
Unusual Temperature Behavior of Stability of Proteinase K Dimer Formed in Crystallization Solution Defined by Molecular Dynamics

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
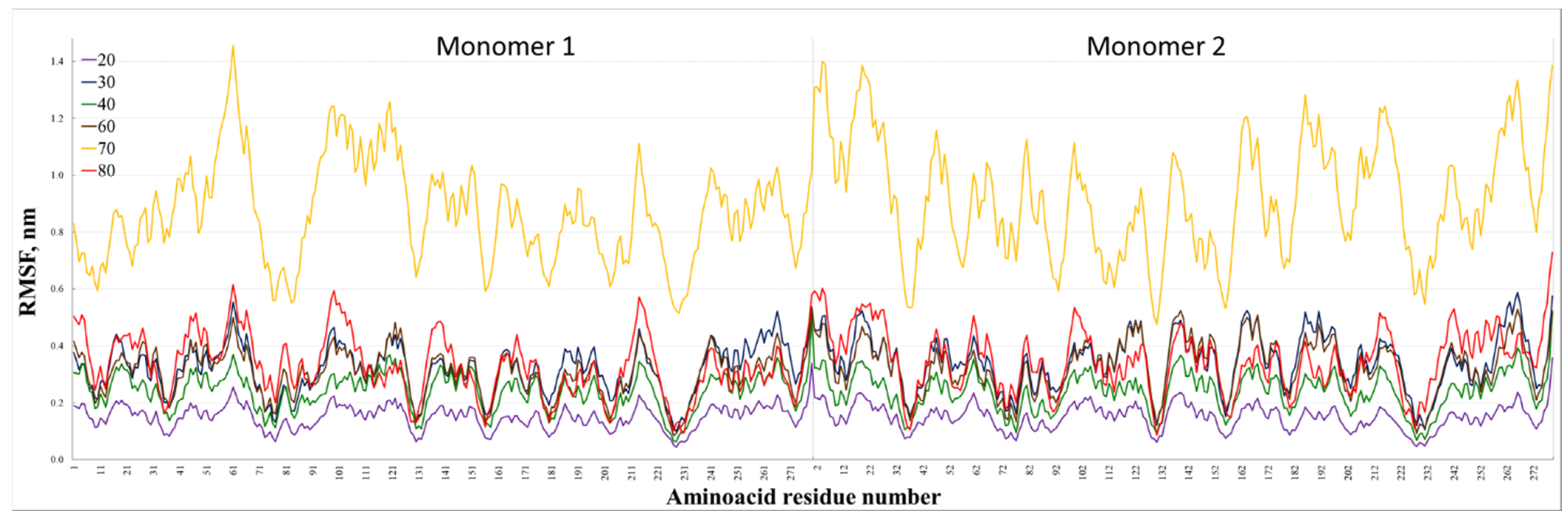
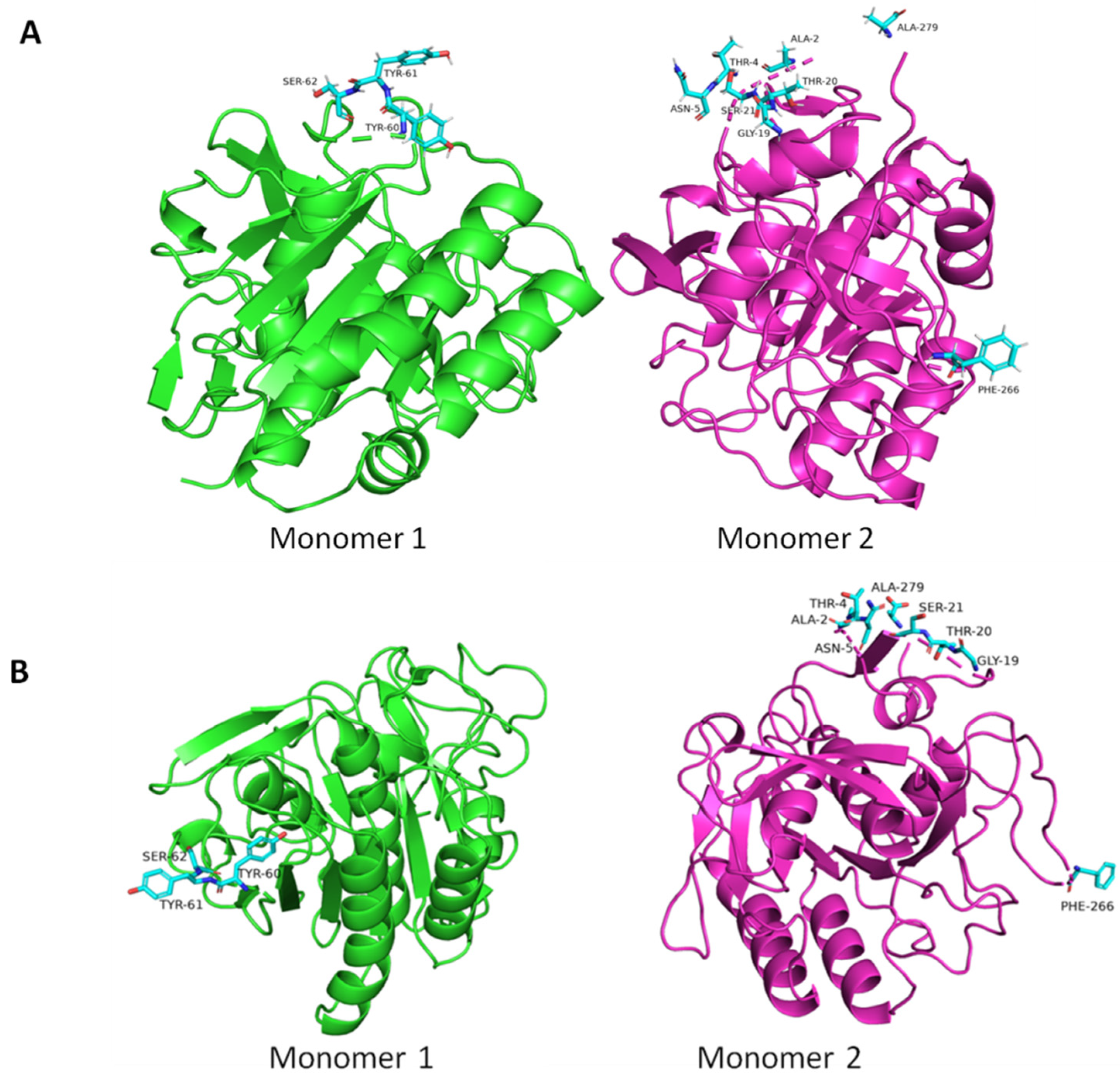
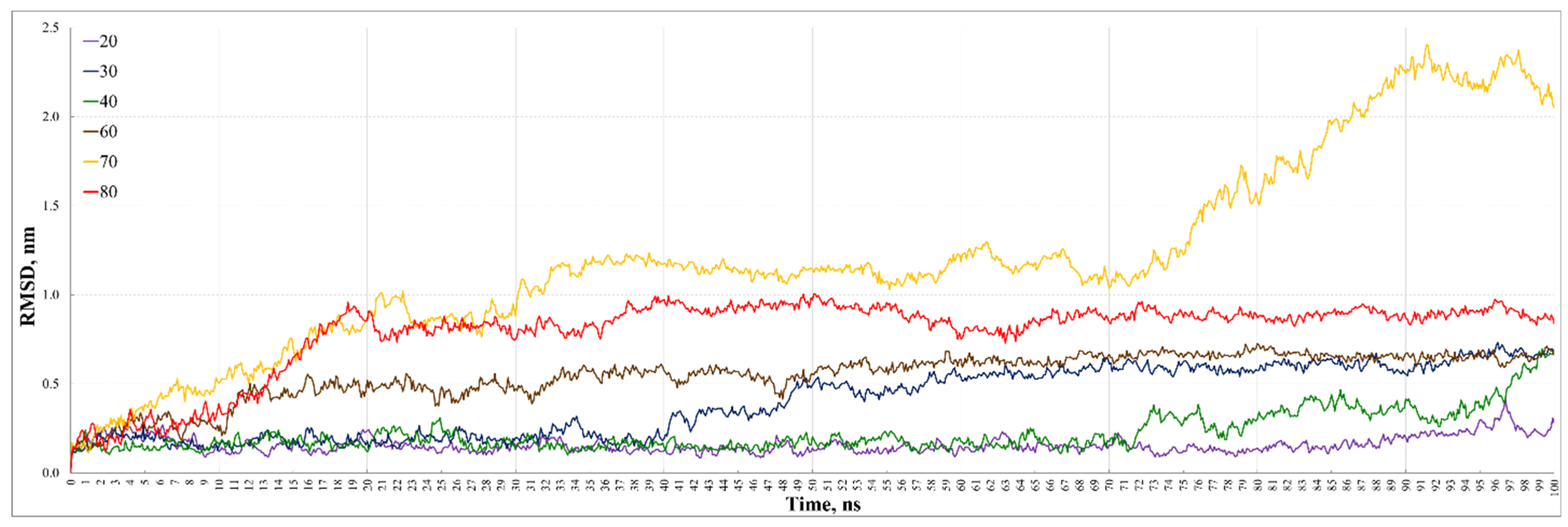
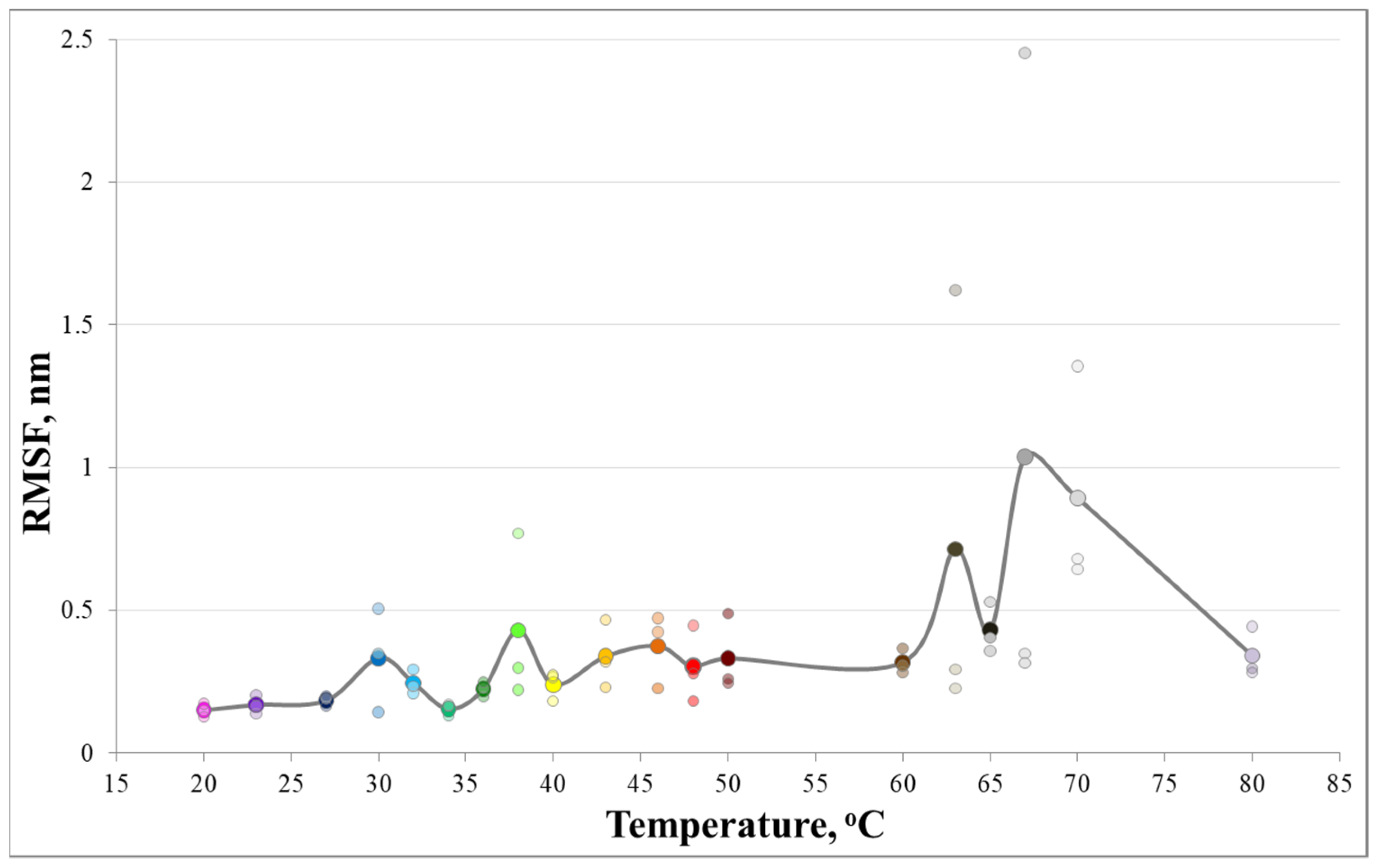
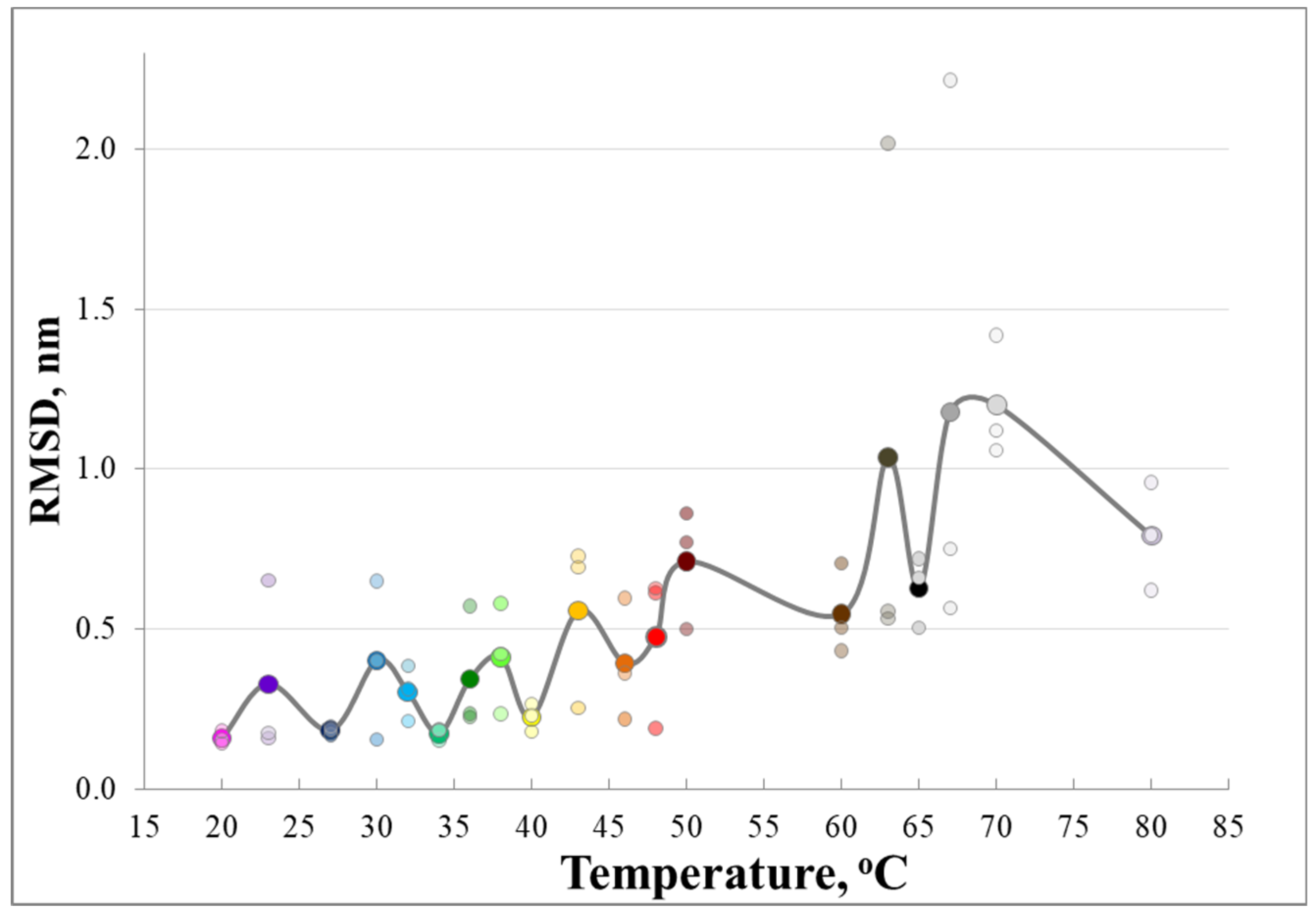
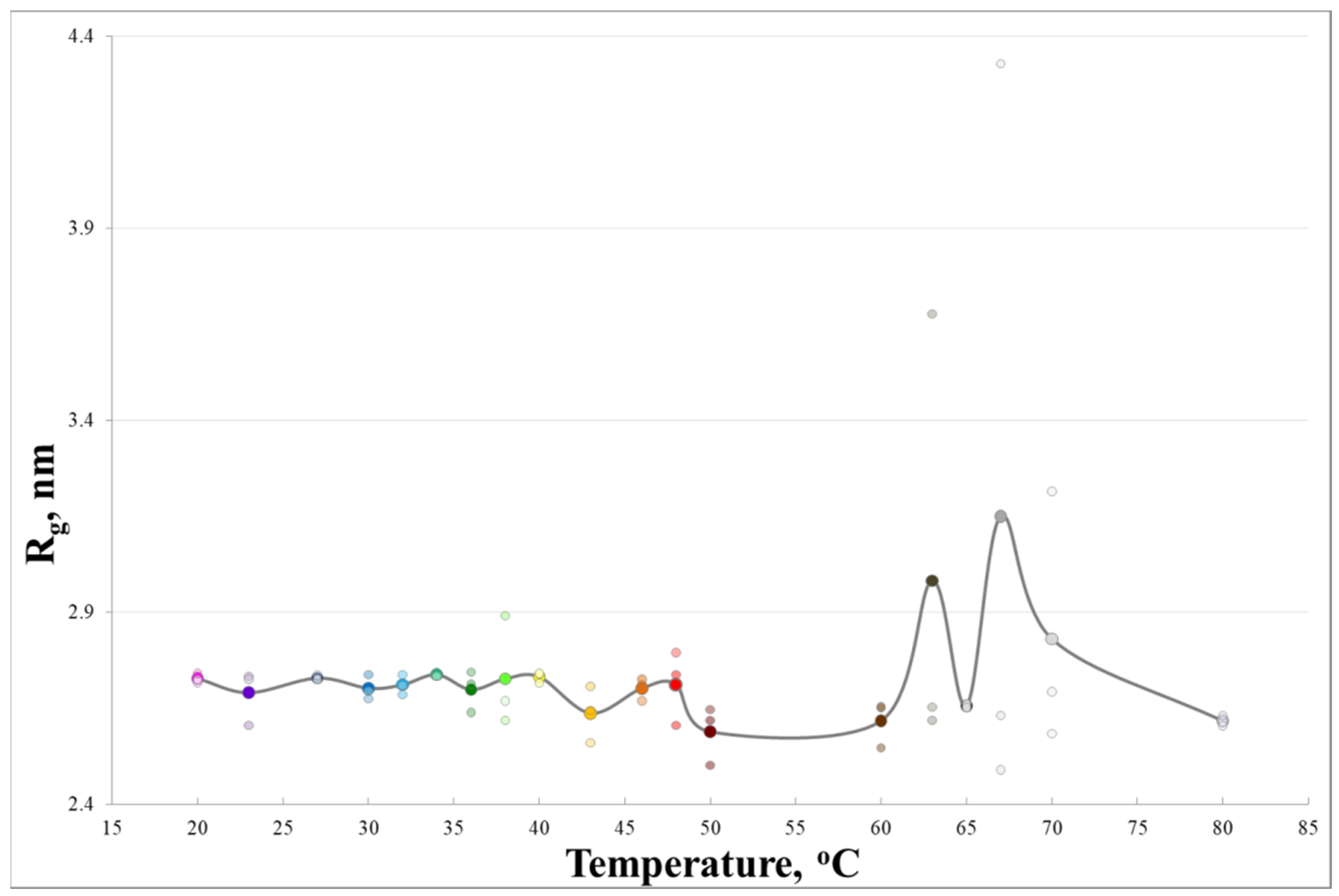
