
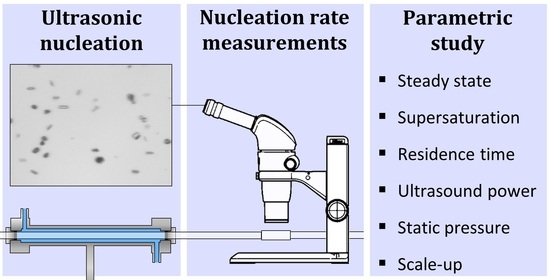
Ultrasound in Continuous Tubular Crystallizers: Parameters Affecting the Nucleation Rate


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
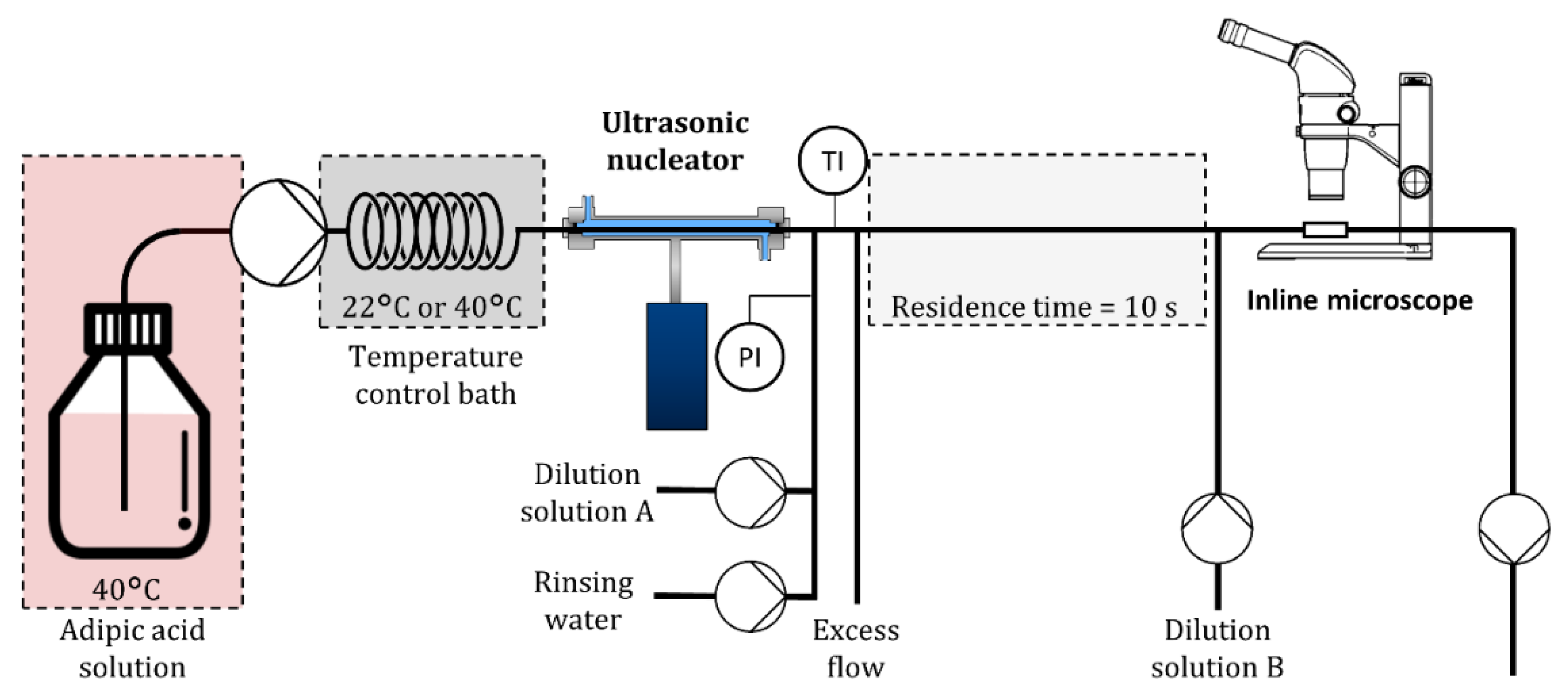
2.2. Experimental Setup
2.3. Nucleation Rate Measurements
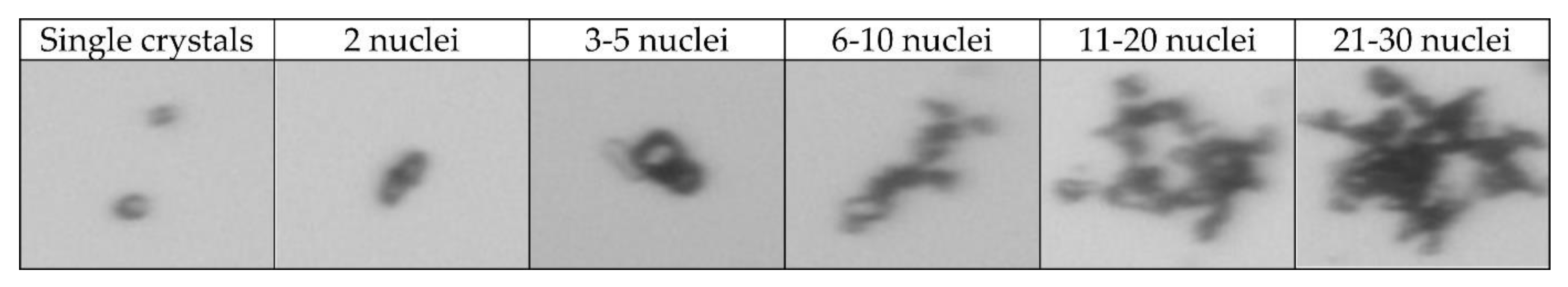
2.4. Aggregation Degree
2.5. Nucleation Rate Experiments
2.6. Calorimetric Measurements
3. Results and Discussion
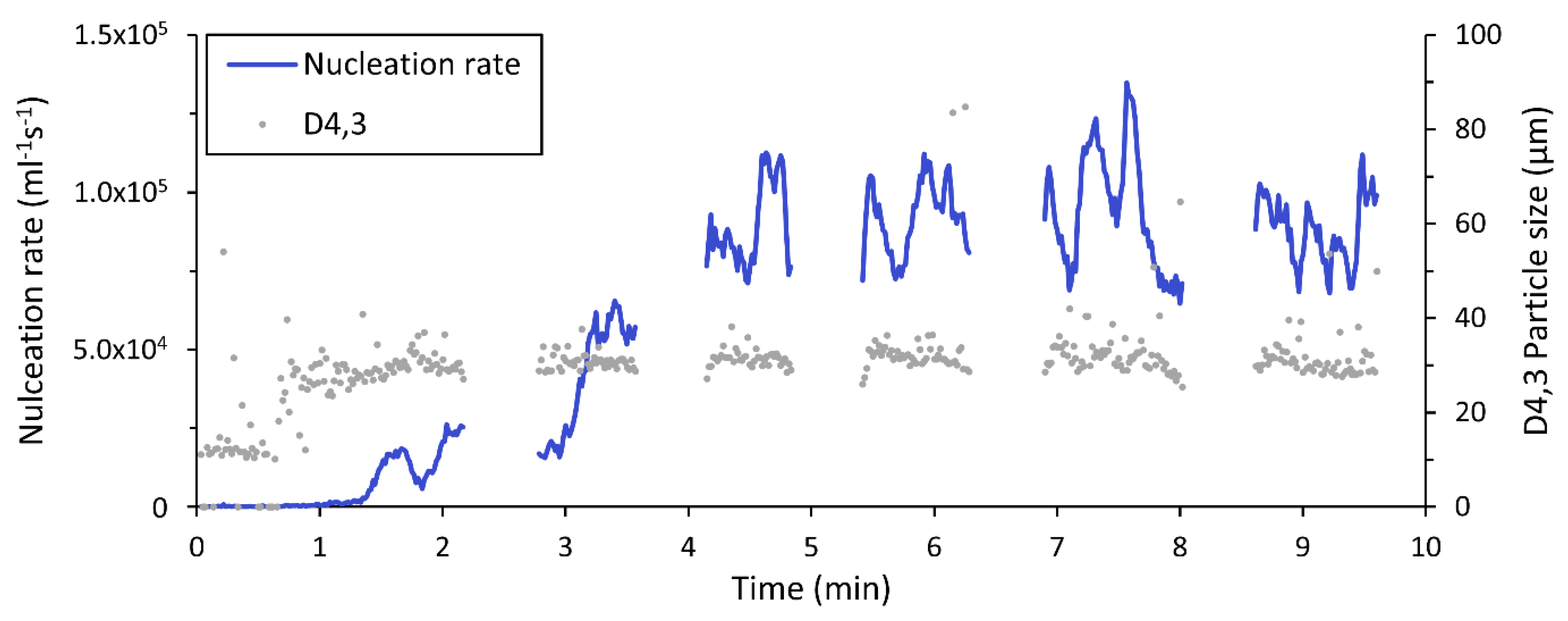
3.1. Steady State
3.2. Influence of Supersaturation
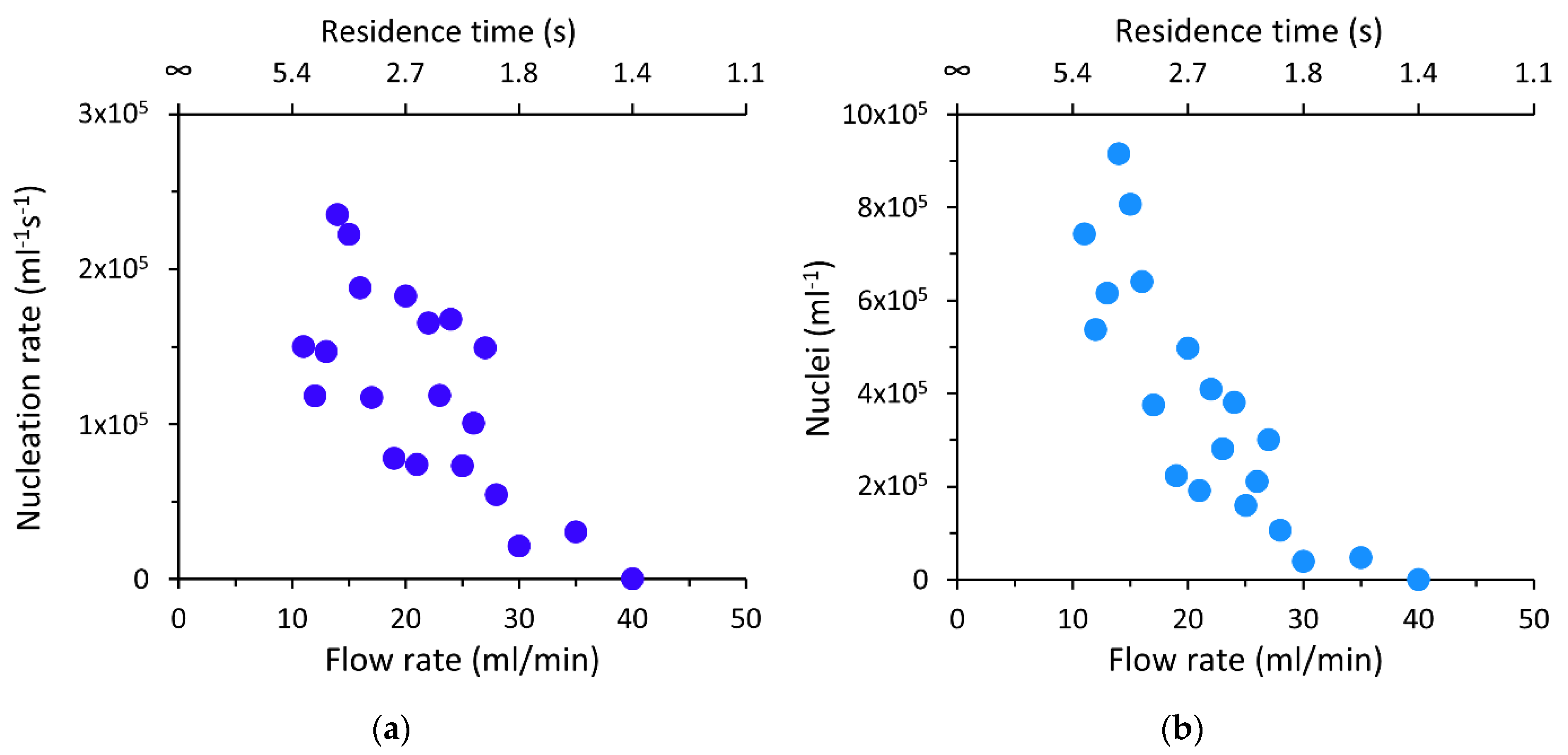
3.3. Influence of Flow Rate and Residence Time
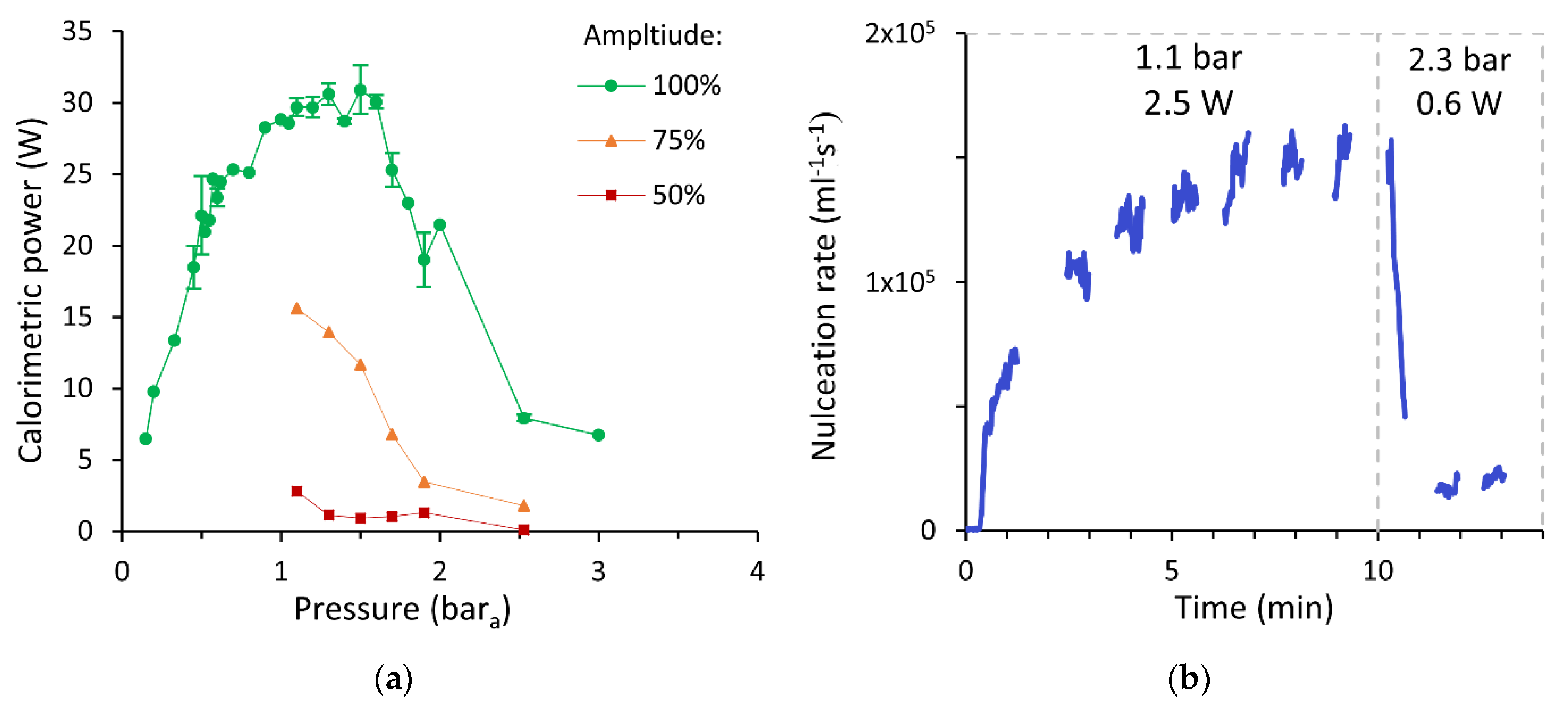
3.4. Influence of Ultrasonic Power
3.5. Scale-Up Parameter
3.6. Influence of Static Pressure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
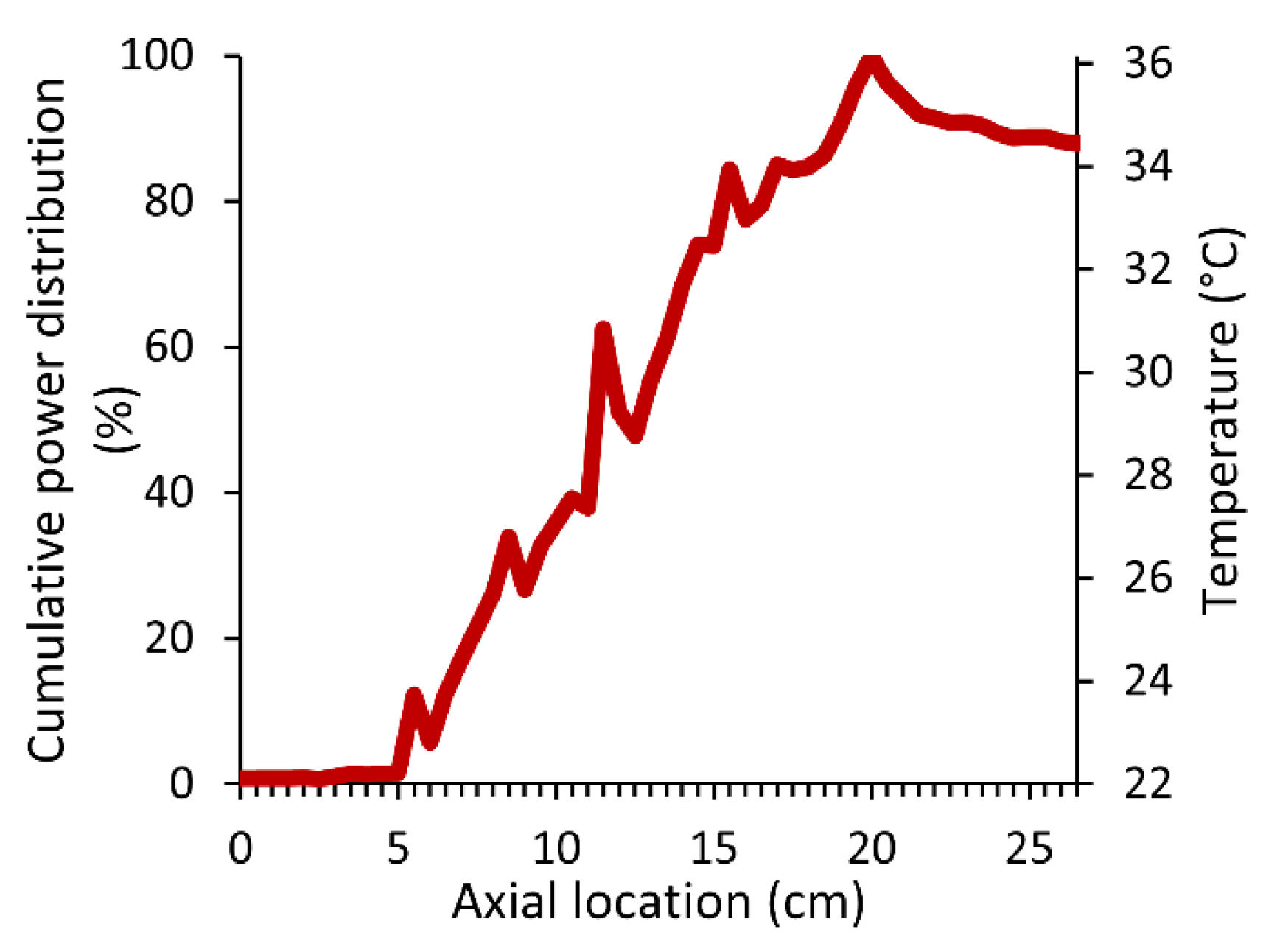
