

Control of 11-Aza:4-X-SalA Cocrystal Polymorphs Using Heteroseeds That Switch On/Off Halogen Bonding
Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of Cocrystal Phases
2.1.1. 11.-Aza:SalA 1
2.1.2. 11.-Aza:4-Cl-SalA 2a
2.1.3. 11.-Aza:4-Cl-SalA 2b
2.1.4. 11.-Aza:4-Br-SalA 3a
2.1.5. 11.-Aza:4-Br-SalA 3b
2.1.6. 11.-Aza2:4-I-SalA 4a
2.1.7. 11.-Aza2:4-I-SalA 4b
2.1.8. X-ray Crystallography
2.2. Differential Scanning Calorimetry
3. Results
3.1. Liquid Assisted Grinding (LAG) of Cocrystal Phases
3.2. Initial Solution Growth of Single Crystals
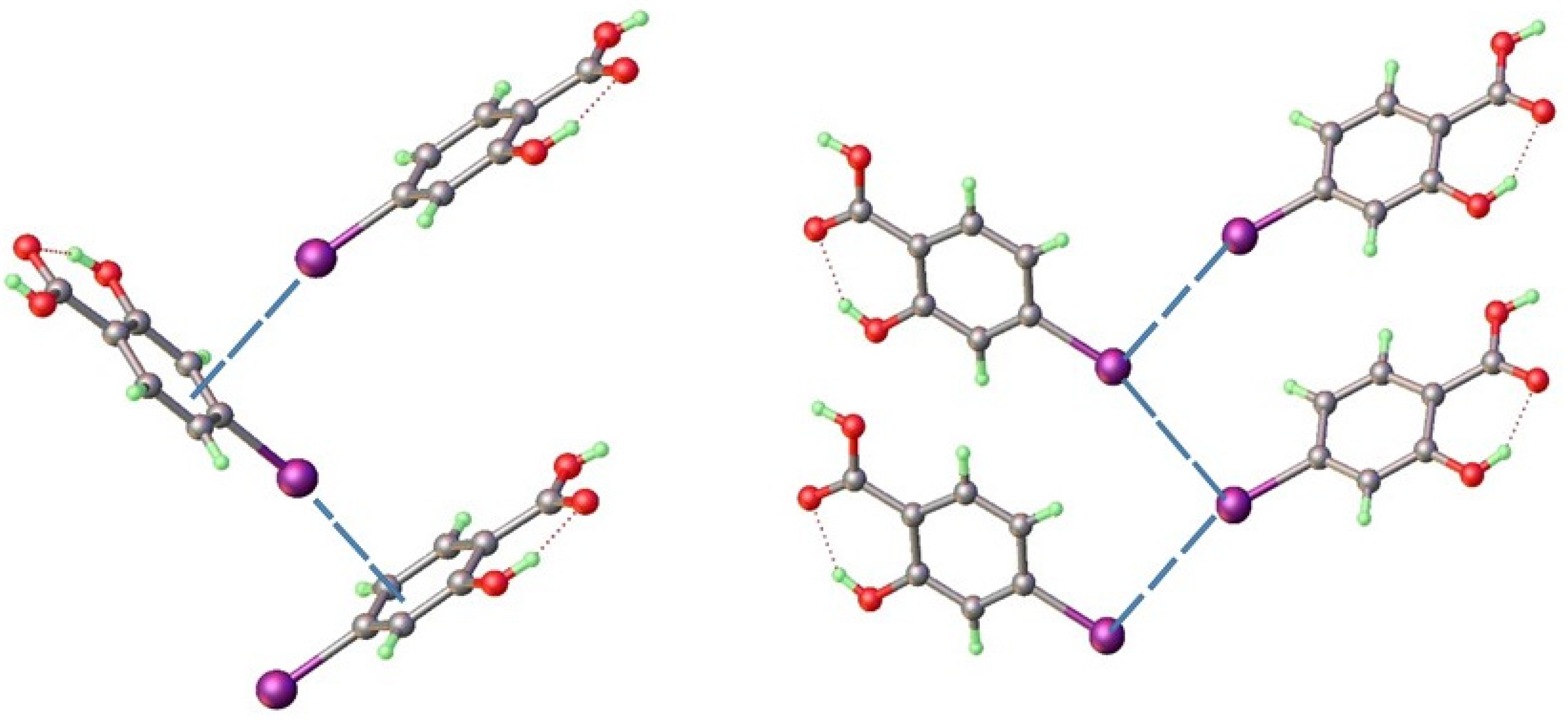
3.3. Homo- and Heteroseed Templating of New Polymorphs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torok, D.S.; Ziffer, H. Synthesis and reactions of 11-azaartemisinin and derivatives. Tetrahedron Lett. 1995, 36, 829–832. [Google Scholar] [CrossRef]
- Harmse, R.; Coertzen, D.; Wong, H.N.; Smit, F.J.; van der Watt, M.E.; Reader, J.; Nondaba, S.H.; Birkholtz, L.-M.; Haynes, R.K.; N’Da, D.D. Activities of 11-Azaartemisinin andN-Sulfonyl Derivatives against Asexual and Transmissible Malaria Parasites. ChemMedChem 2017, 12, 2086–2093. [Google Scholar] [CrossRef]
- Nisar, M.; Haynes, R.K.; Sung, H.H.-Y.; Williams, I.D. Mechanochemical conversion of 11-azaartemisinin into pharmaceutical cocrystals with improved solubility. Acta Crystallogr. Sect. A Found. Adv. 2017, 73, a268. [Google Scholar] [CrossRef]
- Nisar, M.; Sung, H.H.-Y.; Puschmann, H.; Lakerveld, R.; Haynes, R.K.; Williams, I.D. 11-Azaartemisinin cocrystals with preserved lactam: Acid heterosynthons. CrystEngComm 2018, 20, 1205–1219. [Google Scholar] [CrossRef]
- Nisar, M.; Wong, L.W.-Y.; Sung, H.H.-Y.; Haynes, R.K.; Williams, I.D. Cocrystals of the antimalarial drug 11-aza-artemisinin with three alkenoic acids of 1:1 or 2:1 stoichiometry. Acta Crystallogr. C Struct. Chem. 2018, 74, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.; Li, K.; Nisar, M.; Wong, L.W.-Y.; Sung, H.H.-Y.; Haynes, R.K.; Williams, I.D. Varying degrees of homostructurality in a series of cocrystals of antimalarial drug 11-azaartemisinin with salicylic acids. Acta Crystallogr. Sect. C Struct. Chem. 2021, 77, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Williams, I.; Li, K.; Sung, H.H.-Y.; Roy, M.; Nisar, M. Towards engineering the polymorphs of cocrystals. Acta Crystallogr. Sect. A Found. Adv. 2020, 76, a25. [Google Scholar] [CrossRef]
