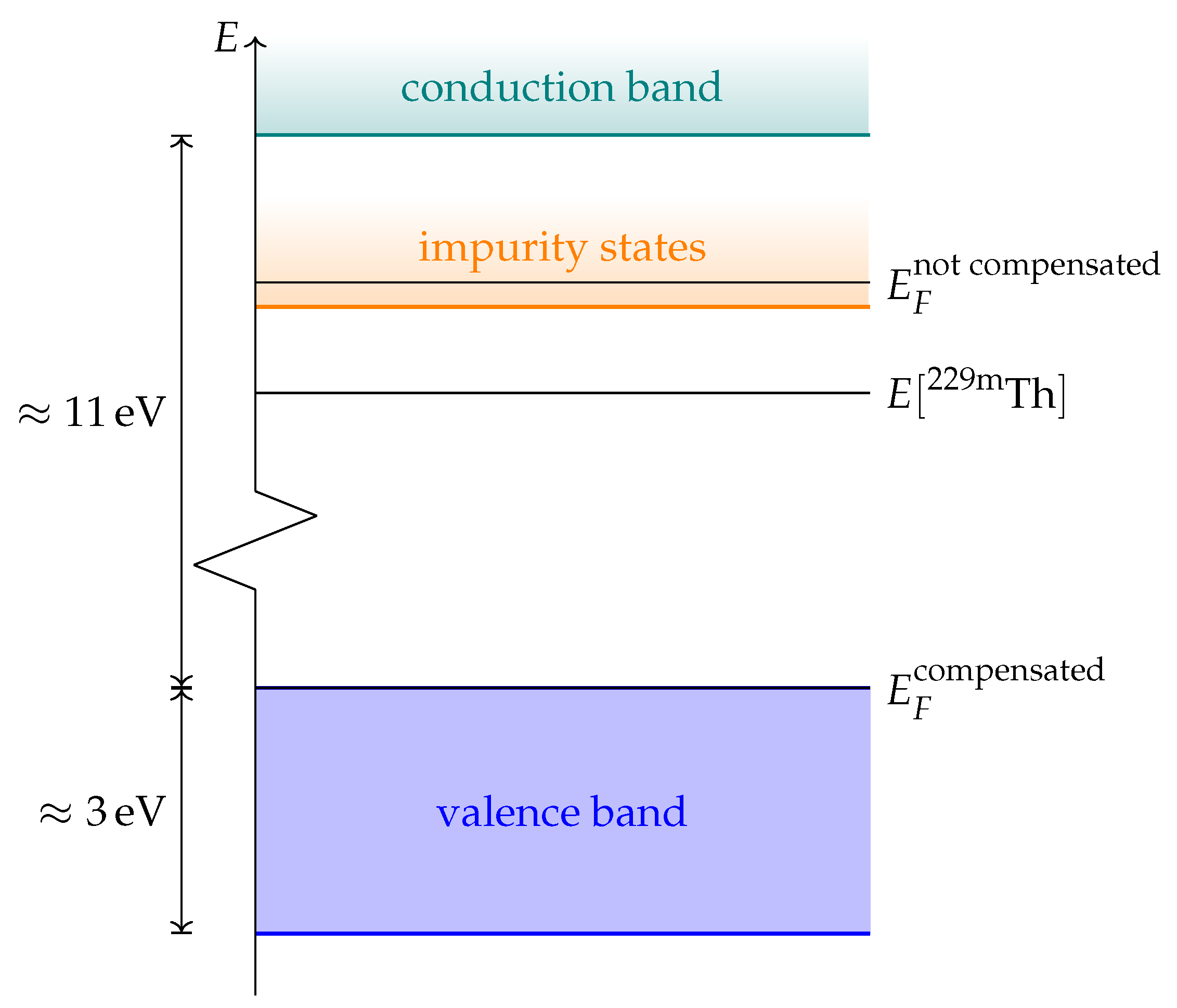
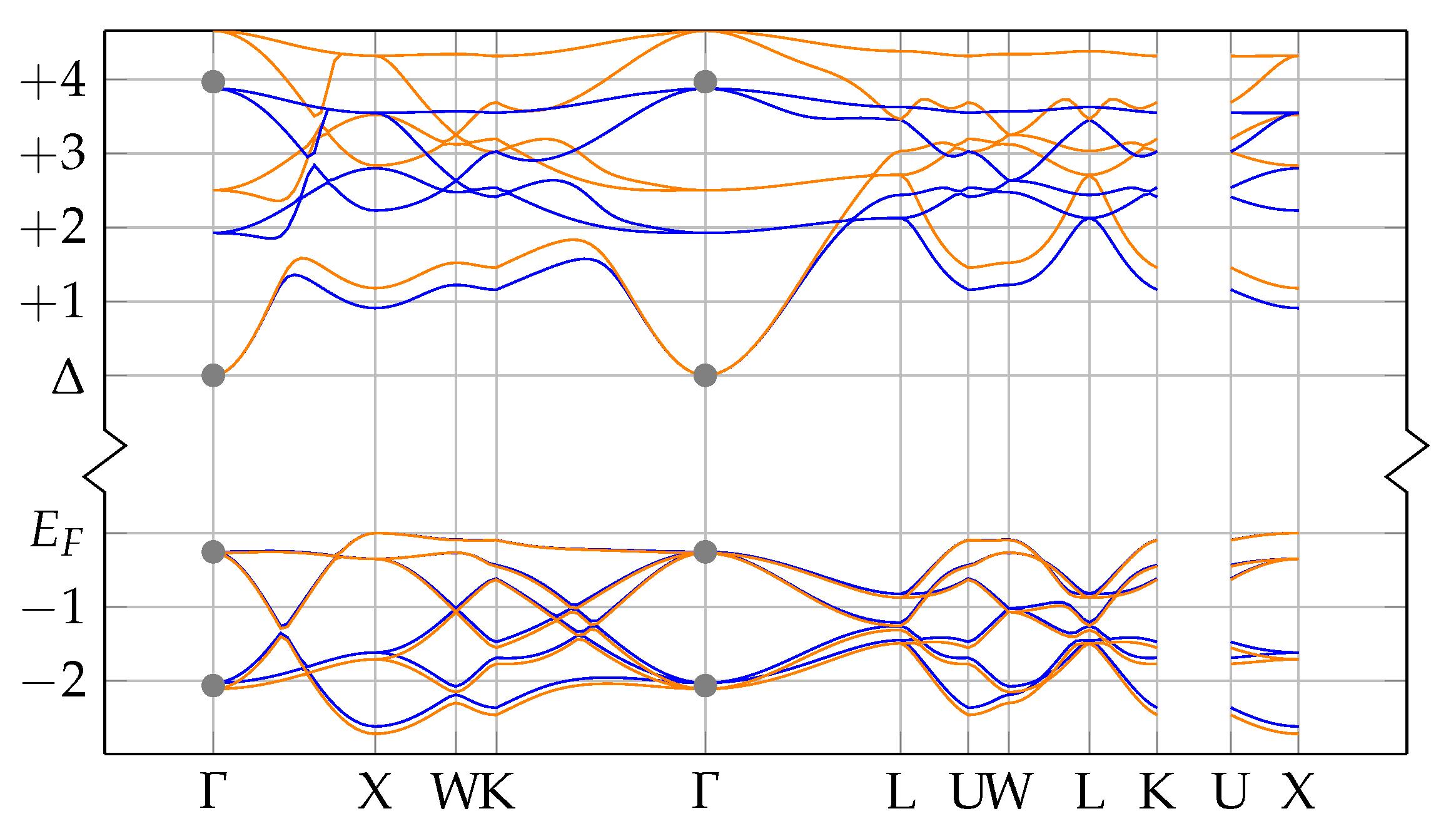
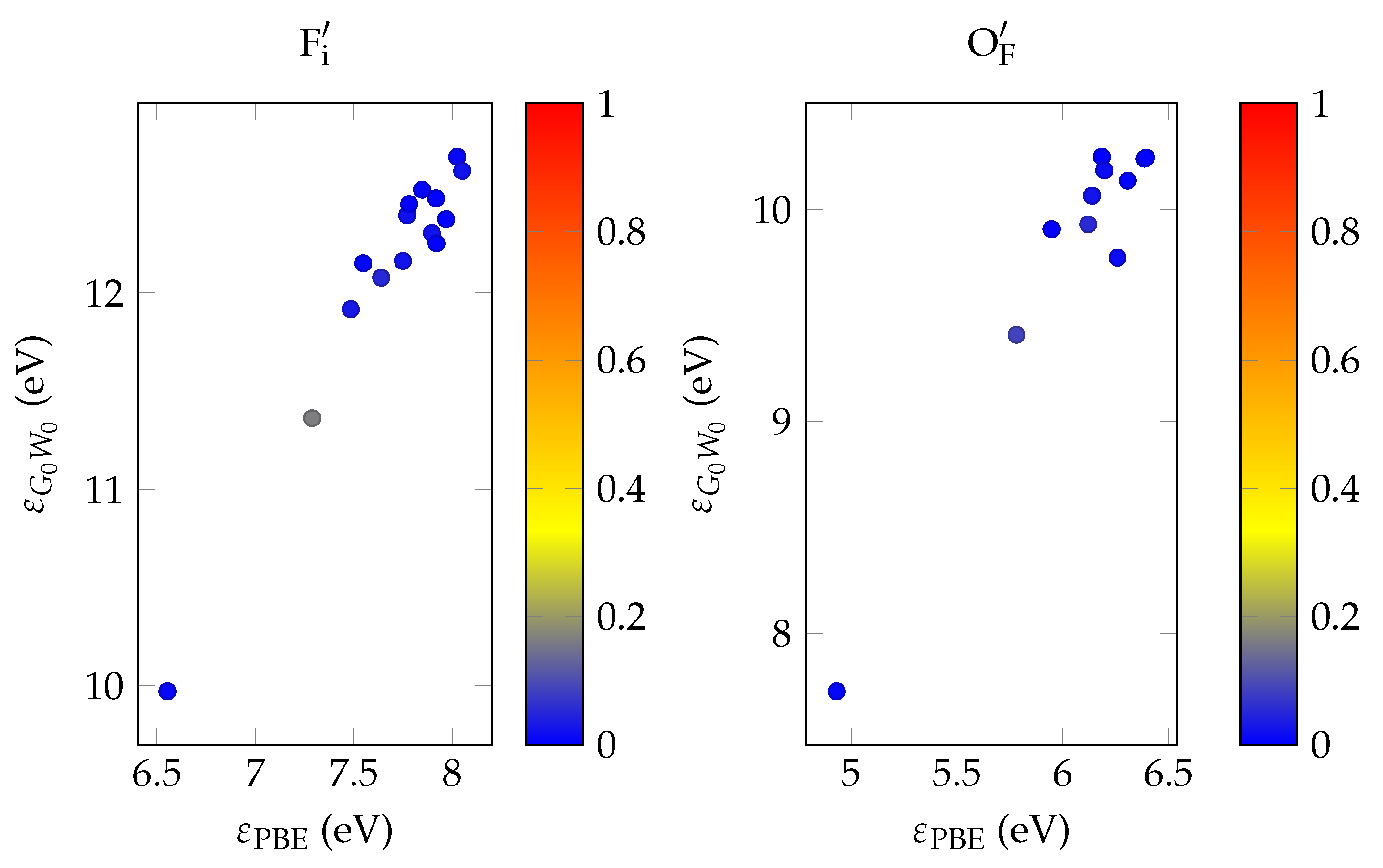
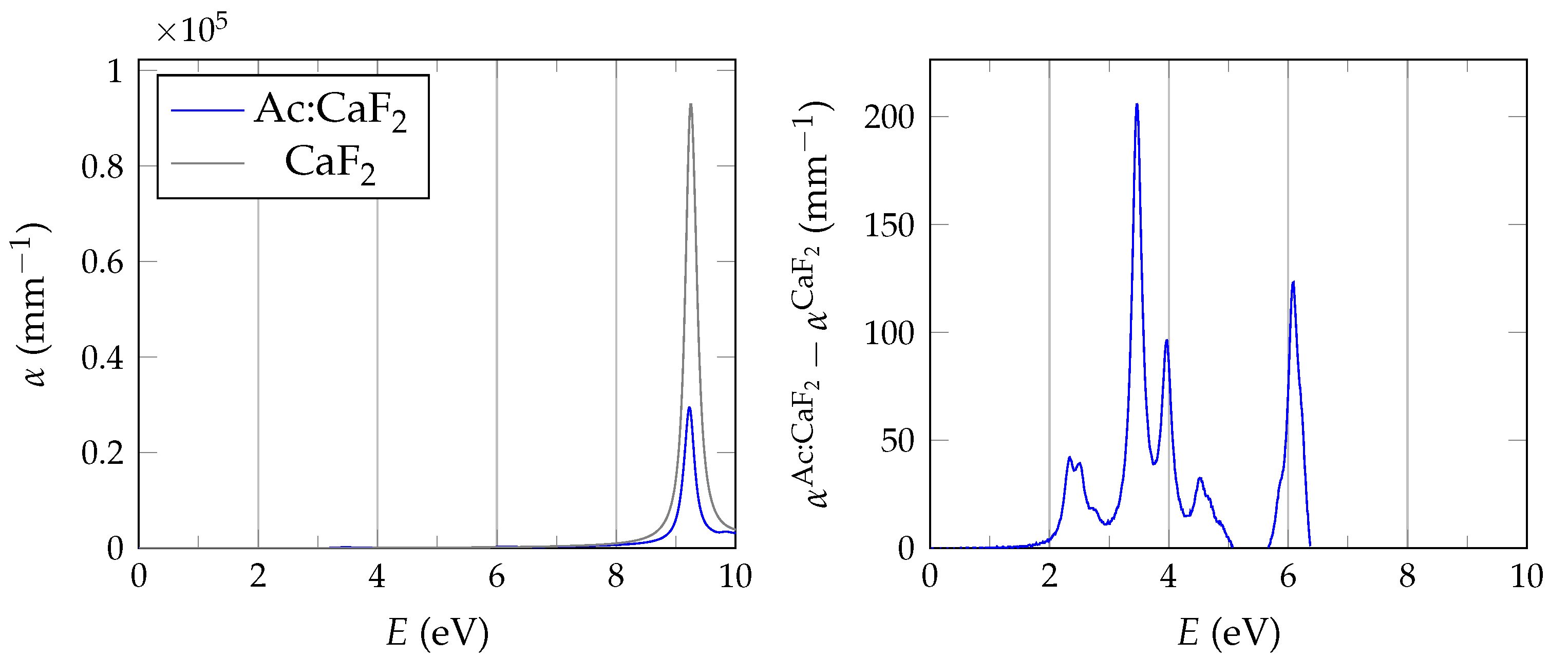
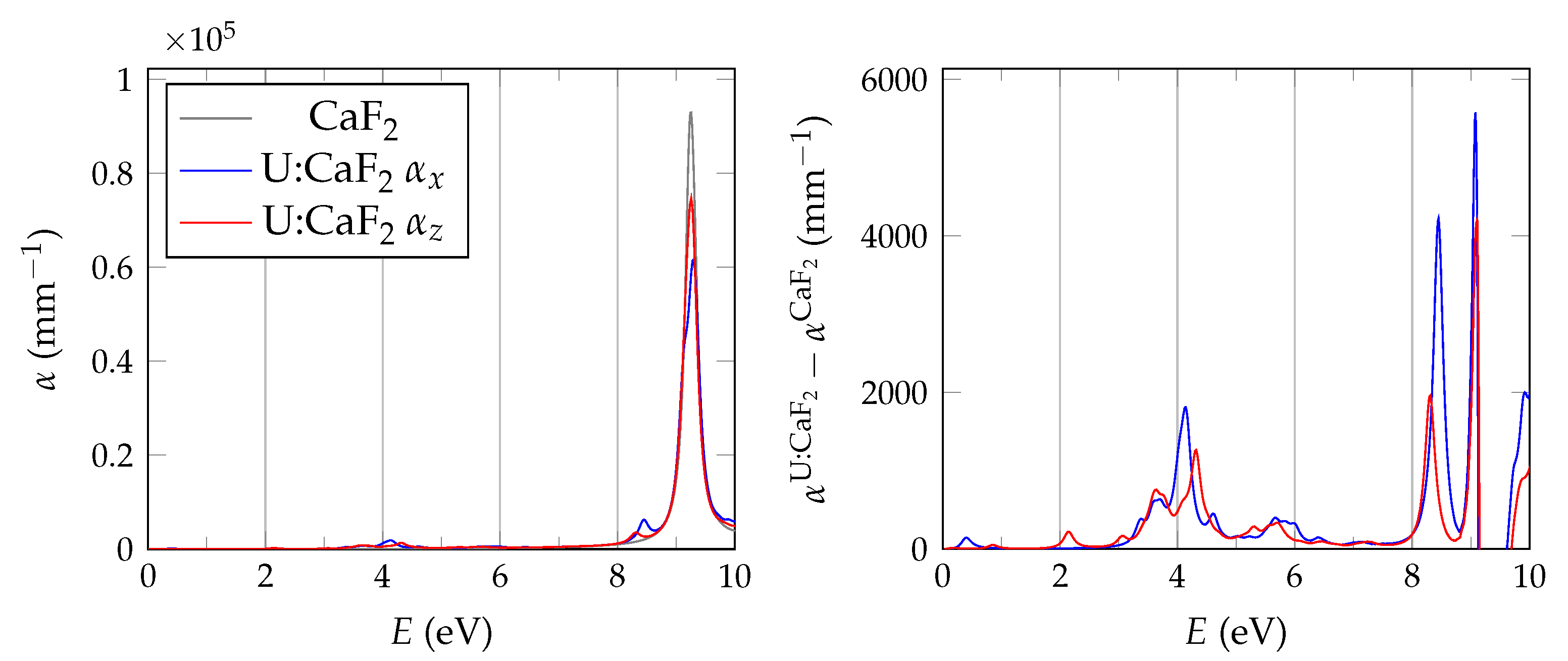
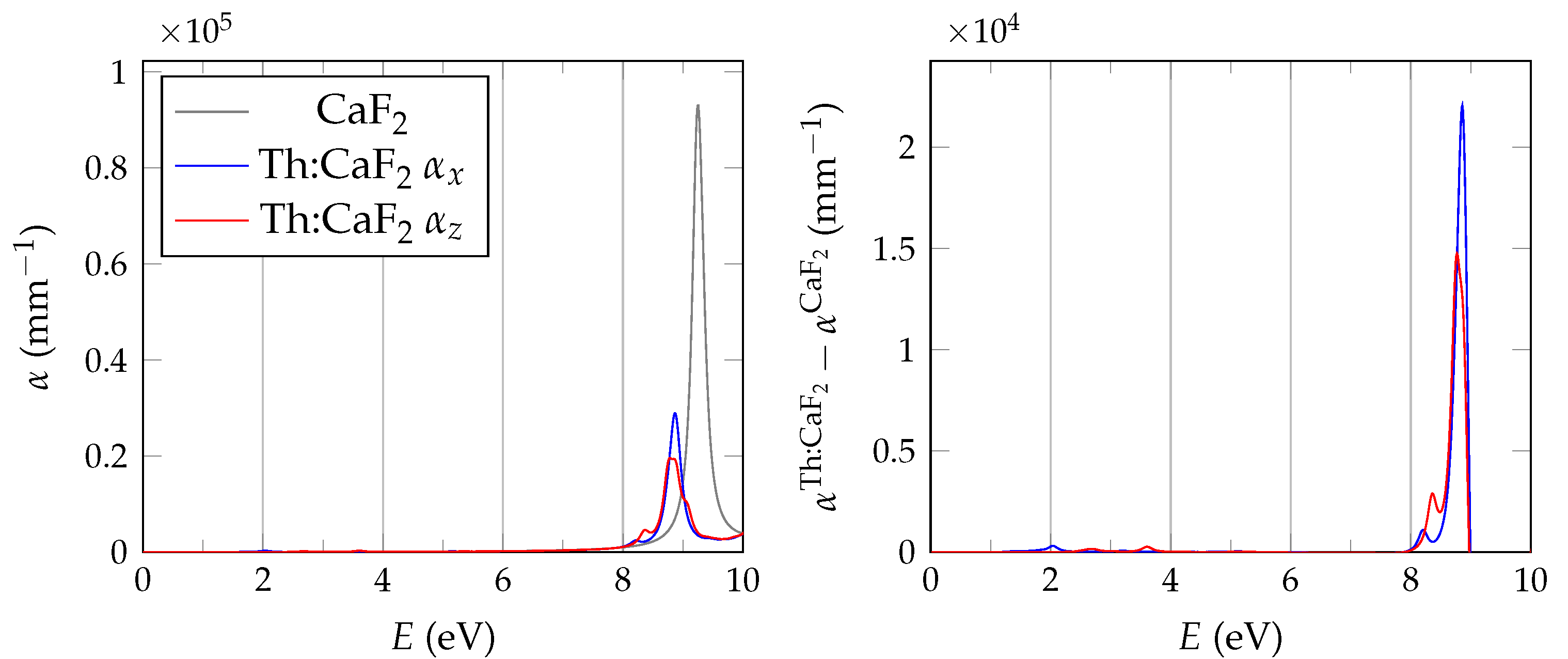
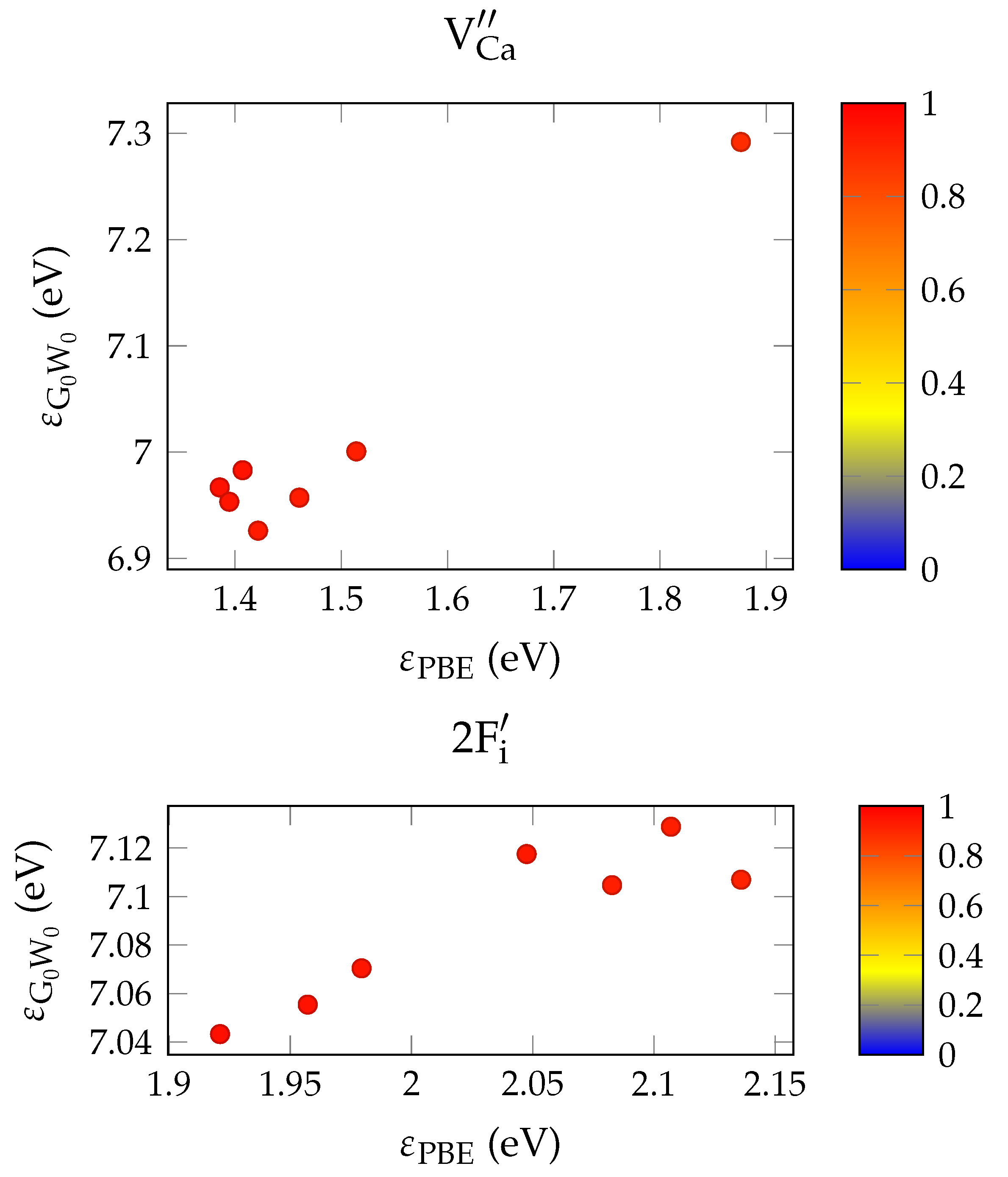
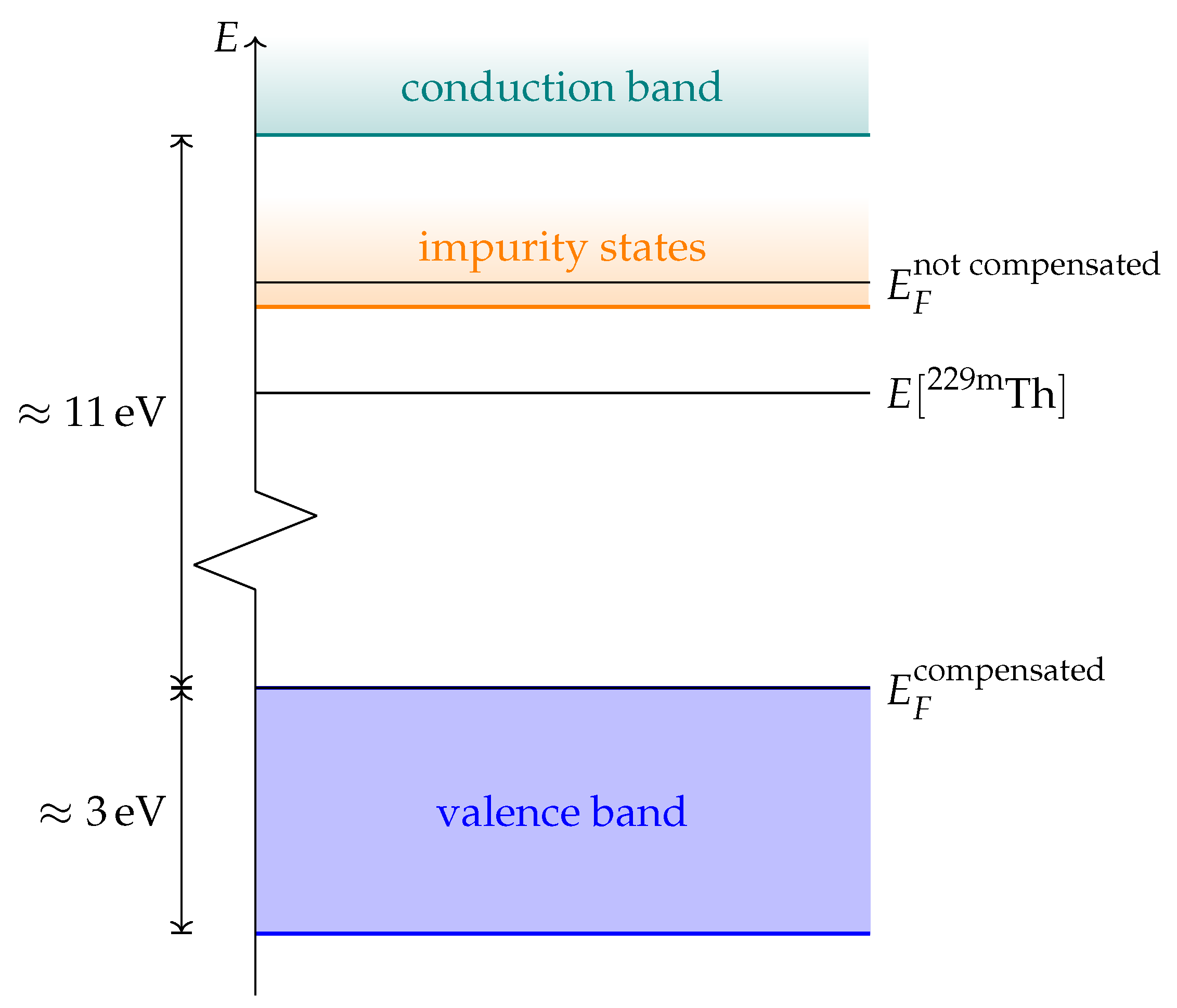
