



Wet Synthesis of Graphene-Polypyrrole Nanocomposites via Graphite Intercalation Compounds



 ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of GBS Products
2.3. Thermal Treatment of GBS Samples
2.4. Microwave Treatment of GBS Samples
2.5. Synthesis of Graphene-Polypyrrole (GP) Nanocomposites
2.6. Characterization
2.7. Electrochemical Measurements
3. Results
3.1. Optical Spectroscopy and SEM Characterization
3.2. XRD Analysis
3.3. Raman Spectroscopy Analysis
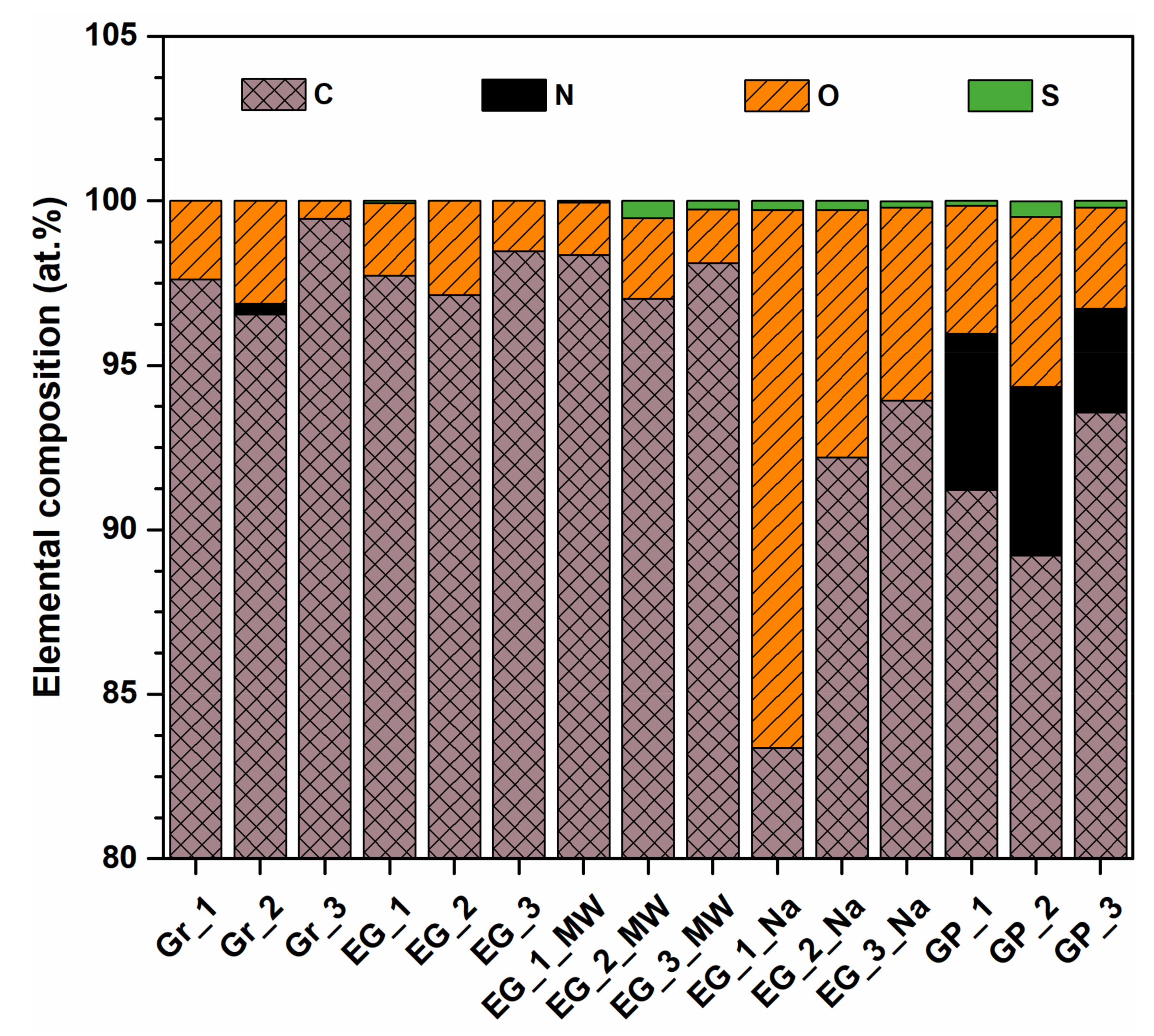
3.4. XPS Analysis
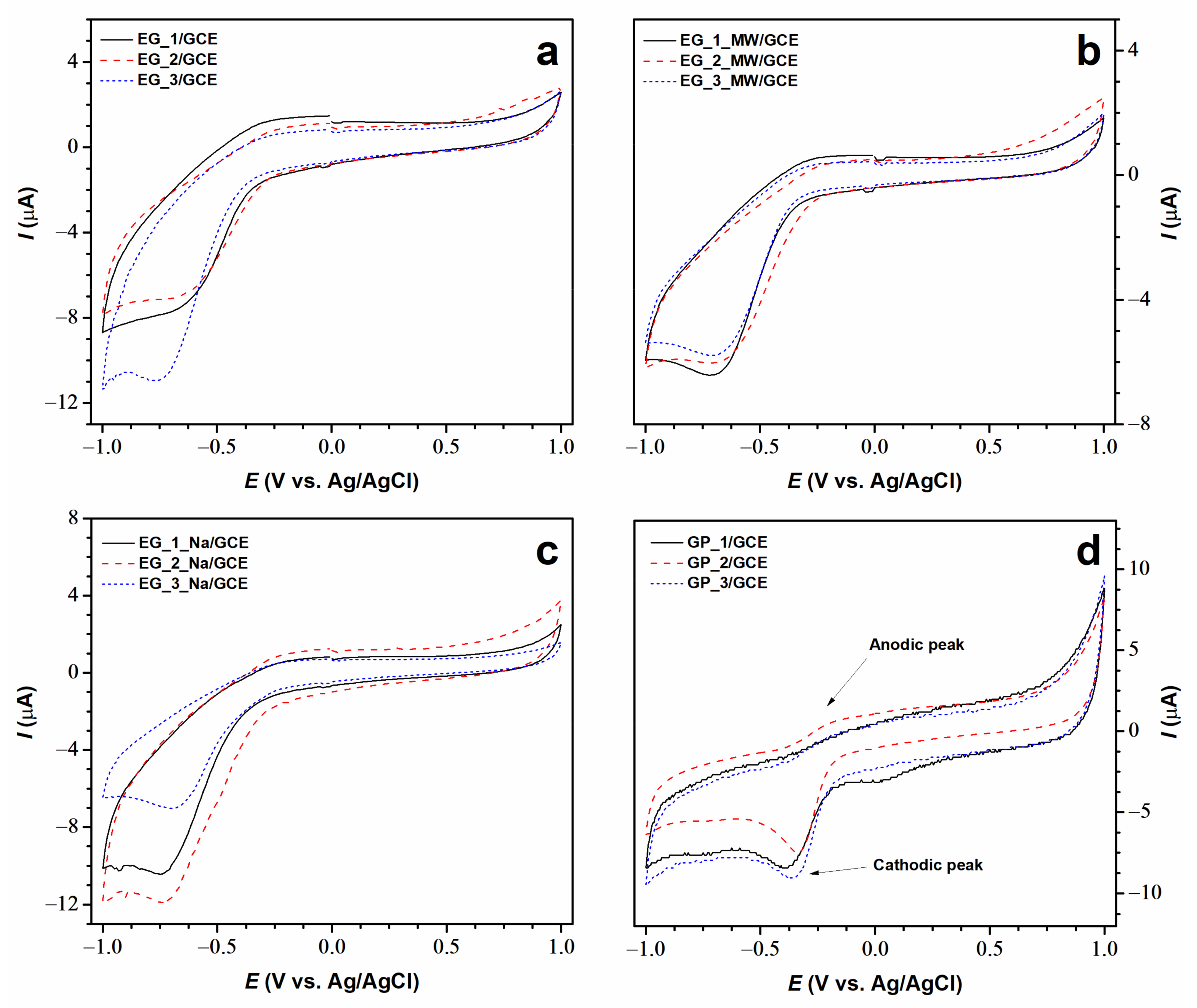
3.5. Electrochemical Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Garcia-Hernandez, M.; Coleman, J. Materials science of graphene: A flagship perspective. 2D Mater. 2016, 3, 010401. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, E.K.; Joo, W.J.; Jang, Y.; Kim, B.S.; Lim, J.Y.; Choi, S.H.; Ahn, S.J.; Ahn, J.R.; Park, M.H.; et al. Wafer-scale growth of single-crystal monolayer graphene on reusable hydrogen-terminated germanium. Science 2014, 344, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Bharathi, M.S.; Wang, L.; Liu, Y.; Chen, H.; Nie, S.; Wang, X.; Chou, H.; Tan, C.; Fallahazad, B.; et al. The role of surface oxygen in the growth of large single-crystal graphene on copper. Science 2013, 342, 720–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eigler, S.; Hirsch, A. Chemistry with graphene and graphene oxide challenges for synthetic chemists. Angew. Chem. Int. Ed. 2014, 53, 7720–7738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgakilas, V.; Otyepka, M.; Bourlinos, A.B.; Chandra, V.; Kim, N.; Kemp, K.C.; Hobza, P.; Zboril, R.; Kim, K.S. Functionalization of graphene: Covalent and non-covalent approaches, derivatives and applications. Chem. Rev. 2012, 112, 6156–6214. [Google Scholar] [CrossRef]
- Mao, H.Y.; Laurent, S.; Chen, W.; Akhavan, O.; Imani, M.; Ashkarran, A.A.; Mahmoudi, M. Graphene: Promises, facts, opportunities, and challenges in nanomedicine. Chem. Rev. 2013, 113, 3407–3424. [Google Scholar] [CrossRef]
- Dreyer, D.R.; Todd, A.D.; Bielawski, C.W. Harnessing the chemistry of graphene oxide. Chem. Soc. Rev. 2014, 43, 5288–5301. [Google Scholar] [CrossRef]
- Dreyer, D.R.; Park, S.; Bielawski, C.W.; Ruoff, R.S. The chemistry of graphene oxide. Chem. Soc. Rev. 2010, 39, 228–240. [Google Scholar] [CrossRef]
