Abstract
Changes in the morphology and structure of the core-shell particles of polymethyl methacrylate-silicon dioxide and hollow SiO2 particles during their heat treatment were studied by electron microscopy, infrared spectroscopy, and X-ray diffraction. The polymeric core of the PMMA-SiO2 hybrid particle was found to undergo an unusual transformation when exposed to the electron microscope beam: its shrinkage occurs through the formation of a spherical cavity. It was shown that the process of silica-shell formation occurs in the temperature range of 200–600 °C and is accompanied by the loss of vinyl- and OH-groups. It was determined by the method of X-ray diffraction, that in the place of the interaction of PMMA and the shell, the degree of ordering of the polymer is higher than that in the volume of the polymer core. It was shown that the frequency of the TO3-vibrational mode (asymmetric stretching vibrations of the Si–O–Si bonds) increases with an increase in the annealing temperature, which is associated with the densification of the silicon dioxide shell.
1. Introduction
Currently, nano- and microencapsulated materials are widely used in metallurgy, health care, cosmetology, pharmacology, and agriculture [1,2,3,4,5,6,7,8]. One of the promising materials for capsule formation, for example, for targeted drug delivery, is silica, due to its heat resistance, chemical inertness, and biocompatibility. The possibility of creating composite materials with improved properties, for example, by incorporating hollow silica particles into polymeric matrices, has great prospects.
Hollow, spherical silica nano/microparticles could be the basis for the development of new types of heat-insulating materials (HIM), due to their high temperature resistance, low thermal conductivity, and chemical inertness as well as the availability of silica. A distinctive advantage of HIM that is based on hollow silica particles is the ability to regulate the thermal properties of materials by modifying the size and porosity of the particles [9,10]. Hollow-silica spheres are a promising insulating material due to their structural analogy with aerogels. Their high thermal-insulating ability has already been shown in measurements on hollow silica-sphere powders [10] and in colloidal ensembles in the air [11]. Nanostructured porous materials can be used to store hydrogen in a molecular form [12].
To create hollow silicon-dioxide particles, a two-step process involving the synthesis of core-shell particles by templating followed by template removal is the most common. This method allows for controlling the shape, size, and homogeneity of the final hollow particles. Polymeric (PMMA, polystyrene) monodisperse particles are used most commonly as “cores” [13,14,15,16,17]. The removal of templates from hybrid particles is usually performed by heat treatment [18,19,20,21]. The properties of capsule shells (density, permeability) depend on the peculiarities of their synthesis and subsequent heat treatment. However, in numerous publications on the synthesis of hollow silica particles, including those using polymeric particles as templates [21,22,23,24,25,26,27,28], generally, there are no data on the effect of synthesis and heat treatment conditions on the size and morphology of particles, as well as on the porosity and permeability of the shells. These characteristics are critical for most practical applications, so research in this area is relevant.
This work aimed to investigate changes in the morphology and structure of PMMA-SiO2 core-shell particles, obtained under different synthesis conditions, and hollow silica particles during their heat treatment by electron microscopy (EM), infrared spectroscopy (IR), and X-ray diffraction (X-ray) methods.
2. Materials and Methods
The synthesis of PMMA-SiO2 core-shell particles and hollow SiO2 particles was performed according to the method described in the previous work [29]. Briefly, monodisperse spherical polymethyl methacrylate (PMMA) particles ~440 nm in diameter, synthesized by the emulsion method, served as templates, on the surface of which organosilicon shells were formed by the hydrolysis of vinyltrimethoxysilane (VTMS), using two different catalysts: ammonium hydroxide (type I particles) and L-arginine amino acid (type II particles). After air drying at 60 °C, the hybrid particles were annealed for 24 h at various temperatures in the range of 100–1100 °C. The morphology and dimensions of the particles were measured by electron microscopy using a Zeiss Supra 50 VP scanning-electron microscope (SEM) and a JEM-2100 high-resolution transmission-electron microscope (HRTEM). The IR transmission spectra of the samples were measured using a VERTEX 80v Fourier spectrometer in the spectral range of 400–4000 cm−1 with a resolution of 4 cm−1. For the measurements, powdered samples were ground in an agate mortar and then applied in a thin layer to a crystalline ground KBr substrate. The X-ray diffraction studies were performed using a Rigaku SmartLab SE diffractometer on the CuKα radiation, λ = 1.54178 Å, 40 kV, 35 mA.
3. Results and Discussion
3.1. Transformation of PMMA-SiO2 Core-Shell Particles under the Action of the Electron Microscope Beam
Figure 1a shows an SEM image of spherical PMMA particles that are 440 ± 21 nm in diameter, which we obtained and used as a template for the formation of hybrid PMMA-SiO2 particles. Their high homogeneity in size (deviation from the average value is less than 5%) determines the monodispersity of the hybrid PMMA-SiO2 particles formed on these templates (Figure 1b).

Figure 1.
SEM images of PMMA particles 440 ± 21 nm in size (a) and hybrid PMMA-SiO2 particles 505 ± 18 nm in diameter obtained from them (b).
During the electron-microscopic studies of PMMA particles, we observed their shrinkage under the action of the electron beam. In [30], the authors showed that the shrinkage of spherical PMMA particles depends on the beam power and can reach 20% of the diameter. In our experiments, particle shrinkage reached 30%. PMMA particles stopped changing their sizes 1–2 min after the beginning of their irradiation and, thereafter, maintained their diameter irrespective of the time of exposure to the electron beam. This testifies to the fact that PMMA decomposition does not occur even under conditions of high vacuum inside the electron-microscope column, during the prolonged heating as a result of exposure to the electron beam. That is, the sample-heating temperature reaches a value sufficient to soften the polymer but insufficient for its depolymerization.
When hybrid submicron PMMA-SiO2 particles were studied, it was found that polymer “cores” exhibit an unusual type of shrinkage under the effect of the electron microscope beam (Figure 2), under which a decrease in the PMMA volume is accompanied by the formation of a spherical cavity, and the polymer is distributed along the inner surface of the SiO2 shell. Due to the high rate of the shrinkage process, the stages of polymer-core transformation can be observed only at low-current densities. Even at an electron-beam-current density of 2 pA/cm2, the transformation of the polymer core expressed in the appearance of regions of lower density is observed in the first seconds of observation. Thus, for the samples of core-shell particles formed on polymer templates ~440 nm in diameter, these areas form a spherical cavity ~310 nm in diameter within ~2 min, after which their further growth stops (Figure 2b). Due to the presence of porosity, spherical PMMA particles without an SiO2 shell undergo shrinkage under the action of the electron beam, compressing to their center [30]. The shrinkage of the polymer core in the hybrid PMMA-SiO2 particle occurs by the formation of an expanding inner cavity with densification of the polymer near the organosilicon shell. This occurs due to the presence of the vinyl groups in the organosilicon shell, providing a strong bond between the shell and the polymer core [31,32].

Figure 2.
SEM images of PMMA-SiO2 particles at the beginning of observation (a), 2 min after exposure to the electron microscope beam at a current density of 2 pA/cm2 (b), and under prolonged exposure to the electron beam at a current density of 10 pA/cm2 (c).