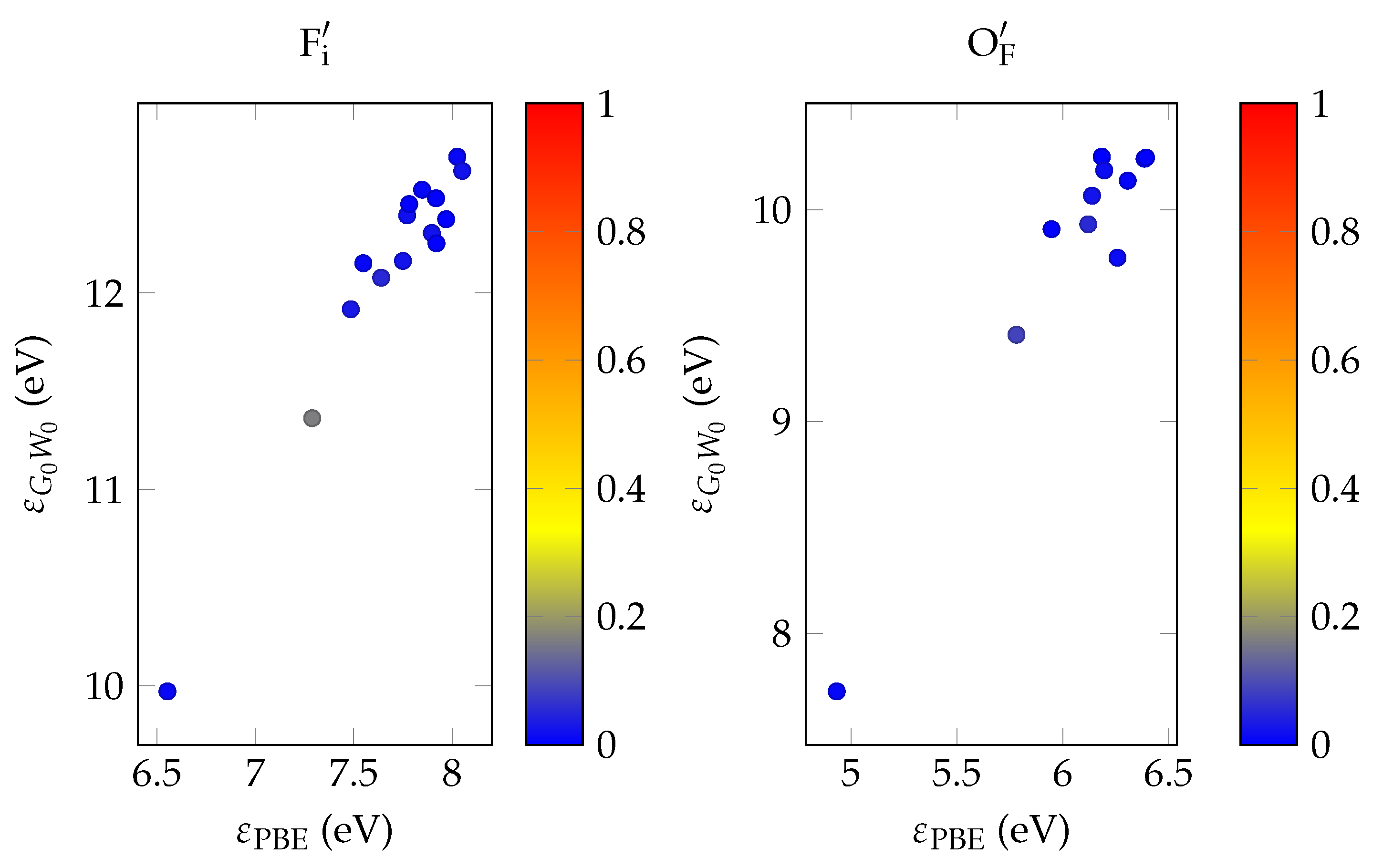
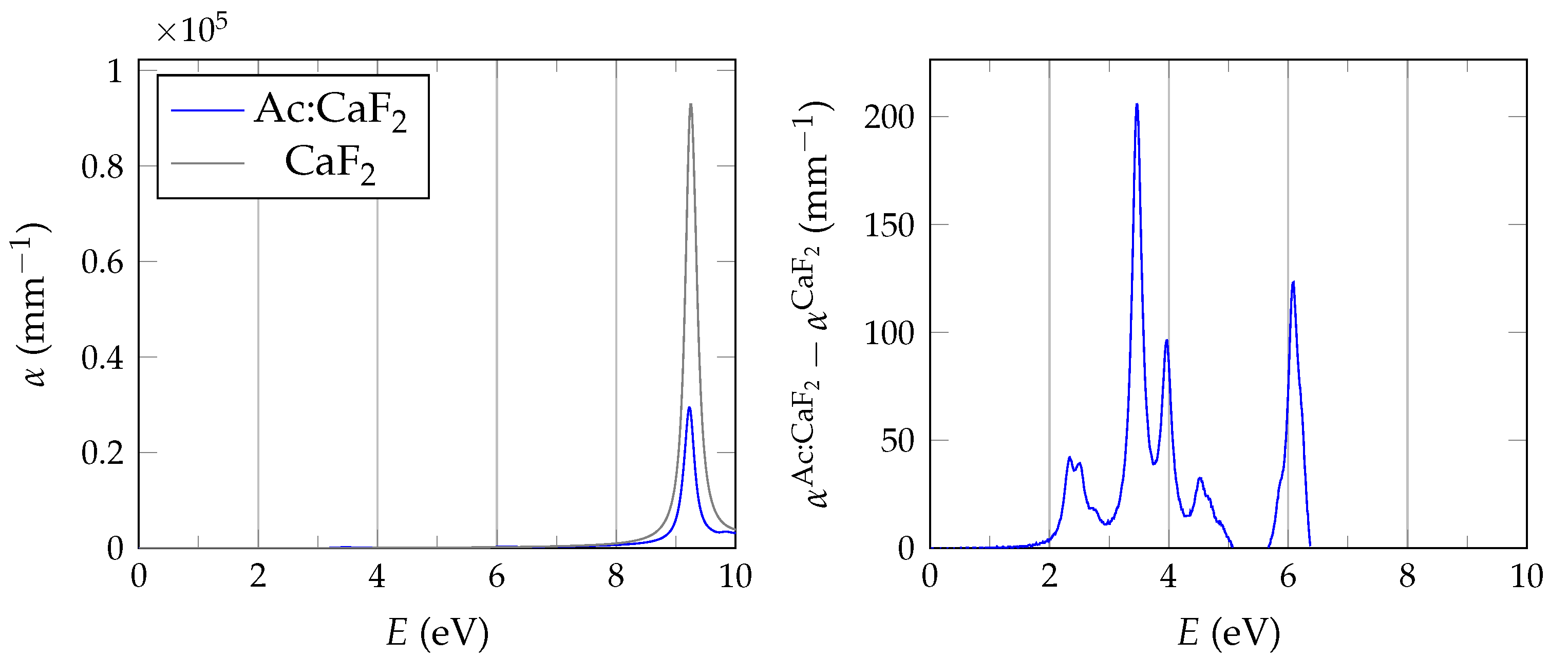
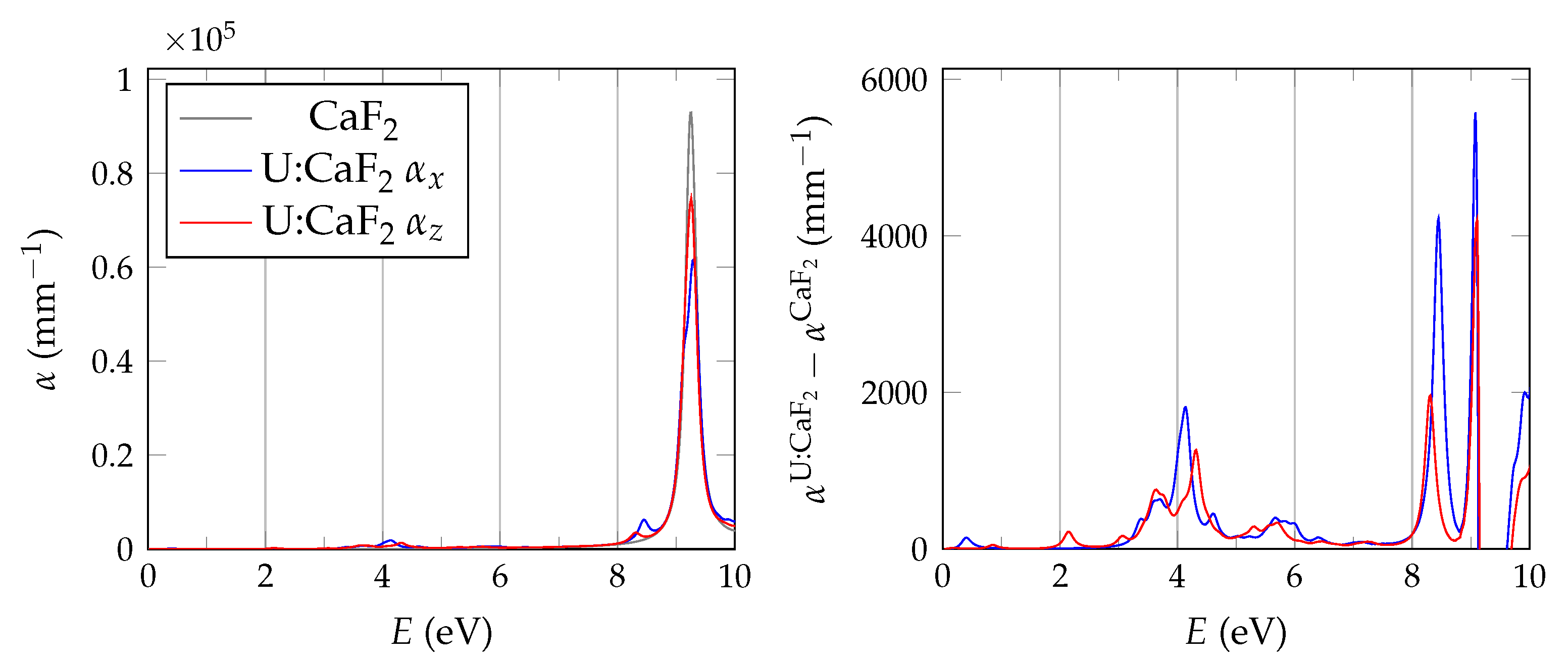
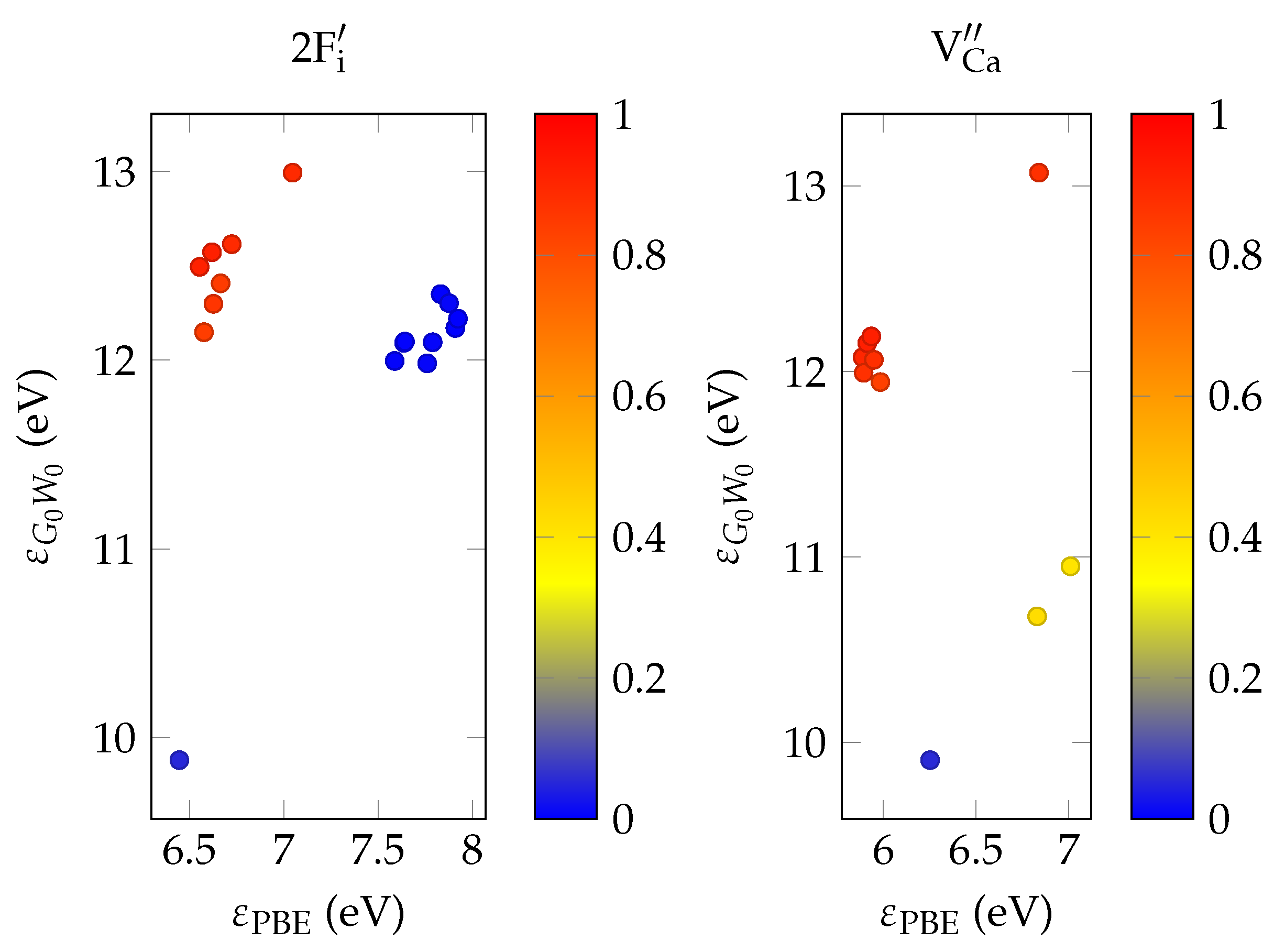
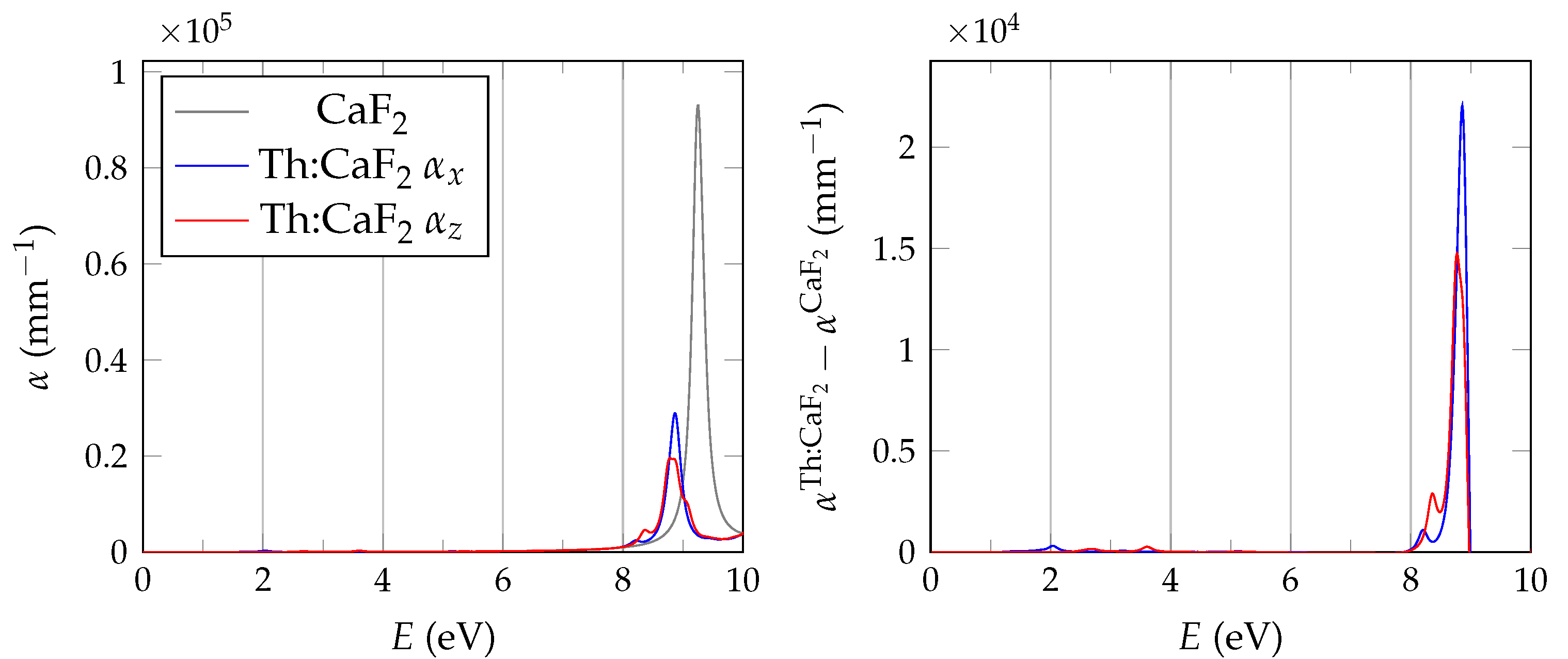
In order to evaluate the important transmissive properties in a fully compensated system, it is sufficient