- Criado, A.; Melchionna, M.; Marchesan, S.; Prato, M. The covalent functionalization of graphene on substrates. Angew. Chem. Int. Ed. 2015, 54, 0734–10750. [Google Scholar] [CrossRef]
- Eigler, S.; Dotzer, C.; Hirsch, A. Visualization of defect densities in reduced graphene oxide. Carbon 2012, 50, 3666–3673. [Google Scholar] [CrossRef]
- Du, W.; Geng, H.; Yang, Y.; Zhang, Y.; Rui, X.; Li, C.C. Pristine graphene for advanced electrochemical energy applications. J. Power Sources 2019, 437, 226899. [Google Scholar] [CrossRef]
- Kovtyukhova, N.I.; Wang, Y.; Berkdemir, A.; Cruz-Silva, R.; Terrones, M.; Crespi, V.H.; Mallouk, T.E. Non-oxidative intercalation and exfoliation of graphite by Brønsted acids. Nat. Chem. 2014, 6, 957–963. [Google Scholar] [CrossRef]
- Aghamohammadi, H.; Eslami-Farsani, R.; Torabian, M.; Amousa, N. Recent advances in one-pot functionalization of graphene using electrochemical exfoliation of graphite: A review study. Synth. Met. 2020, 269, 116549. [Google Scholar] [CrossRef]
- Ruse, E.; Larboni, M.; Lavi, A.; Pyrikov, M.; Leibovitch, Y.; Ohayon-Lavi, A.; Vradman, L.; Regev, O. Molten salt in-situ exfoliation of graphite to graphene nanoplatelets applied for energy storage. Carbon 2021, 176, 168–177. [Google Scholar] [CrossRef]
- Du, W.; Jiang, X.; Zhu, L. From graphite to graphene: Direct liquid-phase exfoliation of graphite to produce single- and few-layered pristine graphene. J. Mater. Chem. A 2013, 1, 10592. [Google Scholar] [CrossRef]
- Xu, Y.; Cao, H.; Xue, Y.; Li, B.; Cai, W. Liquid-phase exfoliation of graphene: An overview on exfoliation media, techniques, and challenges. Nanomaterials 2018, 8, 942. [Google Scholar] [CrossRef] [Green Version]
- Brodie, B. Note sur un nouveau procédé pour la purification et la désagrégation du graphite. Ann. Chim. Phys. 1855, 45, 351–448. [Google Scholar]
- Aronson, S.; Frishberg, C.; Frankl, G. Thermodynamic properties of the graphite-bisulfate lamellar compounds. Carbon 1971, 9, 715–723. [Google Scholar] [CrossRef]
- Daumas, N.; Herold, A. Notes des membres et correspontants et notes présentées ou transmises par leurs soins. C. R. Acad. Sci. Ser. C 1969, 268, 373–375. [Google Scholar]
- Dimiev, A.M.; Ceriotti, G.; Behabtu, N.; Zakhidov, D.; Pasquali, M.; Saito, R.; Tour, J.M. Direct real-time monitoring of stage transitions in graphite intercalation compounds. ACS Nano 2013, 7, 2773–2780. [Google Scholar] [CrossRef]