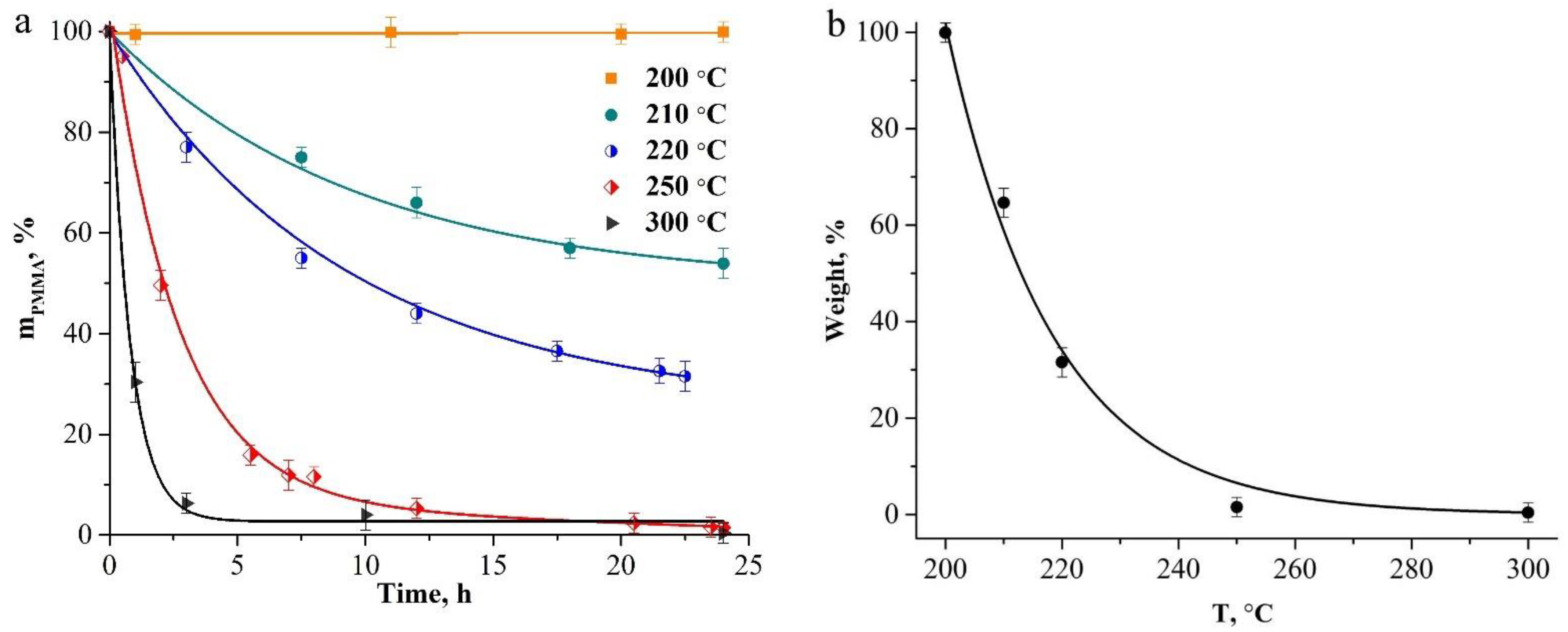
To estimate the initial sizes of the polymer and hybrid particles, we estimated the heating temperature of the core-shell particles under the action of the electron beam. For this purpose, we compared the maximum compression of such particles when exposed to an electron beam of different intensities and under their heating in the furnace. At low-current density (2 pA/cm2), a decrease in the outer diameter of the particles was ~5% (Figure 2b). At a current density of 10 pA/cm2, the maximum compression of the particles reached ~12% (Figure 2c). When comparing these data with the data on the heat treatment of hybrid particles in a wide temperature range (100–900 °C, see Figure 5a in [29]), we assume that the temperature of the core-shell particles under exposure under a low-intensity electron beam does not exceed 150 °C, and at a high-current density it does not exceed 200 °C. At these temperatures, the loss of polymer-core mass does not occur. The thermal-stability studies of the compacts of close-packed spherical PMMA particles at temperatures of 200–300 °C, for 24 h in the air (Figure 3), showed that no weight loss of polymer particles was observed upon heating up to 200 °C inclusive. At temperatures above 200 °C, the beginning of the depolymerization of PMMA particles was observed. When heating the compacts of polymer particles above 200 °C, a gradual increase in the mass-loss rate of the samples occurs, and at 300 °C, the sample evaporates almost completely in 5 h (Figure 3a). Figure 3b illustrates a change in the weight of the compacts with the heating temperature for 24 h. The experimental values of the weight loss (Figure 3a,b) are described well by exponential functions with the coefficient of determination R2 = 0.99 for each curve.
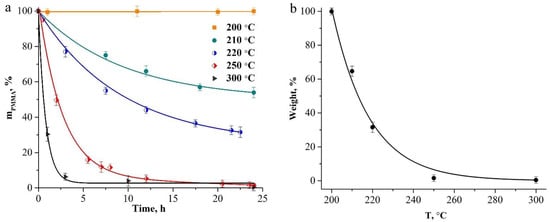
Figure 3.
Dependencies of the weight loss of PMMA particle compacts on temperature and heating duration (a) and on temperature during 24 h annealing (b).
3.2. IR-Transmission Spectra of PMMA-SiO2 Particles
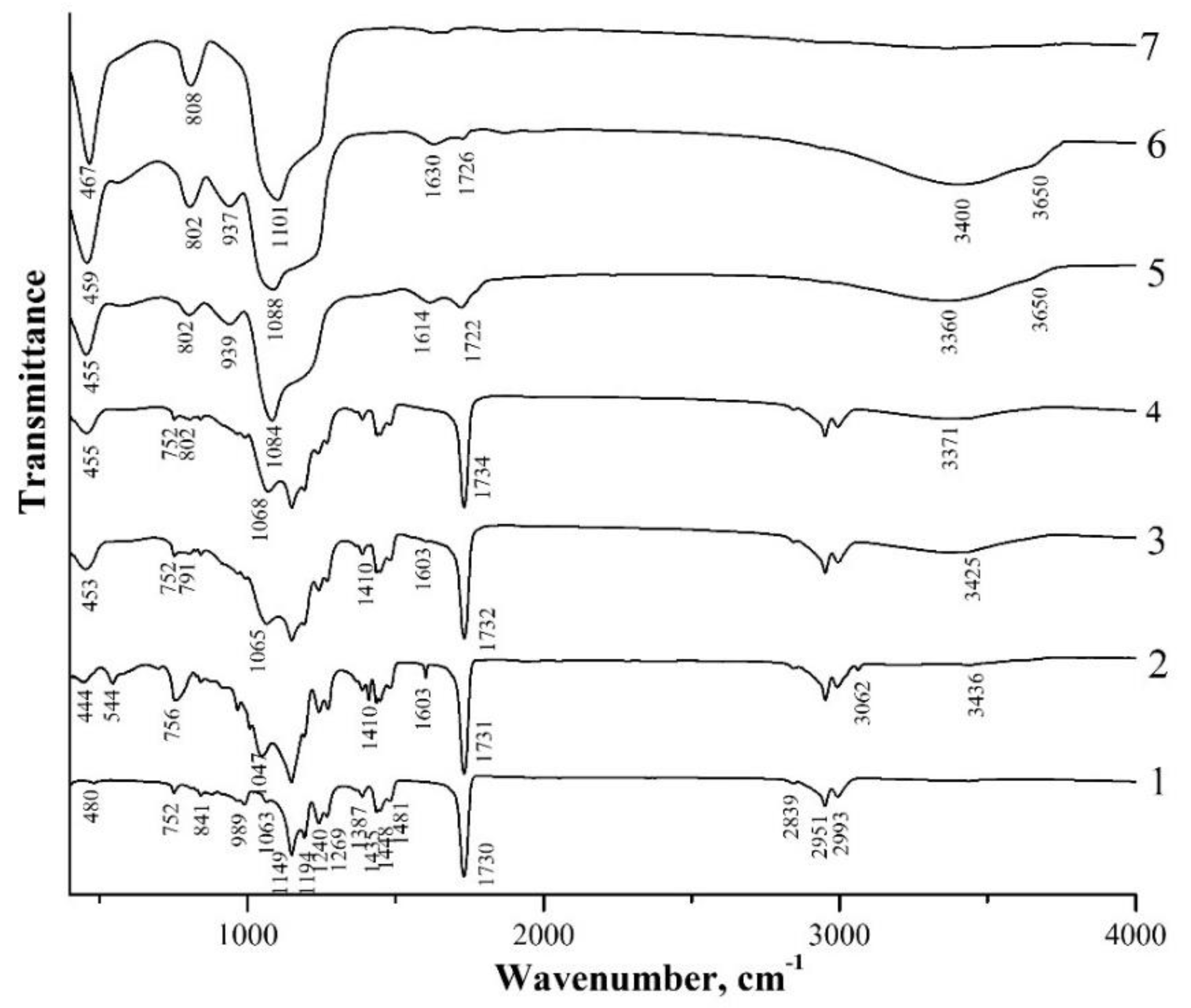
Figure 4 and Figure 5 show IR-transmission spectra of spherical PMMA particles and the initial hybrid core-shell particles obtained using two different catalysts, i.e., ammonium hydroxide (type I particles) and L-arginine amino acid (type II particles), as well as the hybrid particles annealed at different temperatures. The IR spectrum of PMMA (Figure 4 and Figure 5, spectrum 1) agrees well with the PMMA spectra presented in [33,34,35,36]. The absorption bands observed therein, with maxima of 480 cm−1, 752 cm−1, 841 cm−1, 989 cm−1, 1063 cm−1, 1149 cm−1, 1194 cm−1, 1240 cm−1, 1269 cm−1, 1387 cm−1, 1435 cm−1, 1448 cm−1, and 1481 cm−1, are characteristic of the PMMA-absorption spectrum. However, their assignment in the literature sources is not always unambiguous. Thus, in [34,35], intense absorption bands in the range of 1149–1194 cm−1 and 1240–1269 cm−1 are attributed to the stretching vibrations of C–O–C bonds. In [36], bands with maximum frequencies of 1149 cm−1 and 1194 cm−1 are ascribed to the strain torsional and fan vibrations of CH3-groups, respectively. Less-intense bands with a maximum of 1387 cm−1 and those in the range of 1435–1481 cm−1 are due to the bending vibrations of the C–H bonds of the CH3-groups [34,36]. In [35], the 1387 cm−1 band together with the 752 cm−1 band is attributed to the vibrations of the α-methyl group. The 2993 cm−1 and 2839 cm−1 absorption bands are ascribed to the asymmetric and symmetric stretching vibrations of the C–H bonds of the CH3-groups, respectively, and the 2951 cm−1 band is ascribed to the asymmetric stretching vibrations of the C–H bonds of the CH2-groups [33,34,35,36]. The most intense band at 1730 cm−1 is associated with the stretching vibrations of the C=O carbonyl groups [33,34,35,36]. The 989, 841, and 1063 absorption bands are also thought to be characteristic of the PMMA-absorption spectrum [35].