to consider whether the energy differences of the localized defect states to the valence band are larger than the expected isomer energy. However, if not all charges are compensated, the ground state features electrons in localized impurity states, with relatively small energy differences to other localized states or to the conduction band of the host crystal as is depicted in
Figure 1. In this case, transparency at the isomer energy range can only be deduced by considering the energy-dependent absorption coefficient.
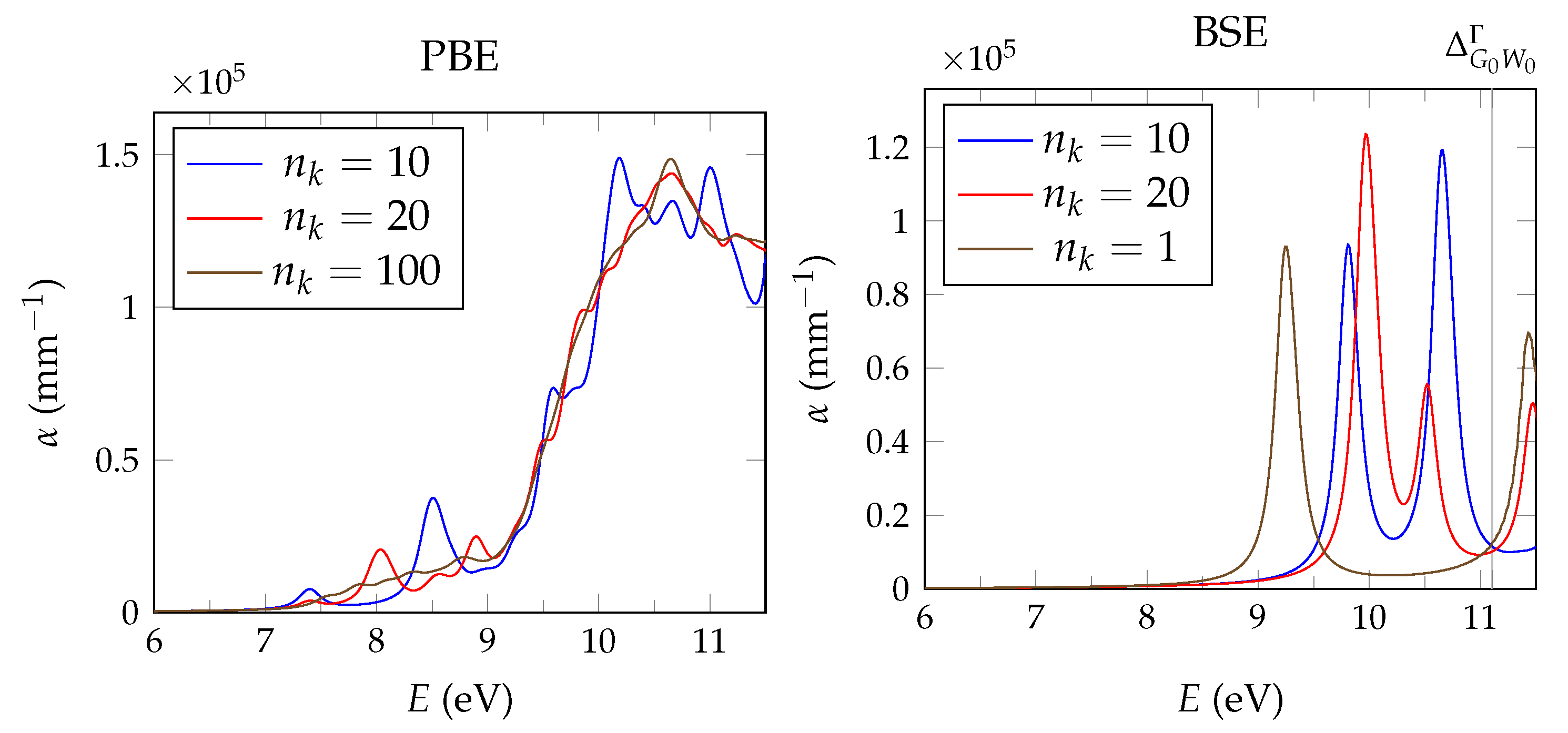
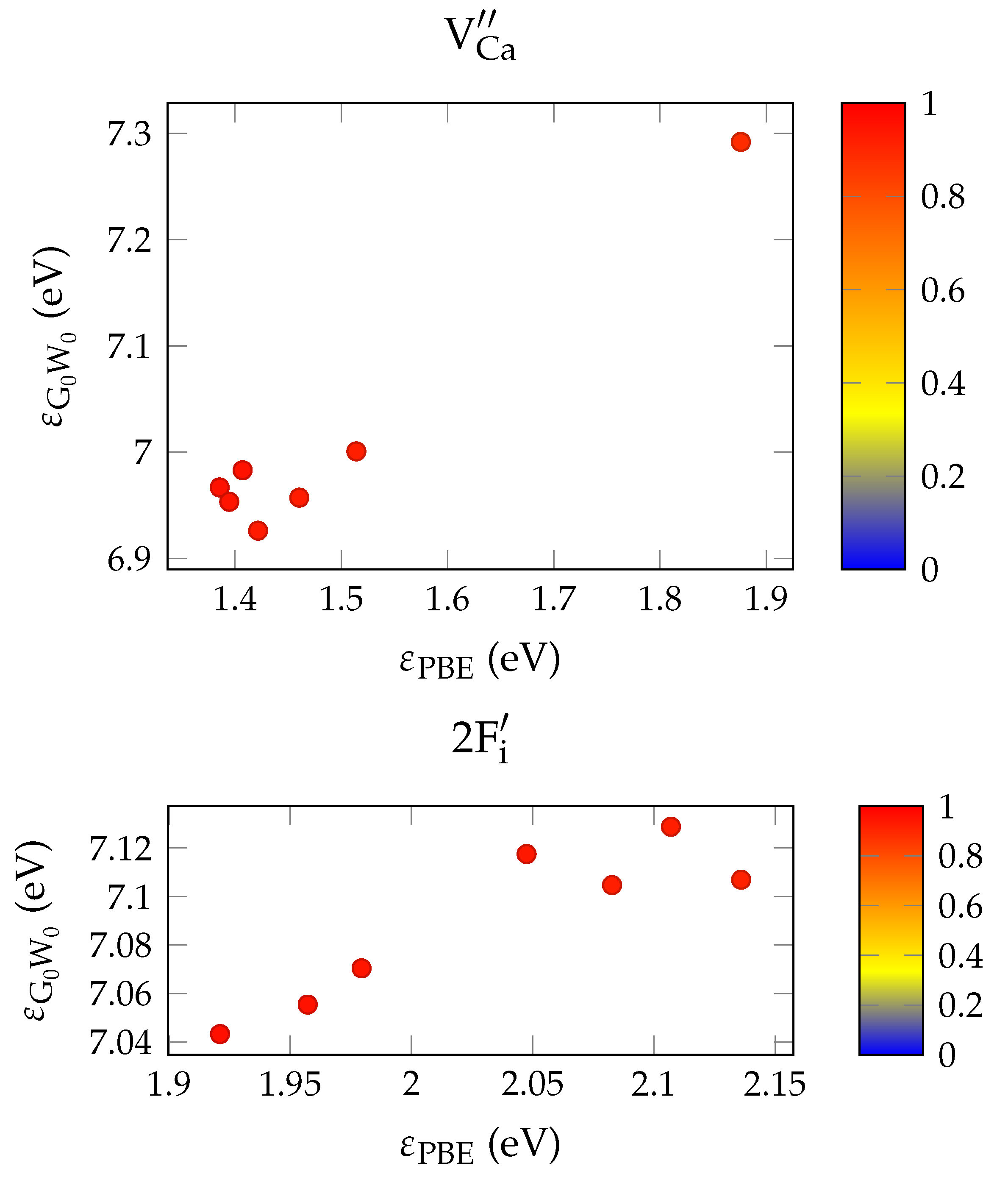
It has been shown that PBE +