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kovalchuk, M.V.; Blagov, A.E.; Dyakova, Y.A.; Gruzinov, A.Y.; Marchenkova, M.A.; Peters, G.S.; Pisarevsky, Y.V.; Timofeev, V.I.; Volkov, V.V. Investigation of the Initial Crystallization Stage in Lysozyme Solutions by Small-Angle X-ray Scattering. Cryst. Growth Des. 2016, 16, 1792–1797. [Google Scholar] [CrossRef]
- Marchenkova, M.A.; Volkov, V.V.; Blagov, A.E.; Dyakova, Y.A.; Ilina, K.B.; Tereschenko, E.Y.; Timofeev, V.I.; Pisarevsky, Y.V.; Kovalchuk, M.V. In Situ Study of the State of Lysozyme Molecules at the Very Early Stage of the Crystallization Process by Small-Angle X-ray Scattering. Crystallogr. Rep. 2016, 61, 5–10. [Google Scholar] [CrossRef]
- Boikova, A.S.; D’yakova, Y.A.; Il’ina, K.B.; Konarev, P.V.; Kryukova, A.E.; Marchenkova, M.A.; Blagov, A.E.; Pisarevskii, Y.V.; Koval’chuk, M.V. Small-Angle X-ray Scattering Study of the Influence of Solvent Replacement (from H2O to D2O) on the Initial Crystallization Stage of Tetragonal Lysozyme. Crystallogr. Rep. 2017, 62, 837–842. [Google Scholar] [CrossRef]
- Boikova, A.S.; D’yakova, Y.A.; Il’ina, K.B.; Konarev, P.V.; Kryukova, A.E.; Marchenkova, M.A.; Pisarevskii, Y.V.; Koval’chuk, M.V. Investigation of the Pre-Crystallization Stage of Proteinase K in Solution (Influence of Temperature and Precipitant Type) by Small-Angle X-ray Scattering. Crystallogr. Rep. 2018, 63, 865–870. [Google Scholar] [CrossRef]
- Kordonskaya, Y.V.; Timofeev, V.I.; Marchenkova, M.A.; Konarev, P.V. Identification of the Precursor Cluster in the Crystallization Solution of Proteinase K Protein by Molecular Dynamics Methods. Crystals 2022, 12, 484. [Google Scholar] [CrossRef]
- Ren, Y.; Luo, H.; Huang, H.; Hakulinen, N.; Wang, Y.; Wang, Y.; Su, X.; Bai, Y.; Zhang, J.; Yao, B.; et al. Improving the Catalytic Performance of Proteinase K from Parengyodontium Album for Use in Feather Degradation. Int. J. Biol. Macromol. 2020, 154, 1586–1595. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, L.; DeLano, W. PyMOL. 2015. Available online: http://www.pymol.org/pymol (accessed on 1 November 2022).