Figure 4.
IR transmission spectra of spherical PMMA particles (1), initial hybrid core-shell particles obtained using ammonium hydroxide (type I particles) (2), and annealed at 150 °C (3), 200 °C (4), 300 °C (5), 400 °C (6) and 600 °C (7).
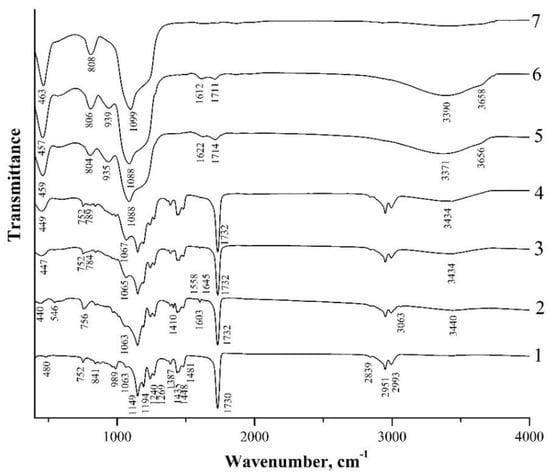
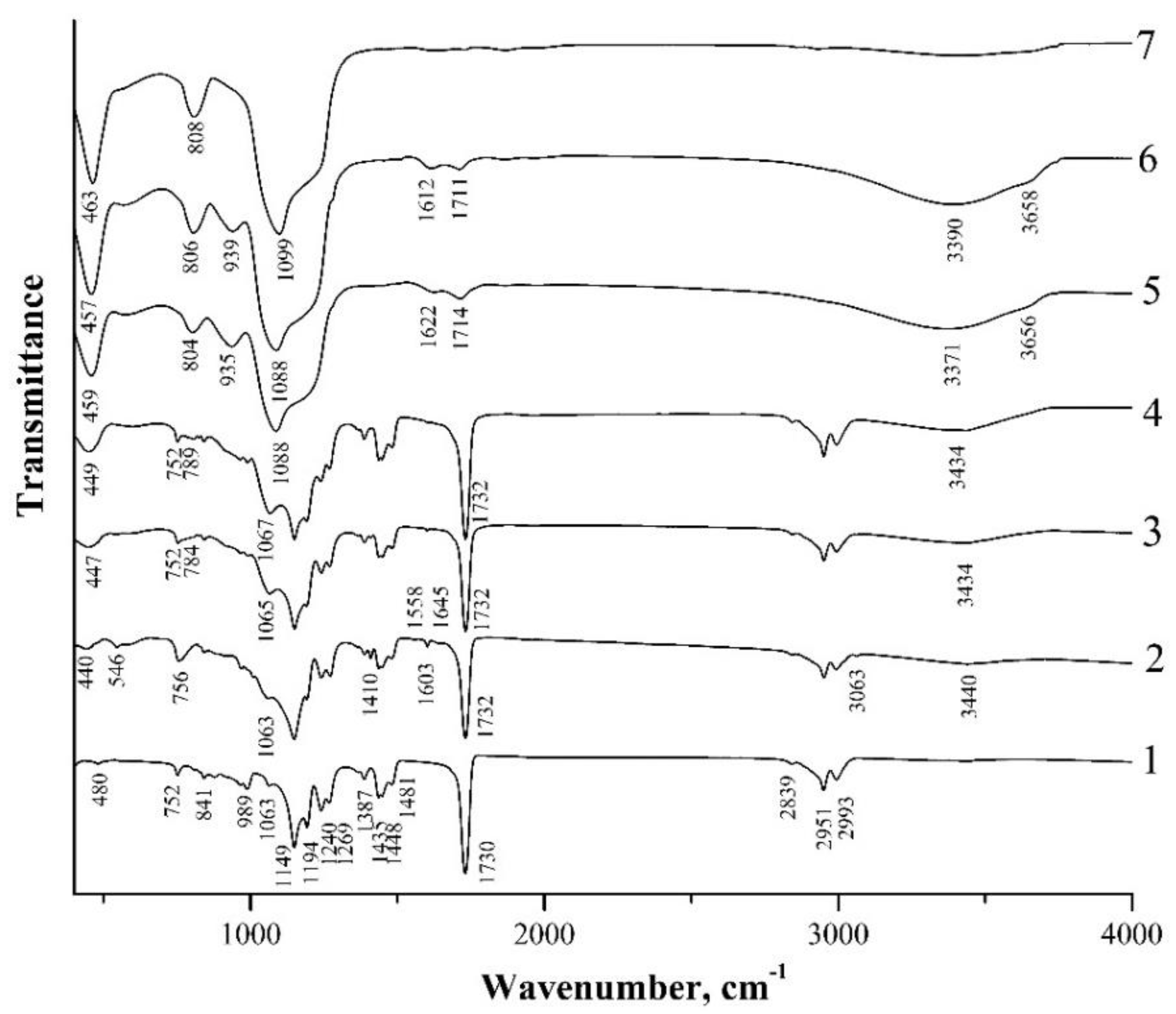
Figure 5.
IR-transmission spectra of spherical PMMA particles (1), initial hybrid core-shell particles obtained using L-arginine (type II particles) (2), and annealed at 150 °C (3), 200 °C (4), 300 °C (5), 400 °C (6) and 600 °C (7).
In the spectra of both types of initial core-shell particles, the PMMA absorption bands indicated above are also clearly present (Figure 4 and Figure 5, spectra 2). At the same time, these spectra contain a number of new bands, among which the weak absorption bands at 544 cm−1, 1410 cm−1, 1603 cm−1, and 3063 cm−1 caused by the presence of vinyl groups in the shell composition of the hybrid particles should be highlighted. The 3063 cm−1 and 1410 cm−1 bands refer to the stretching and bending vibrations of the C–H bonds of the vinyl group (CH2), respectively, the 1603 cm−1 band refers to the stretching vibrations of the C=C bonds of this group, and the 544 band refers to the twisting vibrations of C=CH2 of this group [37,38]. We also note that a number of works discuss different assignments of the last low-frequency band. Thus, in [37], the absorption band at 541 cm–1, which almost coincides in the spectral position, is associated with the deformation vibrations of four-fold siloxane rings. Similarly, in [39,40], weak absorption in the region of 550–640 cm–1 is referred to by such vibrations. The broad absorption band at ~3440 cm−1, in the spectra of both types of the obtained hybrid particles (Figure 4 and Figure 5, spectra 2), appears to be due to the stretching vibrations of O–H-group bonds being present in the shell composition of such particles and, possibly, water molecules adsorbed on the sample surface.