calculations give fairly close estimates to the experimental band gap for a range of semiconductors and insulators [
34]. In the general case, the optical absorption coefficient is reasonably well approximated via the solution of the Bethe–Salpeter by equation using
energies [
29].
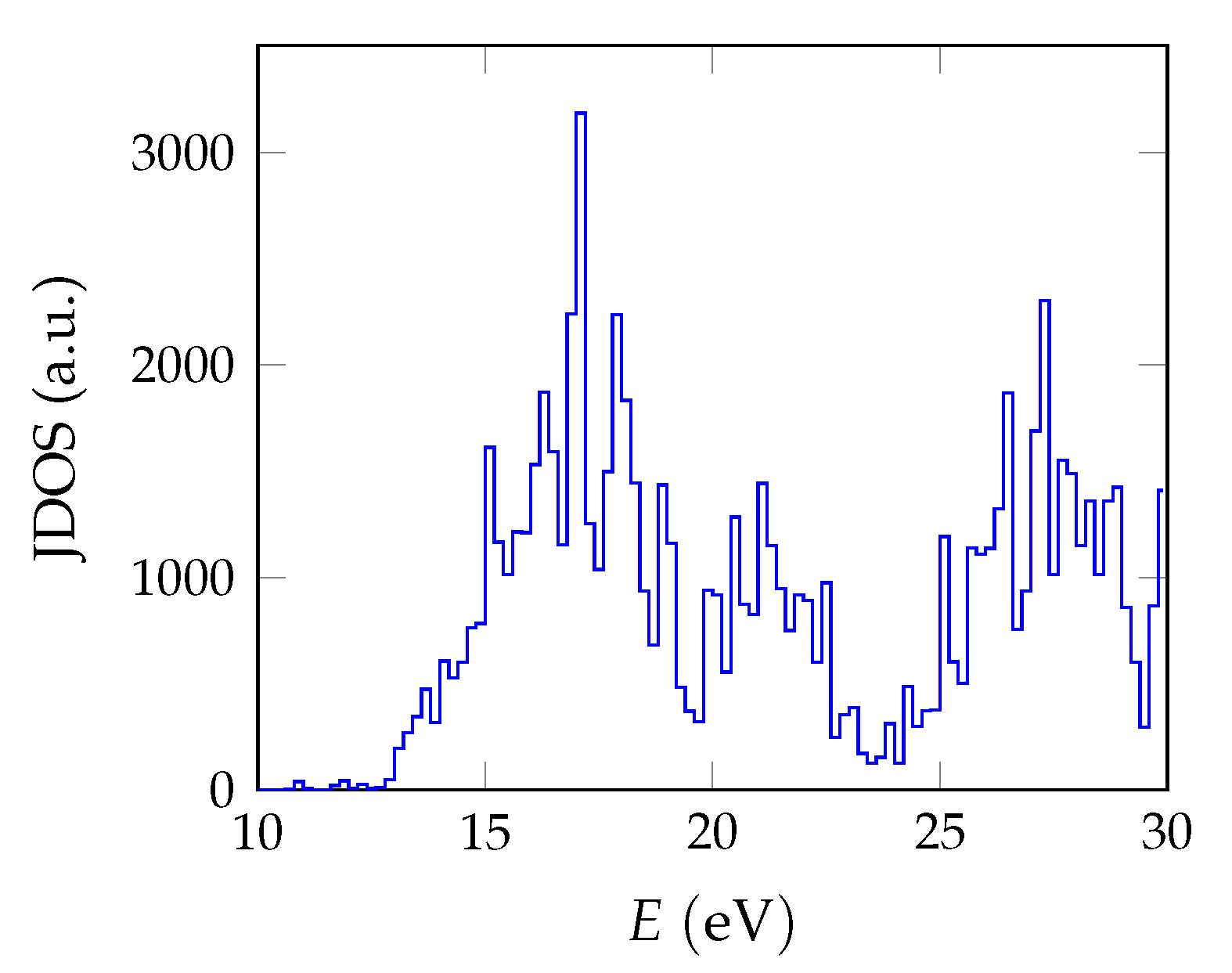