- Chiang, C.K.; Fincher, C., Jr.; Park, Y.W.; Heeger, A.J.; Shirakawa, H.; Louis, E.J.; Gau, S.C.; MacDiarmid, A.G. Electrical conductivity in doped polyacetylene. Phys. Rev. Lett. 1977, 39, 1098. [Google Scholar] [CrossRef]
- Ohta, T.; Bostwick, A.; Seyller, T.; Horn, K.; Rotenberg, E. Controlling the electronic structure of bilayer graphene. Science 2006, 313, 951–954. [Google Scholar] [CrossRef] [PubMed]
- Mouri, S.; Miyauchi, Y.; Matsuda, K. Tunable photoluminescence of monolayer MoS2 via chemical doping. Nano Lett. 2013, 13, 5944–5948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tongay, S.; Zhou, J.; Ataca, C.; Liu, J.; Kang, J.S.; Matthews, T.S.; You, L.; Li, J.; Grossman, J.C.; Wu, J. Broad-range modulation of light emission in two-dimensional semiconductors by molecular physisorption gating. Nano Lett. 2013, 13, 2831–2836. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [Green Version]
- Ryu, S.; Liu, L.; Berciaud, S.; Yu, Y.-J.; Liu, H.; Kim, P.; Flynn, G.W.; Brus, L.E. Atmospheric oxygen binding and hole doping in deformed graphene on a SiO2 substrate. Nano Lett. 2010, 10, 4944–4951. [Google Scholar] [CrossRef] [Green Version]
- Park, K.; Kang, H.; Koo, S.; Lee, D.; Ryu, S. Redox-governed charge doping dictated by interfacial diffusion in two-dimensional materials. Nat. Commun. 2019, 10, 4931. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Ryu, S. Optical imaging of chemically and geometrically controlled interfacial diffusion and redox in 2D van der Waals space. J. Phys. Chem. C 2021, 125, 16819–16826. [Google Scholar] [CrossRef]
- Puech, P.; Hu, T.; Sapelkin, A.; Gerber, I.; Tishkova, V.; Pavlenko, E.; Levine, B.; Flahaut, E.; Bacsa, W. Charge transfer between carbon nanotubes and sulfuric acid as determined by Raman spectroscopy. Phys. Rev. B 2012, 85, 205412. [Google Scholar] [CrossRef] [Green Version]
- Lünsdorf, N.K. Raman spectroscopy of dispersed vitrinite—Methodical aspects and correlation with reflectance. Int. J. Coal Geol. 2016, 153, 75–86. [Google Scholar] [CrossRef]
- Zaaba, N.I.; Foo, K.L.; Hashim, U.; Tan, S.J.; Liu, W.W.; Voon, C.H. Synthesis of graphene oxide using modified hummers method: Solvent influence. Procedia Eng. 2017, 184, 469–477. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Maksimova, N.V.; Kamaev, A.O.; Malakho, A.P.; Avdeev, V.V. Influence of intercalation and exfoliation conditions on macrostructure and microstructure of exfoliated graphite. Mater. Lett. 2018, 8, 403–406. [Google Scholar] [CrossRef]