- Trask, A.V.; van de Streek, J.; Motherwell, W.D.S.; Jones, W. Achieving Polymorphic and Stoichiometric Diversity in Cocrystal Formation: Importance of Solid-State Grinding, Powder X-ray Structure Determination, and Seeding. Cryst. Growth Des. 2005, 5, 2233–2241. [Google Scholar] [CrossRef]
- Srirambhatla, V.K.; Guo, R.; Price, S.L.; Florence, A.J. Isomorphous template induced crystallisation: A robust method for the targeted crystallisation of computationally predicted metastable polymorphs. Chem. Commun. 2016, 52, 7384–7386. [Google Scholar] [CrossRef]
- Case, D.H.; Srirambhatla, V.K.; Guo, R.; Watson, R.E.; Price, L.S.; Polyzois, H.; Cockcroft, J.K.; Florence, A.J.; Tocher, D.A.; Price, S.L. Successful Computationally Directed Templating of Metastable Pharmaceutical Polymorphs. Cryst. Growth Des. 2018, 18, 5322–5331. [Google Scholar] [CrossRef]
- Nisar, M.; Sung, H.H.-Y.; Williams, I.D. Arylsulfonyl derivatives of 11-azaartemisinin: Approaching new polymorphs via seeds of molecular analogues. Acta Crystallogr. Sect. A Found. Adv. 2018, 74, a104. [Google Scholar] [CrossRef]
- Parambil, J.V.; Poornachary, S.K.; Heng, J.Y.Y.; Tan, R.B.H. Template-induced nucleation for controlling crystal polymorphism: From molecular mechanisms to applications in pharmaceutical processing. CrystEngComm 2019, 21, 4122–4135. [Google Scholar] [CrossRef]
- Nangia, A. Conformational Polymorphism in Organic Crystals. Acc. Chem. Res. 2008, 41, 595–604. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J.; Bernstein, J. Conformational Polymorphism. Chem. Rev. 2014, 114, 2170–2191. [Google Scholar] [CrossRef]
- Haynes, R.K.; Wong, H.-N.; Lee, K.-W.; Lung, C.-M.; Shek, L.Y.; Williams, I.D.; Croft, S.L.; Vivas, L.; Rattray, L.; Stewart, L.; et al. Preparation ofN-Sulfonyl- andN-Carbonyl-11-Azaartemisinins with Greatly Enhanced Thermal Stabilities: In vitro Antimalarial Activities. ChemMedChem 2007, 2, 1464–1479. [Google Scholar] [CrossRef]
- Trask, A.V.; Jones, W. Crystal Engineering of Organic Cocrystals by the Solid-State Grinding Approach. Top. Curr. Chem. 2005, 254, 41–70. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment-Olex2 dissected. Acta Cryst. A Found. Crystallogr. 2015, 71, 59–75. [Google Scholar] [CrossRef]
- Parsons, S.; Flack, H.D.; Wagner, T. Use of intensity quotients and differences in absolute structure refinement. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2013, 69, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Douroumis, D.; Ross, S.A.; Nokhodchi, A. Advanced methodologies for cocrystal synthesis. Adv. Drug Deliv. Rev. 2017, 117, 178–195. [Google Scholar] [CrossRef] [PubMed]