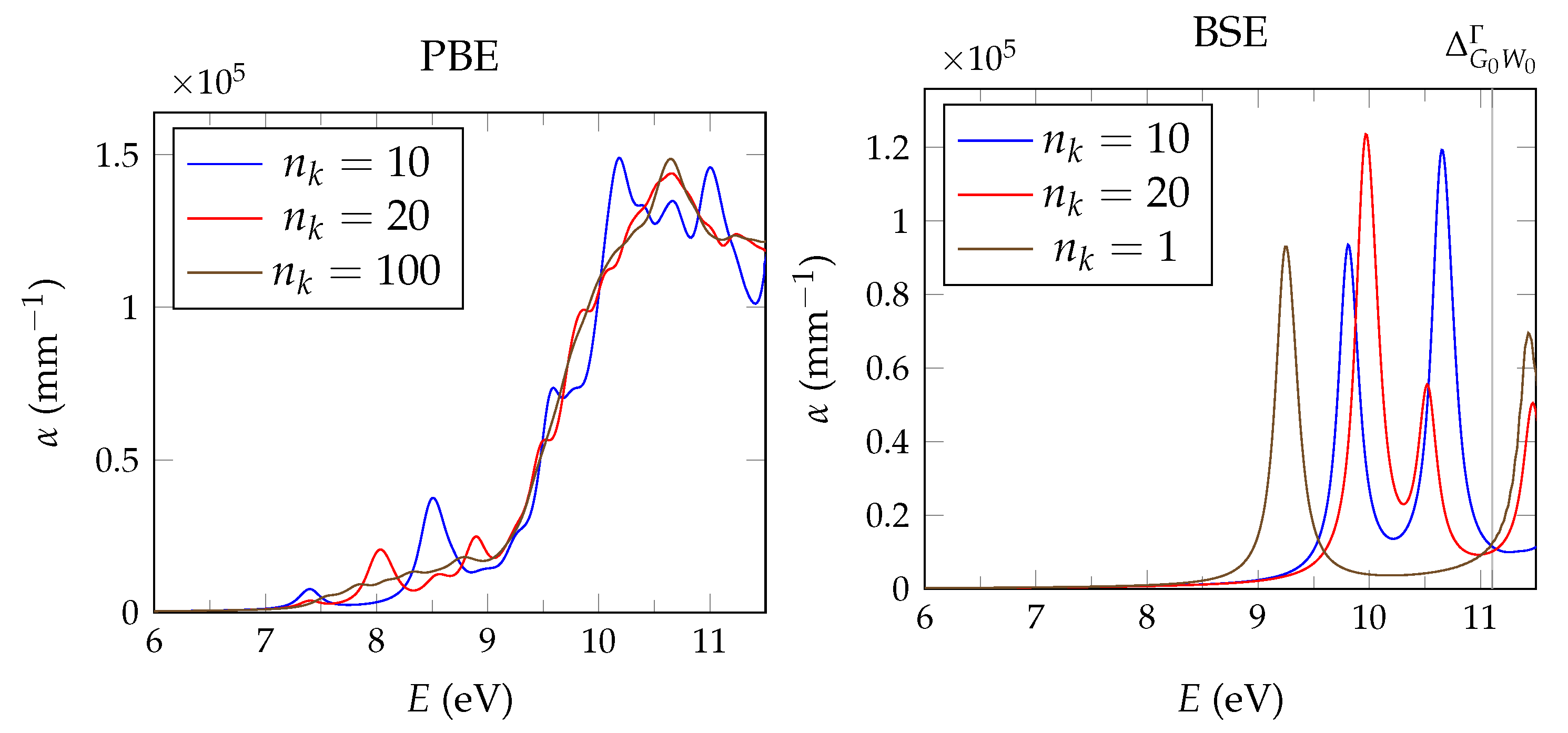
calculations are performed with the spectral method and 24 frequency grid points. Studies in pristine CaF2 use a total of 1536 bands, whereas for doped systems we reduce the computational requirements based on convergence tests by lowering the cutoff to 585 and 270 for the response function and using a total of 34,560 bands.