- Dimiev, A.M.; Shukhina, K.; Behabtu, N.; Pasquali, M.; Tour, J.M. Stage transitions in graphite intercalation compounds: Role of the graphite structure. J. Phys. Chem. C 2019, 123, 19246–19253. [Google Scholar] [CrossRef]
- Bonaccorso, F.; Lombardo, A.; Hasan, T.; Sun, Z.; Colombo, L.; Ferrari, A.C. Production and processing of graphene and 2D crystals. Mater. Today 2012, 15, 564–589. [Google Scholar] [CrossRef]
- Freire, M.G.; Cláudio, A.F.M.; Araújo, J.M.M.; Coutinho, J.A.P.; Marrucho, I.M.; Canongia Lopes, J.N.; Rebelo, L.P.N. Aqueous biphasic systems: A boost brought about by using ionic liquids. Chem. Soc. Rev. 2012, 41, 4966–4995. [Google Scholar] [CrossRef]
- Yao, J.; Liu, C.; Liu, X.; Guo, J.; Zhang, S.; Zheng, J.; Li, S. Azobenzene-assisted exfoliation of 2D covalent organic frameworks into large-area, few-layer nanosheets for high flux and selective molecular separation membrane. J. Membr. Sci. 2020, 601, 117864. [Google Scholar] [CrossRef]
- Wu, W.; Yang, L.; Chen, S.; Shao, Y.; Jing, L.; Zhao, G.; Wei, H. Core–shell nanospherical polypyrrole/graphene oxide composites for high performance supercapacitors. RSC Adv. 2015, 5, 91645–91653. [Google Scholar] [CrossRef]
- Hou, B.; Sun, H.J.; Peng, T.J.; Zhang, X.Y.; Ren, Y.Z. Rapid preparation of expanded graphite at low temperature. New Carbon Mater. 2020, 35, 262–268. [Google Scholar] [CrossRef]
- Cooper, A.J.; Wilson, N.R.; Kinloch, I.A.; Dryfe, R.A.W. Single stage electrochemical exfoliation method for the production of few-layer graphene via intercalation of tetraalkylammonium cations. Carbon 2014, 66, 340–350. [Google Scholar] [CrossRef]
- Trusovas, R.; Ratautas, K.; Račiukaitis, G.; Niaura, G. Graphene layer formation in pinewood by nanosecond and picosecond laser irradiation. Appl. Surf. Sci. 2019, 471, 154–161. [Google Scholar] [CrossRef]
- Seiler, S.; Halbig, C.E.; Grote, F.; Rietsch, P.; Börrnert, F.; Kaiser, U.; Meyer, B.; Eigler, S. Effect of friction on oxidative graphite intercalation and high-quality graphene formation. Nat. Commun. 2018, 9, 836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, A.C.; Robertson, J. Raman spectroscopy of amorphous, nanostructured, diamond-like carbon, and nanodiamond. Philos. Trans. R. Soc. 2004, 362, 2477–2512. [Google Scholar] [CrossRef] [PubMed]