One should note the weak absorption bands at ~1556 cm−1 and 1618–1678 cm−1 in the spectrum of hybrid type II particles obtained in the presence of L-arginine (Figure 5, spectrum 2). These lines are not observed in the spectra of PMMA and the hybrid type I particles obtained in the presence of ammonium hydroxide (Figure 4, spectra 1 and 2) that correspond to the most intense L-arginine-absorption lines. According to [41], the guanidine groups of arginine are firmly bound to the silanol groups of silica. In [29], it was suggested that arginine inclusions in the structure of the organosilicon shell contribute to the porosity of the shells of type II particles, leading to the greater thermal shrinkage of these samples as compared to type I particles.
In the low-frequency region of the spectrum of the sample with initial type I particles, bands with absorption maxima of 444 cm−1, 756 cm−1, and 1047 cm−1 are observed (Figure 4, spectrum 2). According to [40,42], these bands are due to the vibrations of oxygen atoms in the Si–O–Si chains: rocking (TO1), symmetric stretching (TO2), and asymmetric stretching (TO3), respectively. These three lines are also present in the spectrum of the sample with initial type II particles at 440 cm−1, 756 cm−1, and 1063 cm−1, although the 1047 cm−1 line in this spectrum appears only as a low-frequency shoulder of the 1063 cm−1 PMMA-absorption line (Figure 5, spectrum 2).
Summarizing the analysis of the IR spectra of the initial hybrid PMMA-SiO2 particles and the PMMA polymer, we can say that the appearance of the hybrid particles of absorption in the spectra are due to the Si–O–Si bonds, as well as the vinyl groups, which confirms that the process of hydrolysis of VTMS on the surface of the PMMA spheres results in the formation of organosilicon shells.
3.3. IR-Transmission Spectra of PMMA-SiO2 Particles at Different Annealing Temperatures
Figure 4 shows IR spectra of the sample with type I particles annealed at different temperatures (spectra 3–7). The figure demonstrates that up to T = 200 °C, no significant changes in the spectra are observed. At T = 150 °C, absorption bands caused by the vibrations of PMMA, Si–O–Si, and O–H bonds, as well as the weak absorption associated with the vinyl groups, are present in the spectra except for the lowest frequency band at 544 cm−1, which completely disappears. After the annealing of this sample at T = 200 °C (spectrum 4), the remaining absorption bands of the vinyl groups (1410 cm−1, 1603 cm−1, 3062 cm−1) also disappear. The disappearance of the 544 cm−1 band, after the annealing at T = 150 °C, indicates that it is more likely to be attributed to the twisting vibrations of the C=CH2 bond of the vinyl groups than to the deformation vibrations of the four-fold siloxane rings, since in [39] the mentioned mode of the ring vibrations is present in the spectra up to temperatures of 300–500 °C.
The disappearance of the vinyl groups in the IR spectrum of the samples annealed at T = 200 °C indicates the completion of polymerization caused by the opening of the C=C bond of the vinyl groups. Note that in both types of the samples the intensity of the symmetric mode TO2 decreases after annealing at T = 150 °C and T = 200 °C. Similar behavior of this mode was observed during the thermal annealing of the SiO2 films deposited by aerosol–gel method onto a silicon substrate [42].
The radical rearrangement of the transmission spectrum is observed in the spectrum of the sample annealed at T = 300 °C (spectrum 5). The absorption bands of all three modes of Si–O–Si bond oscillations (455 cm−1, 802 cm−1, and 1084 cm−1) are intensified and shifted towards higher frequencies. The combination of these three absorption bands is characteristic of the silica (SiO2) spectrum [43]. In spectrum 5, the 939 cm−1 band appears, which, according to [37,44,45], can be attributed to the vibrations of the Si–(OH) bond. In the high-frequency region of the spectrum in the range of 3100–3700 cm−1, absorption bands of the stretching vibrations of the O-H bonds in H2O (3360 cm−1) and SiOH (3650 cm−1) are observed [44,45]. The absorption line of the C=O carbonyl group (1730→1714 cm−1) weakens and shifts towards low energies, and a weak absorption appears due to the bending vibrations of the O–H bonds in H2O (1614 cm−1) [40]. The comparison of spectra 4 and 5 shows that, whereas PMMA-absorption bands dominate in spectrum 4 after the annealing of the sample at T = 200 °C, in spectrum 5 (T = 300 °C) only one weak-absorption band of the carbonyl group remains, and the absorption band at 1084 cm−1 caused by the stretching vibrations of the Si–O–Si bonds becomes the most intense in this spectrum. The dominance of five intensive absorption bands of the Si–O–Si bonds, characteristic of silica, in the spectrum and the almost complete disappearance of PMMA-absorption bands indicate that annealing at 300 °C leads to almost complete depolymerization of PMMA and its removal from submicron core-shell particles. In this case, the composition of the particle shells is close to pure silica, although this process after T = 300 °C is not complete yet, as evidenced by the presence of the absorption bands of the Si–(OH) and SiO–H bonds. After sample annealing at T = 600 °C (spectrum 7), the Si–(OH)-absorption band disappears as a result of the removal of OH-groups from the silicon-coordination environment; the C=O-absorption band is almost not observed either. The spectrum is characteristic of pure silica (SiO2): only the absorption bands at 467 cm−1, 808 cm−1, and 1101 cm−1, caused by the vibrations of the Si–O–Si bonds, are observed. Their spectral positions almost coincide with the frequencies of the vibrations of these bonds for the SiO2 nanoparticles coated with the PMMA shell [13].
Figure 5 also illustrates IR spectra of the sample, with type II particles annealed at different temperatures (spectra 3–7). After the annealing of this sample at T = 150 °C, the 546 cm−1-twisting-vibration band of the C=CH2 bond of the vinyl groups, as well as the weak-absorption bands (~1556 cm−1 and 1618–1678 cm−1) of L-arginine, which is the catalyst of VTMS hydrolysis (spectrum 3), disappear in its spectrum. Further changes in the spectra of the sample with type II particles after its annealing at higher temperatures are similar to the behavior of the spectra of the sample with type I particles (Figure 4). That is, the absorption lines of the vinyl groups (1410 cm−1, 1603 cm−1, and 3062 cm−1) disappear at T = 200 °C (Figure 5, spectrum 4), and the main changes in the spectra of the samples begin at T = 300 °C (Figure 5, spectrum 5). After the annealing of the sample at T = 600 °C (spectrum 7), only 463 cm−1, 808 cm−1, and 1099 cm−1 absorption bands caused by the vibrations of the Si–O–Si bonds are observed in its spectrum.
A summary of the frequencies of the vibrational modes in the IR spectra is given in Table 1. The assignment of the vibrational modes is made in accordance with the literature data given in the last column of the table.

Table 1.
Vibration frequencies of bonds (cm−1) observed during the transformation of an organosilicon shell into a SiO2 shell during heat treatment.
Thus, the study of the IR spectra of hybrid particles during their heat treatment showed that the polymer core is almost completely removed for 24 h at 300 °C, the hydroxyl groups are removed during annealing at 600 °C, and the process of transformation of core-shell particles into silica spheres is completed.
3.4. Variation of the TO3 Mode-Oscillation Frequency with an Annealing Temperature
IR spectroscopy is widely used to study the microstructure of sol–gel silica films [40]. The degree of condensation of sol–gel silica films largely depends on the synthesis parameters, as well as the duration and temperature of annealing. The condensation is accompanied by shrinkage and stresses. As a consequence, a shift in the frequencies of the Si–O–Si bond vibrations is observed. In [40,42,45,46], a change in the frequency of the most intense absorption line, caused by the motion of an oxygen atom along the line parallel to the Si–Si axis, is described in detail. This motion of the oxygen atom results in the opposite deformation of two neighboring Si–O bonds and is denoted as TO3, which is antisymmetric stretching with a significant contribution from cation motion [40].