- Karki, S.; Friščić, T.; Fábián, L.; Jones, W. New solid forms of artemisinin obtained through cocrystallisation. CrystEngComm 2010, 12, 4038–4041. [Google Scholar] [CrossRef]
- Watson, D.J.; Laing, L.; Gibhard, L.; Wong, H.N.; Haynes, R.K.; Wiesner, L. Toward New Transmission-Blocking Combination Therapies: Pharmacokinetics of 10-Amino-Artemisinins and 11-Aza-Artemisinin and Comparison with Dihydroartemisinin and Artemether. Antimicrob. Agents Chemother. 2021, 65, e00990-21. [Google Scholar] [CrossRef]
- Kavanagh, O.N.; Croker, D.M.; Walker, G.M.; Zaworotko, M.J. Pharmaceutical cocrystals: From serendipity to design to application. Drug Discov. Today 2019, 24, 796–804. [Google Scholar] [CrossRef]
- Schultheiss, N.; Newman, A. Pharmaceutical Cocrystals and Their Physicochemical Properties. Cryst. Growth Des. 2009, 9, 2950–2967. [Google Scholar] [CrossRef]
- Etter, M.C.; Macdonald, J.C.; Bernstein, J. Graph-set analysis of hydrogen-bond patterns in organic crystals. Acta Crystallogr. Sect. B Struct. Sci. 1990, 46, 256–262. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Taylor, C.R.; Day, G.M. Evaluating the Energetic Driving Force for Cocrystal Formation. Cryst. Growth Des. 2018, 18, 892–904. [Google Scholar] [CrossRef]
- Steed, J.W. 21st century developments in the understanding and control of molecular solids. Chem. Commun. 2018, 54, 13175–13182. [Google Scholar] [CrossRef]
- la Vega, A.S.-D.; Duarte, L.J.; Silva, A.F.; Skelton, J.M.; Rocha-Rinza, T.; Popelier, P.L.A. Towards an atomistic understanding of polymorphism in molecular solids. Phys. Chem. Chem. Phys. 2022, 24, 11278–11294. [Google Scholar] [CrossRef]
- Thakore, S.D.; Sood, A.; Bansal, A.K. Emerging role of primary heterogeneous nucleation in pharmaceutical crystallization. Drug Dev. Res. 2020, 81, 3–22. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 11-Aza:4-X-SalA | X = H 1 | X = Cl 2a | X = Br 3a | X = I 4a |
|---|---|---|---|---|
| Reference | Ref. [4] | This work | Ref. [6] | This work |
| CCDC Number | 2194192 | 2194193 | ||
| Empirical formula | C22H29NO7 | C22H28ClNO7 | C22H28BrNO7 | C22H28INO7 |
| Formula weight | 419.47 | 453.90 | 498.36 | 545.35 |
| Temperature/K | 100.01(10) | 100.15 | 100.01(10) | 100.15 |
| Crystal system | monoclinic | monoclinic | monoclinic | monoclinic |
| Space group | P21 | P21 | P21 | P21 |