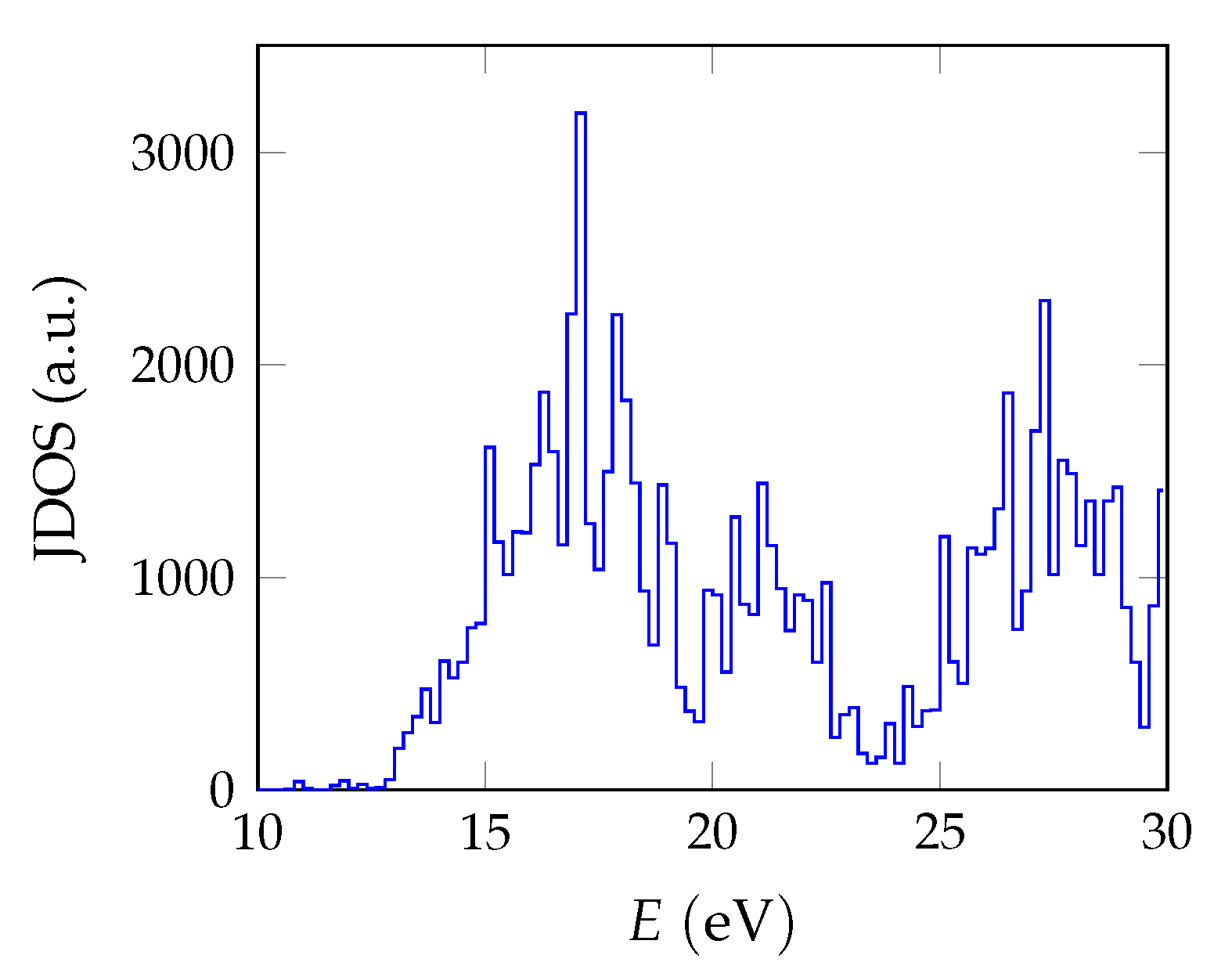
The BSE absorption coefficients are computed by considering the full valence band of the system and an even greater number of conduction band states for electron-hole pairs, as is explained in
Section 3.1.
Unfortunately, the + BSE approach demands considerable memory resources. Even ≈ 6 TB of random-access memory limit the calculation of doped super structures to the -point in reciprocal space. As a consequence, absorption coefficients are not fully converged and must be interpreted with care.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}