Appendix A. Characterization of the Ultrasonic Field

References
- Jiang, M.; Zhu, Z.; Jimenez, E.; Papageorgiou, C.D.; Waetzig, J.; Hardy, A.; Langston, M.; Braatz, R.D. Continuous-flow tubular crystallization in slugs spontaneously induced by hydrodynamics. Cryst. Growth Des. 2014, 14, 851–860. [Google Scholar] [CrossRef]
- Yang, Y.; Song, L.; Zhang, Y.; Nagy, Z.K. Application of wet milling-based automated direct nucleation control in continuous cooling crystallization processes. Ind. Eng. Chem. Res. 2016, 55, 4987–4996. [Google Scholar] [CrossRef]
- Shim, J.U.; Cristobal, G.; Link, D.R.; Thorsen, T.; Fraden, S. Using microfluidics to decouple nucleation and growth of protein crystals. Cryst. Growth Des. 2007, 7, 2192–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Papageorgiou, C.D.; Waetzig, J.; Hardy, A.; Langston, M.; Braatz, R.D. Indirect ultrasonication in continuous slug-flow crystallization. Cryst. Growth Des. 2015, 15, 2486–2492. [Google Scholar] [CrossRef]
- Yazdanpanah, N.; Nagy, Z.K. Handbook of Continuous Crystallization; Royal Society of Chemistry: London, UK, 2019; Volume 53, ISBN 9788578110796. [Google Scholar]
- Vancleef, A.; Seurs, S.; Jordens, J.; Van Gerven, T.; Thomassen, L.C.J.; Braeken, L. Reducing the Induction Time Using Ultrasound and High-Shear Mixing in a Continuous Crystallization Process. Crystals 2018, 8, 326. [Google Scholar] [CrossRef] [Green Version]
- Hussain, M.N.; Jordens, J.; John, J.J.; Braeken, L.; Van Gerven, T. Enhancing pharmaceutical crystallization in a flow crystallizer with ultrasound: Anti-solvent crystallization. Ultrason. Sonochem. 2019, 59, 104743. [Google Scholar] [CrossRef]