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An Automated Pipeline for the Setup of Poisson-Boltzmann Electrostatics Calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved Side-Chain Torsion Potentials for the Amber Ff99SB Protein Force Field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Horn, H.W.; Swope, W.C.; Pitera, J.W.; Madura, J.D.; Dick, T.J.; Hura, G.L.; Head-Gordon, T. Development of an Improved Four-Site Water Model for Biomolecular Simulations: TIP4P-Ew. J. Chem. Phys. 2004, 120, 9665–9678. [Google Scholar] [CrossRef] [PubMed]
- Sousa Da Silva, A.W.; Vranken, W.F. ACPYPE-AnteChamber PYthon Parser InterfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Strain Fluctuations and Elastic Constants. J. Chem. Phys. 1982, 76, 2662–2666. [Google Scholar] [CrossRef]
- Van Gunsteren, W.F.; Berendsen, H.J.C. A Leap-Frog Algorithm for Stochastic Dynamics. Mol. Simul. 1988, 1, 173–185. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8592. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Kordonskaya, Y.V.; Marchenkova, M.A.; Timofeev, V.I.; Dyakova, Y.A.; Pisarevsky, Y.V.; Kovalchuk, M.V. Precipitant ions influence on lysozyme oligomers stability investigated by molecular dynamics simulation at different temperatures. J. Biomol. Struct. Dyn. 2021, 39, 7223–7230. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kordonskaya, Y.V.; Timofeev, V.I.; Dyakova, Y.A.; Marchenkova, M.A.; Pisarevsky, Y.V.; Silvestrova, S.Y.; Kovalchuk, M.V. Unusual Temperature Behavior of Stability of Proteinase K Dimer Formed in Crystallization Solution Defined by Molecular Dynamics. Crystals 2022, 12, 1645. https://doi.org/10.3390/cryst12111645
Kordonskaya YV, Timofeev VI, Dyakova YA, Marchenkova MA, Pisarevsky YV, Silvestrova SY, Kovalchuk MV. Unusual Temperature Behavior of Stability of Proteinase K Dimer Formed in Crystallization Solution Defined by Molecular Dynamics. Crystals. 2022; 12(11):1645. https://doi.org/10.3390/cryst12111645
Chicago/Turabian StyleKordonskaya, Yuliya V., Vladimir I. Timofeev, Yulia A. Dyakova, Margarita A. Marchenkova, Yury V. Pisarevsky, Svetlana Yu. Silvestrova, and Mikhail V. Kovalchuk. 2022. "Unusual Temperature Behavior of Stability of Proteinase K Dimer Formed in Crystallization Solution Defined by Molecular Dynamics" Crystals 12, no. 11: 1645. https://doi.org/10.3390/cryst12111645
APA StyleKordonskaya, Y. V., Timofeev, V. I., Dyakova, Y. A., Marchenkova, M. A., Pisarevsky, Y. V., Silvestrova, S. Y., & Kovalchuk, M. V. (2022). Unusual Temperature Behavior of Stability of Proteinase K Dimer Formed in Crystallization Solution Defined by Molecular Dynamics. Crystals, 12(11), 1645. https://doi.org/10.3390/cryst12111645