- ChacónChac, J.C.; Wirtz, L.; Pichler, T. Raman spectroscopy of graphite intercalation compounds: Charge transfer, strain, and electron-phonon coupling in graphene layers. Phys. Status Solidi B 2014, 251, 2337–2355. [Google Scholar] [CrossRef] [Green Version]
- Gurzęda, B.; Buchwald, T.; Krawczyk, P. Thermal exfoliation of electrochemically synthesized graphite intercalation compound with perrhenic acid. J. Solid State Electrochem. 2020, 24, 1363–1370. [Google Scholar] [CrossRef]
- Eigler, S. Graphite sulphate—A precursor to graphene. Chem. Commun. 2015, 51, 3162–3165. [Google Scholar] [CrossRef] [Green Version]
- Pei, S.; Wei, Q.; Huang, K.; Cheng, H.M.; Ren, W. Green synthesis of graphene oxide by seconds timescale water electrolytic oxidation. Nat. Commun. 2018, 9, 145. [Google Scholar] [CrossRef] [Green Version]
- Trusovas, R.; Račiukaitis, G.; Niaura, G.; Barkauskas, J.; Valušis, G.; Pauliukaite, R. Recent advances in laser utilization in the chemical modification of graphene oxide and its applications. Adv. Opt. Mater. 2016, 4, 37–65. [Google Scholar] [CrossRef]
- Ferrari, A.C. Raman spectroscopy of graphene and graphite: Disorder, electron-phonon coupling, doping and nonadiabatic effects. Solid State Commun. 2007, 143, 47–57. [Google Scholar] [CrossRef]
- Douda, J.; González Vargas, C.R.; Basiuk, E.V.; Díaz Cano, A.I.; Fuentes García, J.A.; Hernández Contreras, X.A. Optical properties of amine-functionalized graphene oxide. Appl. Nanosci. 2019, 9, 567–578. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, Q.; Jiang, C.; Ji, B.; Ji, X.; Tang, Y.; Cheng, H.-M. Strategies towards low-cost dual-ion batteries with high performance. Angew. Chem. Int. Ed. 2020, 59, 3802–3832. [Google Scholar] [CrossRef]
- Zou, J.; Sole, C.; Drewett, N.E.; Velický, M.; Hardwick, L.J. In situ study of li intercalation into highly crystalline graphitic flakes of varying thicknesses. J. Phys. Chem. Lett. 2016, 7, 4291–4296. [Google Scholar] [CrossRef] [Green Version]
- Salvatore, M.; Carotenuto, G.; de Nicola, S.; Camerlingo, C.; Ambrogi, V.; Carfagna, C. Synthesis and characterization of highly intercalated graphite bisulfate. Nanoscale Res. Lett. 2017, 12, 167. [Google Scholar] [CrossRef]