| a/Å | 9.79451 (14) | 11.0607(2) | 11.0814 (2) | 11.1497(2) |
| b/Å | 9.30687 (13) | 9.2616(2) | 9.28293 (18) | 9.3275(2) |
| c/Å | 11.51291 (16) | 11.0879(2) | 11.1404 (2) | 11.2726(2) |
| α/° | 90 | 90 | 90 | 90 |
| β/° | 92.3070 (12) | 97.941(2) | 98.638 (2) | 99.692(2) |
| γ/° | 90 | 90 | 90 | 90 |
| Volume/Å3 | 1048.62 (3) | 1124.95(4) | 1132.99 (4) | 1155.60(4) |
| Z | 2 | 2 | 2 | 2 |
| ρcalcg/cm3 | 1.340 | 1.567 | ||
| μ/mm−1 | 1.873 | 11.249 | ||
| F(000) | 480.0 | 552.0 | ||
| Crystal size/mm3 | 0.10 × 0.08 × 0.08 | 0.08 × 0.06 × 0.05 | ||
| Radiation | CuKα (λ = 1.5418) | CuKα (λ = 1.5418) | ||
| 2Θ range/° | 8.052 to 153.55 | 7.956 to 153.236 | ||
| Index ranges | −11 ≤ h ≤ 13, −11 ≤ k ≤ 11, −13 ≤ l ≤ 13 | −14 ≤ h ≤ 13, −10 ≤ k ≤ 11, −14 ≤ l ≤ 12 | ||
| Reflections collected | 8337 | 7263 | ||
| Independent reflecs. Rint, Rsigma | 4542 0.0278, 0.0381 | 3859 0.0258, 0.0345 | ||
| Data/restr./params. | 4542/1/285 | 3859/1/289 | ||
| Goodness-of-fit F2 | 1.029 | 1.033 | ||
| Final R1, wR2, I ≥ σ(I) | 0.0300, 0.0738 | 0.0229, 0.0620 | ||
| Final R [all data] | 0.0326, 0.0762 | 0.0237, 0.0629 | ||
| Res. peak/hole/e Å−3 | 0.23/−0.17 | 0.34/−0.59 | ||
| Flack parameter | −0.012(10) | −0.015(5) |
| 11-Aza:4-X-SalA | X = Cl 2b | X = Br 3b | X = I 4b |
|---|---|---|---|
| Reference | This work | This work | This work |
| CCDC Number | 2194194 | 2194677 | 2194195 |
| Empirical formula | C22H28ClNO7 | C22H28BrNO7 | C22H28INO7 |
| Formula weight | 453.90 | 498.36 | 545.35 |
| Temperature/K | 100.01(10) | 100.01(10) | 100.01(10) |
| Crystal system | orthorhombic | orthorhombic | orthorhombic |
| Space group | P212121 | P212121 | P212121 |
| a/Å | 5.59110(10) | 5.58020(10) | 5.55980(10) |
| b/Å | 15.0890(2) | 15.2471(2) | 15.7714(2) |
| c/Å | 25.5977(3) | 25.6542(3) | 25.6306(3) |
| α/° | 90 | 90 | 90 |
| β/° | 90 | 90 | 90 |
| γ/° | 90 | 90 | 90 |
| Volume/Å3 | 2159.53(5) | 2182.71(6) | 2247.44(6) |
| Z | 4 | 4 | 4 |
| ρcalcg/cm3 | 1.396 | 1.517 | 1.612 |
| μ/mm−1 | 1.952 | 2.950 | 11.568 |
| F(000) | 960.0 | 1032.0 | 1104.0 |
| Crystal size/mm3 | 0.15 × 0.15 × 0.1 | 0.15 × 0.10 × 0.08 | 0.2 × 0.06 × 0.02 |
| Radiation | Cu Kα (λ = 1.54184) | CuKα (λ = 1.54184) | CuKα (λ = 1.54184) |
| 2Θ range/° | 6.8 to 145.046 | 6.71 to 141.78 | 6.58 to 144.876 |
| Index ranges | −6 ≤ h ≤ 6, −16 ≤ k ≤ 18, −29 ≤ l ≤ 31 | −4 ≤ h ≤ 6, −17 ≤ k ≤ 18, −26 ≤ l ≤ 31 | −6 ≤ h ≤ 5, −19 ≤ k ≤ 19, −28 ≤ l ≤ 31 |