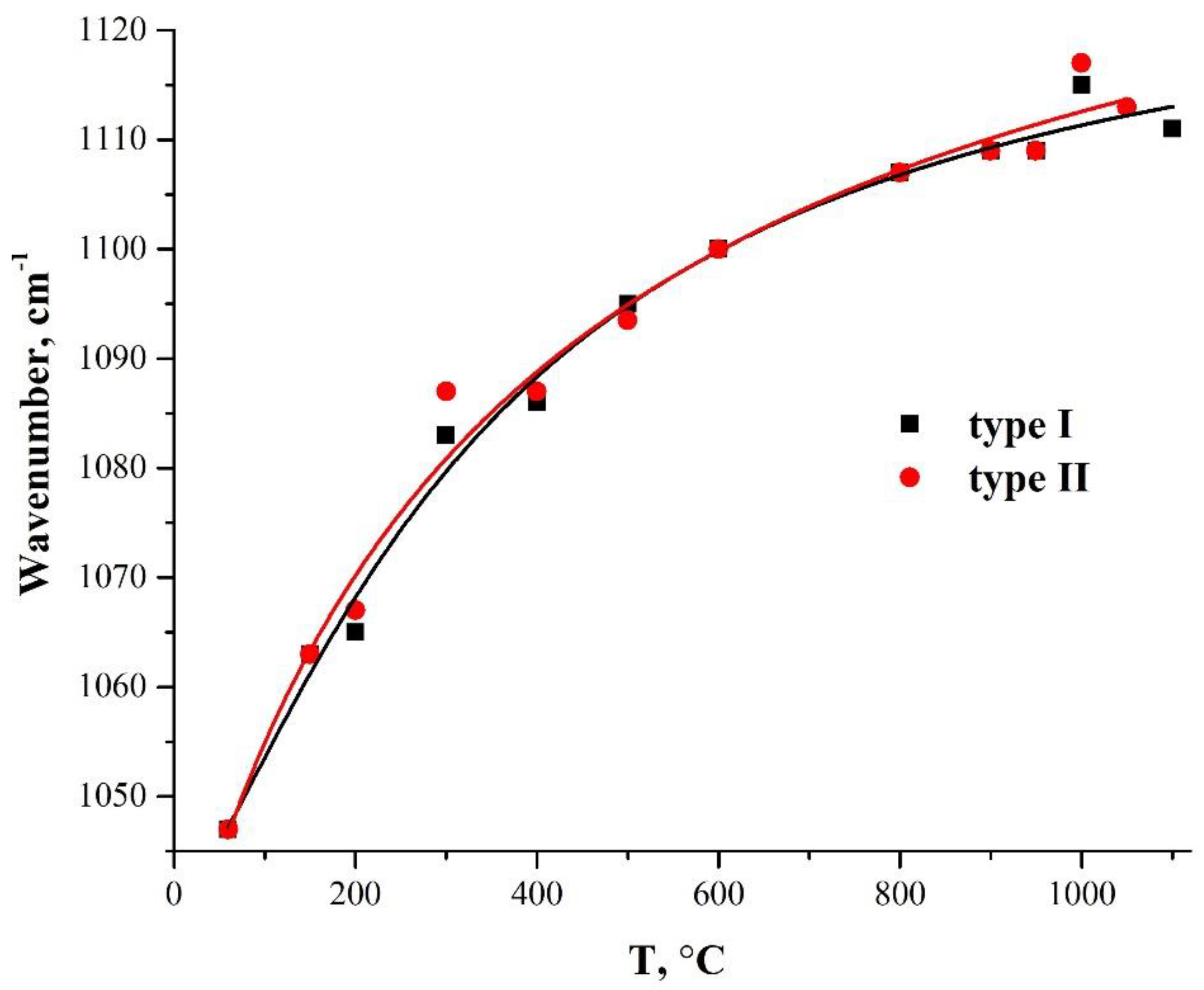
Figure 6 shows the dependencies of the TO3-mode oscillation frequency in two types of PMMA-SiO2 hybrid particles on their annealing temperature. Both samples exhibit a smooth increase in the frequency of TO3 oscillations with an increasing annealing temperature, which is due to the densification of the silica-shell structure [40,42,45,46]. This character of the temperature dependence of the vibrational mode of the Si–O–Si bonds indicates the microstructural homogeneity of the synthesized-silica shell.
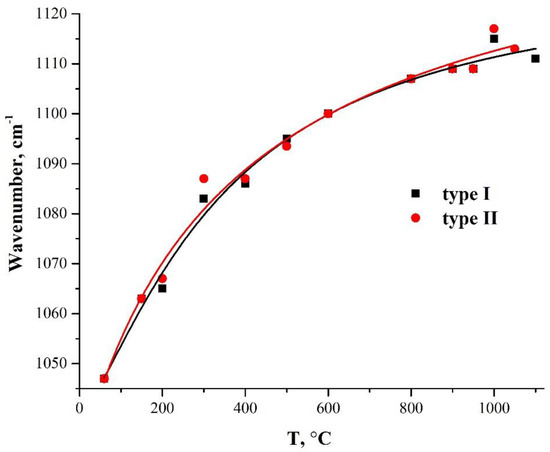
Figure 6.
Dependence of the frequency of the TO3-mode oscillations on the annealing temperature in hybrid PMMA-SiO2 particles obtained using two different catalysts: ammonium hydroxide (type I) and L-arginine amino acid (type II).
Publications [42,45,46] show that the wave number of the TO3 mode during heat treatment of the silica samples passes through the minimum at 450–600 °C, and then increases with further temperature increase. The authors attribute the explanation of such behavior to additional porosity, due to the removal of alkoxy groups during annealing. The absence of a characteristic sag on the curve of the dependence of the TO3-mode frequency, on the annealing temperature in the range of 450–600 °C (Figure 6), can be explained by the absence of unreacted alkoxy groups in our samples.
The authors of [42] did not observe the specified minimum after the deconvolution of the absorption bands in the 1300–900 cm−1 region. The frequency of the TO3 mode oscillations remained constant (1060 cm−1) up to the annealing temperature of 250 °C, and, after annealing at 300 °C, the frequency increased sharply to 1080 cm−1. Next, in the temperature range of 300–500 °C, the frequency dependence of the TO3 mode has an almost horizontal shelf with a weak decrease in the frequency in this area, followed by a sharp increase in the frequencies with an increase in the temperature above 500 °C. The authors of [42] explain this behavior by the presence of an unresolved TEOS (Si–O–C) band at 1080 cm−1, which obscures the TO3 band and disappears by 300 °C.
Note that the lowest TO3 oscillation frequency was also observed by the authors of [42] after the deconvolution of the absorption bands in the region of 1300–900 cm−1, also for the initial unannealed samples. This may mean that the passage of the wave number of the TO3 mode through the minimum at 450–600 °C observed in [45,46] reflects not the nature of the real dependence of the TO3-mode frequency on the annealing temperature, but the effect of the method of obtaining samples, leading to the presence of a significant amount of unreacted alkoxy groups in the structure of the obtained films, on the IR spectra. This leads to additional absorption of the unresolved TEOS (Si–O–C) band at 1080 cm−1, shading the TO3 band [40,42].