- Chung, D.D.L. A review of exfoliated graphite. J. Mater. Sci. 2015, 51, 554–568. [Google Scholar] [CrossRef]
- Okan, B.S.; Yürüm, A.; Gorgülü, N.; Gürsel, S.A.; Yürüm, Y. Polypyrrole coated thermally exfoliated graphite nanoplatelets and the effect of oxygen surface groups on the interaction of platinum catalysts with graphene-based nanocomposites. Ind. Eng. Chem. Res. 2011, 50, 12562–12571. [Google Scholar] [CrossRef]
- Kulandaivalu, S.; Suhaimi, N.; Sulaiman, Y. Unveiling high specific energy supercapacitor from layer-by-layer assembled polypyrrole/graphene oxide|polypyrrole/manganese oxide electrode material. Sci. Rep. 2019, 9, 4884. [Google Scholar] [CrossRef] [Green Version]
- Shams, S.S.; Zhang, R.; Zhu, J. Graphene synthesis: A Review. Mater. Sci. Pol. 2015, 33, 566–578. [Google Scholar] [CrossRef] [Green Version]
- Hardwick, L.J.; Buqa, H.; Novák, P. Graphite surface disorder detection using in situ Raman microscopy. Solid State Ion. 2006, 177, 2801–2806. [Google Scholar] [CrossRef]
- Krishnamoorthy, K.; Veerapandian, M.; Yun, K.; Kim, S.J. The chemical and structural analysis of graphene oxide with different degrees of oxidation. Carbon 2013, 53, 38–49. [Google Scholar] [CrossRef]
- Shao, Y.; Zhang, S.; Engelhard, M.H.; Li, G.; Shao, G.; Wang, Y.; Liu, J.; Aksay, I.A.; Lin, Y. Nitrogen-doped graphene and its electrochemical applications. J. Mater. Chem. 2010, 20, 7491–7496. [Google Scholar] [CrossRef]
- Lai, L.; Potts, J.R.; Zhan, D.; Wang, L.; Poh, C.K.; Tang, C.; Gong, H.; Shen, Z.; Lin, J.; Ruoff, R.S. Exploration of the active center structure of nitrogen-doped graphene-based catalysts for oxygen reduction reaction. Energy Environ. Sci. 2012, 5, 7936–7942. [Google Scholar] [CrossRef]
- Lee, Y.H.; Chang, K.H.; Hu, C.C. Differentiate the pseudocapacitance and double-layer capacitance contributions for nitrogen-doped reduced graphene oxide in acidic and alkaline electrolytes. J. Power Sources 2013, 227, 300–308. [Google Scholar] [CrossRef]
- Huang, Z.; Zhou, H.; Yang, W.; Fu, C.; Chen, L.; Kuang, Y. Three-dimensional hierarchical porous nitrogen and sulfur-codoped graphene nanosheets for oxygen reduction in both alkaline and acidic media. ChemCatChem 2017, 9, 987–996. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, M.; Yang, J.; Huang, W.; Dong, X. Graphene-based three-dimensional hierarchical sandwich-type architecture for high performance supercapacitors. RSC Adv. 2014, 4, 8466–8471. [Google Scholar] [CrossRef]