| Reflections collected | 12817 | 4709 | 12937 |
| Independent reflecs. Rint, Rsigma | 4213 0.0298, 0.0287 | 3350 0.0264, 0.0420 | 4375 0.0306, 0.0283 |
| Data/restr./params. | 4213/0/285 | 3350/0/289 | 4375/0/285 |
| Goodness-of-fit on F2 | 1.019 | 1.041 | 1.037 |
| Final R1, wR2 [I ≥ σ(I)] | 0.0288, 0.0755 | 0.0270, 0.0652 | 0.0284, 0.0664 |
| Final R [all data] | 0.0303, 0.0770 | 0.0285, 0.0663 | 0.0288, 0.0666 |
| Res. peak/hole/e Å−3 | 0.23/−0.20 | 0.36/0.19 | 0.94/−1.04 |
| Flack parameter | 0.002(7) | 0.017(15) | −0.011(3) |
| Distance(Å)/Angle(o) | 2a 2b | 3a 3b | 4a 4b |
|---|---|---|---|
| N(11)H---O(21) | 2.930 2.852 | 2.930 2.841 | 2.926 2.839 |
| O(10)---HO(20) | 2.555 2.570 | 2.551 2.564 | 2.561 2.553 |
| X---X 2 | 4.632 3.724 | 4.643 3.726 | 4.668 3.871 |
| X---pi 3 | 3.532 NA | 3.495 NA | 3.496 NA |
| C(10)-N(11)---O(21)-C(20) | 51.4 −59.8 | 50.9 −59.4 | 49.5 −55.8 |
| C(10)-O(10)---O(20)-C(20) | 40.9 −37.6 | 40.7 −37.6 | 41.6 −35.9 |
| Specific Volume (Å3) | 562.5 539.9 | 566.5 545.7 | 577.8 561.9 |
| Difference ΔV (Å3, %) | 22.5, −4.0% | 20.8, −3.7% | 14.9, −2.8% |
 | 1 Atom labels refer to the inset figure on the left and apply to both type-I and -II forms. 2 X---X represents shortest halogen-halogen contact. 3 X--pi represents halogen displacement from neighboring aromatic ring plane for type-I only. | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, K.; Roy, M.; Nisar, M.; Wong, L.W.-Y.; Sung, H.H.-Y.; Haynes, R.K.; Williams, I.D. Control of 11-Aza:4-X-SalA Cocrystal Polymorphs Using Heteroseeds That Switch On/Off Halogen Bonding. Crystals 2022, 12, 1368. https://doi.org/10.3390/cryst12101368
Li K, Roy M, Nisar M, Wong LW-Y, Sung HH-Y, Haynes RK, Williams ID. Control of 11-Aza:4-X-SalA Cocrystal Polymorphs Using Heteroseeds That Switch On/Off Halogen Bonding. Crystals. 2022; 12(10):1368. https://doi.org/10.3390/cryst12101368
Chicago/Turabian StyleLi, Keyao, Monalisa Roy, Madiha Nisar, Lawrence W-Y. Wong, Herman H-Y. Sung, Richard K. Haynes, and Ian D. Williams. 2022. "Control of 11-Aza:4-X-SalA Cocrystal Polymorphs Using Heteroseeds That Switch On/Off Halogen Bonding" Crystals 12, no. 10: 1368. https://doi.org/10.3390/cryst12101368
APA StyleLi, K., Roy, M., Nisar, M., Wong, L. W.-Y., Sung, H. H.-Y., Haynes, R. K., & Williams, I. D. (2022). Control of 11-Aza:4-X-SalA Cocrystal Polymorphs Using Heteroseeds That Switch On/Off Halogen Bonding. Crystals, 12(10), 1368. https://doi.org/10.3390/cryst12101368