3.5. Evolution of the Shell Structure during Heat Treatment
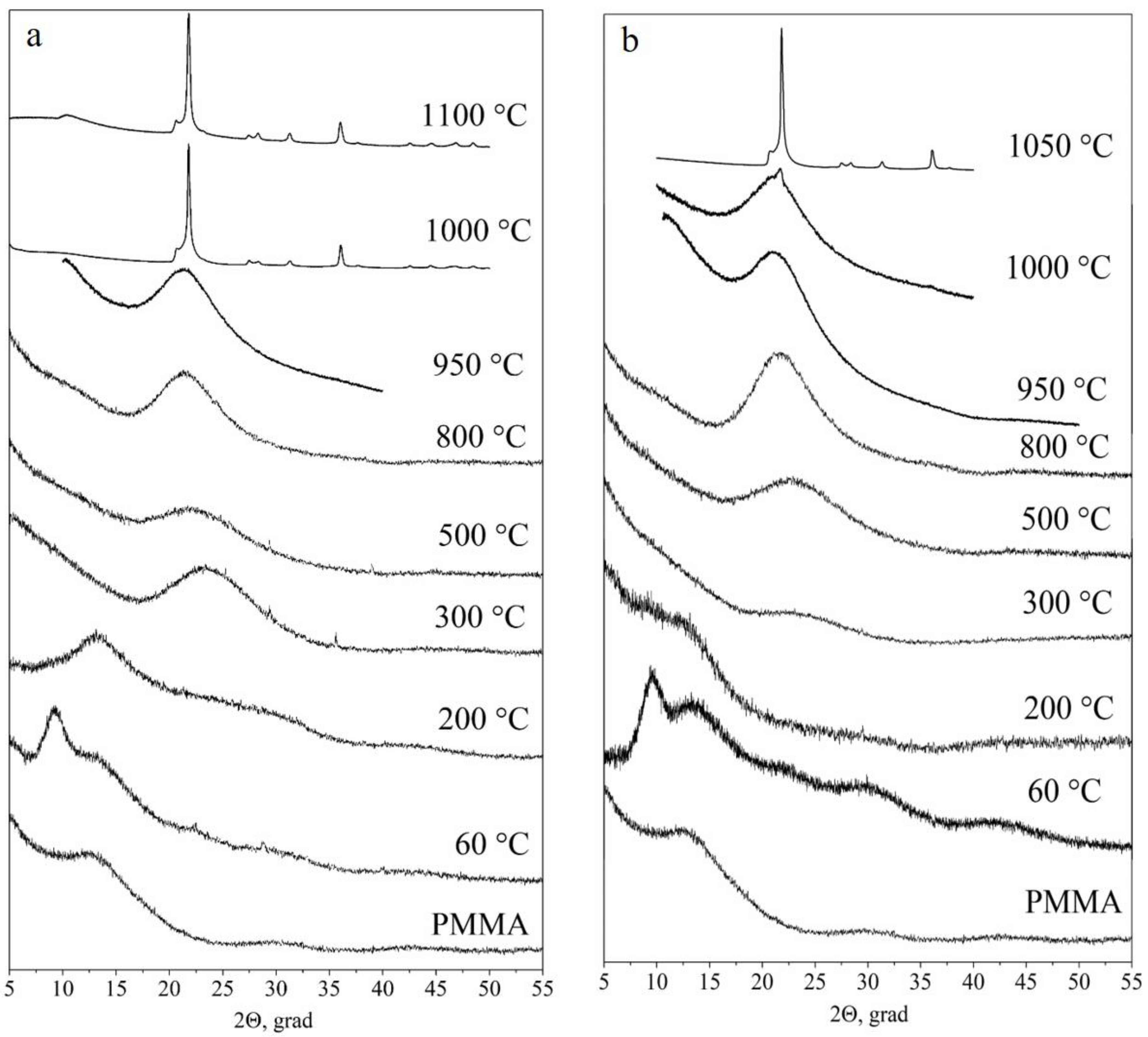
Figure 7 shows X-ray diffraction patterns of initial polymer particles (used as cores) and PMMA-SiO2-hybrid particles of type I (a) and type II (b) annealed at 200 °C, 300 °C, 500 °C, 800 °C, 950 °C, 1000 °C, and 1100 °C and 200 °C, 300 °C, 500 °C, 800 °C, 950 °C, 1000 °C, and 1050 °C, respectively. The X-ray diffraction pattern of the polymer is a series of wide bands, which is typical for an amorphous material, with the first small-angle peak clearly distinguished in the intensity at the 2θ angle of ~13.5° [47,48]. The shape of the first peak reflects the degree of polymer-chain ordering. At a qualitative level, the peak width can be used to estimate the degree of polymer amorphism. The smaller the peak width, the less randomly the macromolecular chains are arranged.
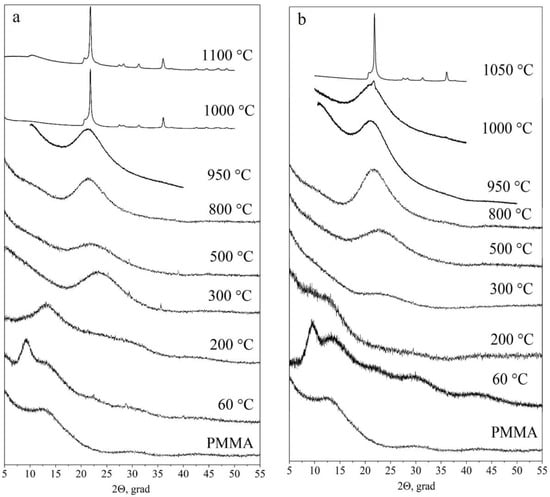
Figure 7.
X-ray diffraction patterns of polymer particles and hybrid PMMA-SiO2 particles based on them, during their transition into hollow SiO2 particles of type I (a) and type II (b), annealed at 60 °C, 200 °C, 300 °C, 500 °C, 800 °C, 950 °C, 1000 °C, and 1100 °C and at 60 °C, 200 °C, 300 °C, 500 °C, 800 °C, 950 °C, 1000 °C, and 1050 °C, respectively.
The X-ray diffraction pattern of both types of initial PMMA-SiO2 particles (60 °C) has an additional peak at the 2θ angle of ~9.3° (Figure 7), as compared to the X-ray diffraction pattern of the initial polymer. In addition, at the 2θ angle of ~23°, a weakly pronounced broad peak (hint) associated with the presence of amorphous silica is observed. Taking into account the multiple superiorities of the polymer core over the SiO2 shell in the mass and volume in the PMMA-SiO2 hybrid particle, it can be assumed that this low-angle peak (9.3°) refers to the polymer core (PMMA). The disappearance of this peak after annealing at 200 °C confirms this assumption. The shape and position of this peak give reason to associate it with the peak at the 2θ angle of ~13.5°. Since this peak arose after the growth of the organosilicon shell on the polymer-core surface, we assume that it is caused by their interaction.
The comparison of the polymer structure in the regions of 2θ angles of ~13.5° and ~9.3°, in terms of the degree of ordering and intermolecular distance, demonstrates an increase in both characteristics for the polymer in the region of shell-core coupling. According to the estimate made for the type I sample by the formula R = 5∕8λ/sinθ, where R is the distance between macromolecular chains, λ is the X-ray wavelength, and θ is the angle corresponding to the diffraction-peak maximum [47,48], the intermolecular distance increased from R ~8.0 Å for the peak at the 2θ angle of ~13.5° to R ~12.0 Å for the peak at the 2θ angle of ~9.3°.
The degree of ordering in the polymer was estimated from the average size of “crystallites” using the formula Lc = 57.3 Kλ/β cosθ, where K = 0.87 is the Scherrer constant, λ = 1.54 Å is the X-ray wavelength, and β is the peak width at half the maximum (2θ) [47,48]. For the peak at the 2θ angle of ~13.5°, the value of Lc is ~16.8 Å, and for the peak at the 2θ angle of ~9.3°, the value of Lc is ~36.2 Å. Thus, the degree of ordering in the polymer in the region of shell-core coupling increased significantly. Note that the values of R and Lc are close for both samples (types I and II).
After the annealing of the samples at 200 °C, the peak at the 2θ angle of ~9.3° almost disappears. The peak at the 2θ angle of ~13.5° remains the most intense in the X-ray diffraction pattern. In this case, there is an increase in the plateau intensity in the regions of 2θ angles of 22–25°, which corresponds to silicon dioxide. Annealing at 300 °C leads to the complete disappearance of the polymer peak at the 2θ angle of ~13.5°. These data are consistent with the results obtained by IR spectroscopy (Figure 4 and Figure 5). At the same time, an intense peak appears in the X-ray diffraction pattern, centered near the 2θ angle of ~23°, which corresponds to amorphous silicon dioxide. Subsequent annealing of the samples at 500 °C and 800 °C leads to insignificant changes in the X-ray diffraction patterns. Thus, the only remaining peak shifts with an increasing temperature towards smaller angles, and its width decreases. The position of the peak center after 500 °C shifts to the 2θ angle of ~22°, and that at 800 °C shifts to the 2θ angle of ~21°. At an annealing temperature of ~1000 °C, the loss of a spherical shape and sintering of both types of the particles occur [29]. The X-ray diffraction studies show that, at this temperature, type I particles pass into the crystalline phase (α-cristobalite SiO2) (Figure 7a). Type II particles remain predominantly amorphous: reflections of the crystalline phase of α-cristobalite SiO2 are barely visible against the background of the halo (Figure 7b).