- Gaidukevic, J.; Aukstakojyte, R.; Barkauskas, J.; Niaura, G.; Murauskas, T.; Pauliukaite, R. A novel electrochemical sensor based on thermally reduced graphene oxide for the sensitive determination of dopamine. Appl. Surf. Sci. 2022, 592, 153257. [Google Scholar] [CrossRef]
- Oliveira, L.S.; Alba, J.F.G.; Silva, V.L.; Ribeiro, R.T.; Falcão, E.H.L.; Navarro, M. The effect of surface functional groups on the performance of graphite powders used as electrodes. J. Electroanal. Chem. 2018, 818, 106–113. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | 2θ (deg) | d002 (nm) | L (nm) | ID/IG |
|---|---|---|---|---|
| Gr_1 | 26.54 | 0.336 | 31.29 | 0.297 |
| Gr_2 | 26.52 | 0.336 | 33.80 | 0.104 |
| Gr_3 | 26.50 | 0.336 | 62.87 | 0.028 |
| EG_1 | 26.38 | 0.338 | 20.54 | 0.185 |
| EG_2 | 26.45 | 0.337 | 40.21 | 0.114 |
| EG_3 | 26.52 | 0.336 | 39.09 | 0.093 |
| EG_1_MW | 26.43 | 0.337 | 23.68 | 0.259 |
| EG_2_MW | 26.46 | 0.337 | 24.10 | 0.114 |
| EG_3_MW | 26.42 | 0.337 | 16.97 | 0.118 |
| EG_1_Na | 26.81 | 0.332 | 24.03 | 0.196 |
| EG_2_Na | 26.74 | 0.333 | 36.92 | 0.100 |
| EG_3_Na | 26.76 | 0.333 | 44.18 | 0.065 |
| GP_1 | 26.41 | 0.337 | 26.35 | 0.580 |
| GP_2 | 26.46 | 0.337 | 36.42 | 0.483 |
| GP_3 | 26.45 | 0.337 | 32.32 | 0.504 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rimkutė, G.; Niaura, G.; Pauliukaitė, R.; Gaidukevič, J.; Barkauskas, J. Wet Synthesis of Graphene-Polypyrrole Nanocomposites via Graphite Intercalation Compounds. Crystals 2022, 12, 1793. https://doi.org/10.3390/cryst12121793
Rimkutė G, Niaura G, Pauliukaitė R, Gaidukevič J, Barkauskas J. Wet Synthesis of Graphene-Polypyrrole Nanocomposites via Graphite Intercalation Compounds. Crystals. 2022; 12(12):1793. https://doi.org/10.3390/cryst12121793
Chicago/Turabian StyleRimkutė, Gintarė, Gediminas Niaura, Rasa Pauliukaitė, Justina Gaidukevič, and Jurgis Barkauskas. 2022. "Wet Synthesis of Graphene-Polypyrrole Nanocomposites via Graphite Intercalation Compounds" Crystals 12, no. 12: 1793. https://doi.org/10.3390/cryst12121793
APA StyleRimkutė, G., Niaura, G., Pauliukaitė, R., Gaidukevič, J., & Barkauskas, J. (2022). Wet Synthesis of Graphene-Polypyrrole Nanocomposites via Graphite Intercalation Compounds. Crystals, 12(12), 1793. https://doi.org/10.3390/cryst12121793