At an annealing temperature of 1050 °C, type II particles crystallize in the α-cristobalite SiO2 phase (Figure 7b), similarly to type I particles (Figure 7a). The beginning of the crystallization of hollow type II particles at a higher temperature can be associated with a lower density of their shells compared to the shells of type I particles [29].
4. Conclusions
In this work, changes in the morphology and structure of PMMA-SiO2 core-shell particles and hollow SiO2 particles during their heat treatment have been studied by electron microscopy, infrared spectroscopy, and X-ray diffraction. An unusual transformation of the polymer core in PMMA-SiO2 hybrid particles has been found: spherical cavities are formed inside the polymer cores under the action of an electron-microscope beam. This behavior, as well as the compression of PMMA particles without the silica shell under the action of an electron beam, indicates the porosity of the initial polymer particles. In this case, there is no loss of polymer mass because, according to the estimates, the heating of the samples did not exceed 200 °C.
The dependence of the frequency of the Si–O–Si bond vibrations (TO3 mode) in the samples on the temperature of their treatment has been measured. The samples demonstrate an increase in the TO3-oscillation frequency with an increase in the annealing temperature. An analysis of the diffraction patterns has revealed the formation of a new diffraction maximum of the polymer in the initial hybrid PMMA-SiO2 particles at the 2θ angle of ~9.3°, which arose as a result of the interaction between the organosilicon shell and the polymer-core surface. It is shown that in the region of the interaction between the polymer and the silica shell, the polymer structure changes. The distance R between the macromolecular chains in this region and the average size of “crystallites” Lc increased, compared with the initial polymer. After the annealing of the samples at 200 °C, the peak at the 2θ angle of ~9.3° almost disappears. Further annealing at 300 °C leads to the complete removal of the polymer from the samples. The X-ray diffraction studies show that type I particles heated to 1000 °C for 24 h pass into the crystalline phase (α-cristobalite SiO2), while type II particles remain predominantly amorphous at this temperature.
Note that when using VTMS as a precursor of silicon dioxide in the synthesis of hollow particles by the template method, it is preferable to use the thermal removal of polymer templates. During heat treatment, unreacted organic residues are removed, which is important for their use in pharmaceutical and food industries. In this case, the change in the size and structure of the particle shells that occurs during annealing should be taken into account.
Author Contributions
Conceptualization, G.A.E. and N.S.S.; methodology, V.M.M.; investigation, T.N.F., I.I.K. and I.I.Z.; formal analysis, A.A.Z.; writing—original draft preparation, G.A.E. and T.N.F.; writing—review and editing, V.M.M.; visualization, N.S.S.; supervision, G.A.E. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The work was supported by ISSP RAS—Russian government contracts using the equipment of the Research Facility Center of ISSP RAS and the Center for Collective Use in Chernogolovka.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Zhang, X.; Wang, P.; Sun, D.; Li, X.; An, J.; Yu, T.; Yang, E.-H.; Yang, J. Dynamic plastic deformation and failure mechanisms of individual microcapsule and its polymeric composites. J. Mech. Phys. Solids 2020, 139, 103933. [Google Scholar] [CrossRef]
- Eisinaite, V.; Juraite, D.; Schroen, K.; Leskauskaite, D. Food-grade double emulsions as effective fat replacers in meat systems. J. Food Eng. 2017, 213, 54–59. [Google Scholar] [CrossRef]
- Ouarga, A.; Noukrati, H.; Iraola-Arregui, I.; Elaissari, A.; Barroug, A.; Ben Youcef, H. Development of anti-corrosion coating based on phosphorylated ethyl cellulose microcapsules. Prog. Org. Coat. 2020, 148, 105885. [Google Scholar] [CrossRef]
- Huang, L.; Zhou, J.; Chen, Y.; Li, W.; Han, X.; Wang, L. Engineering Microcapsules for Simultaneous Delivery of Combinational Therapeutics. Adv. Mater. Technol. 2020, 5, 2000623. [Google Scholar] [CrossRef]
- Ma, G.; Yue, H. Advances in Uniform Polymer Microspheres and Microcapsules: Preparation and Biomedical Applications. Chin. J. Chem. 2020, 38, 911–923. [Google Scholar] [CrossRef]
- Shang, X.; Zhan, B.; Li, J.; Zhong, R. Novel microcapsules for internal curing of high-performance cementitious system. Sci. Rep. 2020, 10, 8318. [Google Scholar] [CrossRef]
- Iqbal, M.; Zafar, N.; Fessi, H.; Elaissari, A. Double emulsion solvent evaporation techniques used for drug encapsulation. Int. J. Pharm. 2015, 496, 173–190. [Google Scholar] [CrossRef]
- Hofmeister, I.; Landfester, K.; Taden, A. pH-Sensitive Nanocapsules with Barrier Properties: Fragrance Encapsulation and Controlled Release. Macromolecules 2014, 47, 5768–5773. [Google Scholar] [CrossRef]
- Gao, T.; Sandberg, L.I.C.; Jelle, B.P.; Gustavsen, A. Fuelling the Future: Advances in Science and Technologies for Energy Generation, Transmission and Storage; Mendez-Vilas, A.A., Ed.; Brown Walker Press: Boca Raton, FL, USA, 2012; pp. 535–539. [Google Scholar]
- Gao, T.; Jelle, B.P.; Sandberg, L.I.C.; Gustavsen, A. Monodisperse Hollow Silica Nanospheres for Nano Insulation Materials: Synthesis, Characterization, and Life Cycle Assessment. ACS Appl. Mater. Interfaces 2013, 5, 761–767. [Google Scholar] [CrossRef]
- Ruckdeschel, P.; Philipp, A.; Retsch, M. Understanding Thermal Insulation in Porous, Particulate Materials. Adv. Funct. Mater. 2017, 27, 1702256–1702266. [Google Scholar] [CrossRef]
- Barthelemy, H.; Weber, M.; Barbier, F. Hydrogen storage: Recent improvements and industrial perspectives. Int. J. Hydrog. Energy 2017, 42, 7254–7262. [Google Scholar] [CrossRef]
- Zhu, A.; Shi, Z.; Cai, A.; Zhao, F.; Liao, T. Synthesis of core–shell PMMA–SiO2 nanoparticles with suspension–dispersion–polymerization in an aqueous system and its effect on mechanical properties of PVC composites. Polym. Test. 2008, 27, 540–547. [Google Scholar] [CrossRef]
- Choi, W.S.; Koo, H.Y.; Kim, D.-Y. Facile Fabrication of Core-in-Shell Particles by the Slow Removal of the Core and Its Use in the Encapsulation of Metal Nanoparticles. Langmuir 2008, 24, 4633–4636. [Google Scholar] [CrossRef]
- Liu, H.; Li, H.; Ding, Z.; Fu, A.; Wang, H.; Guo, P.; Yu, J.; Wang, C.; Zhao, X.S. Preparation of Porous Hollow SiO2 Spheres by a Modified Stöber Process Using MF Microspheres as Templates. J. Clust. Sci. 2012, 23, 273–285. [Google Scholar] [CrossRef]
- Huang, Z.F.; Qu, X.Y.; Chen, Z. Nano-SiO2/PMMA-PU composite particles with core-shell structure via emulsion polymerization and their application in epoxy resin. J. Appl. Polym. Sci. 2015, 132, 41919. [Google Scholar] [CrossRef]
- Mostafa, H.Y.; Hussain, A.I.; El-Masry, A.M.; Maher, A. Novel Core–Shell of Polymethyl Methacrylate/butyl Acrylate/vinyl Silica Nanocomposite Particles through Seed Emulsion Polymerization. Polym. Technol. Eng. 2017, 56, 411–420. [Google Scholar] [CrossRef]
- Yamada, Y.; Mizutani, M.; Nakamura, T.; Yano, K. Mesoporous Microcapsules with Decorated Inner Surface: Fabrication and Photocatalytic Activity. Chem. Mater. 2010, 22, 1695–1703. [Google Scholar] [CrossRef]
- Cao, S.; Zhao, Z.; Jin, X.; Sheng, W.; Li, S.; Ge, Y.; Dong, M.; Wu, W.; Fang, L. Unique double-shelled hollow silica microspheres: Template-guided self-assembly, tunable pore size, high thermal stability, and their application in removal of neutral red. J. Mater. Chem. 2011, 21, 19124–19131. [Google Scholar] [CrossRef]
- Ernawati, L.; Ogi, T.; Balgis, R.; Okuyama, K.; Stucki, M.; Hess, S.C.; Stark, W.J. Hollow Silica as an Optically Transparent and Thermally Insulating Polymer Additive. Langmuir 2016, 32, 338–345. [Google Scholar] [CrossRef]
- Tissot, I.; Reymond, J.P.; Lefebvre, F.; Bourgeat-Lami, E. SiOH-Functionalized Polystyrene Latexes. A Step toward the Synthesis of Hollow Silica Nanoparticles. Chem. Mater. 2002, 14, 1325–1331. [Google Scholar] [CrossRef]
- Chen, M.; Wu, L.; Zhou, S.; You, B. Synthesis of Raspberry-like PMMA/SiO2 Nanocomposite Particles via a Surfactant-Free Method. Macromolecules 2004, 37, 9613–9619. [Google Scholar] [CrossRef]
- Cheng, X.; Chen, M.; Zhou, S.; Wu, L. Preparation of SiO2/PMMA composite particles via conventional emulsion polymerization. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 3807–3816. [Google Scholar] [CrossRef]
- Jiu, H.; Jia, W.; Zhang, L.; Huang, C.; Jiao, H.; Chang, J. Synthesis, luminescent and drug-release properties of SiO2@Y2O3:Eu hollow mesoporous microspheres. J. Porous Mater. 2015, 22, 1511–1518. [Google Scholar] [CrossRef]
- Kato, N.; Ishii, T.; Koumoto, S. Synthesis of Monodisperse Mesoporous Silica Hollow Microcapsules and Their Release of Loaded Materials. Langmuir 2010, 26, 14334–14344. [Google Scholar] [CrossRef] [PubMed]
- Nandiyanto, A.B.D.; Akane, Y.; Ogi, T.; Okuyama, K. Mesopore-Free Hollow Silica Particles with Controllable Diameter and Shell Thickness via Additive-Free Synthesis. Langmuir 2012, 28, 8616–8624. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Fang, L.; Zhao, Z.; Ge, Y.; Piletsky, S.; Turner, A.P.F. Hierachically Structured Hollow Silica Spheres for High Efficiency Immobilization of Enzymes. Adv. Funct. Mater. 2013, 23, 2162–2167. [Google Scholar] [CrossRef]
- Wang, J.; Pan, M.; Yuan, J.; Lin, Q.; Zhang, X.; Liu, G.; Zhu, L. Hollow mesoporous silica with a hierarchical shell from in situ synergistic soft–hard double templates. Nanoscale 2020, 12, 10863–10871. [Google Scholar] [CrossRef]
- Masalov, V.M.; Sukhinina, N.S.; Khodos, I.I.; Zverkova, I.I.; Zhokhov, A.A.; Emelchenko, G.A. Effect of Heat Treatment on the Physical Properties and Morphology of Hollow Submicron SiO2 Particles. J. Surf. Investig. 2021, 15, 1174–1180. [Google Scholar] [CrossRef]
- Nemtsev, I.V.; Shabanov, A.V.; Shabanova, O.V. Electron Microscopy Investigation of Polymethylmethacrylate Spherical Particles & Artificial Opals Based on it. Sib. J. Sci. Technol. 2012, 1, 126–129. [Google Scholar]
- Soucek, M.D.; He, J. UV-Curable Vinyl Functionalized Siloxane Colloids. Radtech Rep. 2007, 21, 10–16. [Google Scholar]
- Gunji, T.; Kawaguchi, Y.; Okonogi, H.; Sakan, T.; Arimitsu, K.; Abe, Y. Preparation and Properties of Organic-Inorganic Hybrid Gel Films Based on Polyvinylpolysilsesquioxane Synthesized from Trimethoxy(vinyl)silane. J. Sol-Gel Sci. Tech. 2005, 33, 9–13. [Google Scholar] [CrossRef]
- Tommasini, F.J.; Ferreira, L.; Tienne, L.G.P.; de Oliveira Aguiar, V.; Prado da Silva, M.H.; da Mota Rocha, L.F.; Vieira Marques, M.D.F. Poly (Methyl Methacrylate)-SiC Nanocomposites Prepared Through in situ Polymerization. Mater. Res. 2018, 21, e20180086. [Google Scholar] [CrossRef]
- Tham, D.Q.; Hoang, T.; Giang, N.V.; Dung, N.T.K.; Chung, I. Synthesis and characterization of (4-arm-star-PMMA)/PMMA-g-SiO2 hybrid nanocomposites. Green Process. Synth. 2018, 7, 391–398. [Google Scholar] [CrossRef]
- Duan, G.; Zhang, C.; Li, A.; Yang, X.; Lu, L.; Wang, X. Preparation and Characterization of Mesoporous Zirconia Made by Using a Poly (methyl methacrylate) Template. Nanoscale Res. Lett. 2008, 3, 118–122. [Google Scholar] [CrossRef] [Green Version]
- Mas Haris, M.R.H.; Kathiresan, S.; Mohan, S. FT-IR and FT-Raman Spectra and Normal Coordinate Analysis of Poly methyl methacrylate. Der Pharma Chem. 2010, 2, 316–323. [Google Scholar]
- Ou, D.L.; Seddon, A. Near- and mid-infrared spectroscopy of sol–gel derived ormosils: Vinyl and phenyl silicates. J. Non-Cryst. Solids 1997, 210, 187–203. [Google Scholar] [CrossRef]
- Chen, S.; Osaka, A.; Hayakawa, S.; Shirosaki, Y.; Tsuru, K. Morphology and structure of organosilica hybrid particles derived from tetramethoxysilane and vinyltrimethoxysilane via a catalyst-free sol–gel route. J. Mater. Chem. 2010, 20, 7337–7339. [Google Scholar] [CrossRef]
- Yoshino, H.; Kamiya, K.; Nasu, H. IR study on the structural evolution of sol-gel derived SiO2 gels in the early stage of conversion to glasses. J. Non-Cryst. Solids 1990, 126, 68–78. [Google Scholar] [CrossRef]
- Innocenzi, P. Infrared spectroscopy of sol–gel derived silica-based films: A spectra-microstructure overview. J. Non-Cryst. Solids 2003, 316, 309–319. [Google Scholar] [CrossRef]
- Vlasova, N.N.; Golovkova, L.P. The adsorption of amino acids on the surface of highly dispersed silica. Colloid J. 2004, 66, 657–662. [Google Scholar] [CrossRef]
- Primeau, N.; Vautey, C.; Langlet, M. The effect of thermal annealing on aerosol-gel deposited SiO2 films: A FTIR deconvolution study. Thin Solid Film. 1997, 310, 47–56. [Google Scholar] [CrossRef]
- Kirk, C.T. Quantitative analysis of the effect of disorder-induced mode coupling on infrared absorption in silica. Phys. Rev. B 1988, 38, 1255–1273. [Google Scholar] [CrossRef] [PubMed]
- Parrill, T. Heat treatment of spun-on acid-catalyzed sol-gel silica films. J. Mater. Res. 1994, 9, 723–730. [Google Scholar] [CrossRef]
- Parrill, T. Transmission infrared study of acid-catalyzed sol-gel silica coatings during room ambient drying. J. Mater. Res. 1992, 7, 2230–2239. [Google Scholar] [CrossRef]
- Almeida, R.M.; Vasconcelos, H.C.; Ilharco, L.M. Relationship between infrared absorption and porosity in silica-based sol-gel films. SPIE 1994, 2288, 678–687. [Google Scholar] [CrossRef]
- Hussain, R.; Mohammad, D. X-ray Diffraction Study of the Changes Induced During the Thermal Degradation of Poly (Methyl Methacrylate) and Poly (Methacryloyl Chloride). Turk. J. Chem. 2004, 28, 725–729. [Google Scholar]
- Shobhana, E. X-ray Diffraction and UV-Visible Studies of PMMA Thin Films. Int. J. Mod. Eng. Res. 2012, 2, 1092–1095. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).