- Ezeanowi, N.; Pajari, H.; Laitinen, A.; Koiranen, T. Monitoring the Dynamics of a Continuous Sonicated Tubular Cooling Crystallizer. Cryst. Growth Des. 2020, 20, 1458–1466. [Google Scholar] [CrossRef]
- Dennehy, R.D. Particle Engineering Using Power Ultrasound. Org. Process Res. Dev. 2003, 7, 1002–1006. [Google Scholar] [CrossRef]
- Narducci, O.; Jones, A.G.; Kougoulos, E. Continuous crystallization of adipic acid with ultrasound. Chem. Eng. Sci. 2011, 66, 1069–1076. [Google Scholar] [CrossRef]
- Liu, Y.; Van Den Berg, M.H.; Alexander, A.J. Supersaturation dependence of glycine polymorphism using laser-induced nucleation, sonocrystallization and nucleation by mechanical shock. Phys. Chem. Chem. Phys. 2017, 19, 19386–19392. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Song, L.; Gao, T.; Nagy, Z.K. Integrated Upstream and Downstream Application of Wet Milling with Continuous Mixed Suspension Mixed Product Removal Crystallization. Cryst. Growth Des. 2015, 15, 5879–5885. [Google Scholar] [CrossRef]
- Rimez, B.; Debuysschère, R.; Scheid, B. On the effect of flow restrictions on the nucleation behavior of molecules in tubular flow Nucleators. J. Flow Chem. 2020, 10, 241–249. [Google Scholar] [CrossRef]
- Conte, J.; Lecomte-norrant, E.; Gourdon, C.; Rimez, B.; Debuyssche, R.; Cognet, P.; Scheid, B. Continuous-Flow Tubular Crystallization To Discriminate between Two Competing Crystal Polymorphs. 1. Cooling Crystallization. Cryst. Growth Des. 2018, 18, 6431–6439. [Google Scholar] [CrossRef]
- Jamshidi, R.; Rossi, D.; Saffari, N.; Gavriilidis, A.; Mazzei, L. Investigation of the Effect of Ultrasound Parameters on Continuous Sonocrystallization in a Millifluidic Device. Cryst. Growth Des. 2016, 16, 4607–4619. [Google Scholar] [CrossRef]
- Jordens, J.; Gielen, B.; Xiouras, C.; Hussain, M.N.; Stefanidis, G.D.; Thomassen, L.C.J.; Braeken, L.; Van Gerven, T. Sonocrystallisation: Observations, theories and guidelines. Chem. Eng. Process.-Process Intensif. 2019, 139, 130–154. [Google Scholar] [CrossRef]
- Kudo, S.; Takiyama, H. Production of fine organic crystalline particles by using milli segmented flow crystallizer. J. Chem. Eng. Jpn. 2012, 45, 305–309. [Google Scholar] [CrossRef]
- Furuta, M.; Mukai, K.; Cork, D.; Mae, K. Continuous crystallization using a sonicated tubular system for controlling particle size in an API manufacturing process. Chem. Eng. Process. Process Intensif. 2016, 102, 210–218. [Google Scholar] [CrossRef]
- Eder, R.J.P.; Schrank, S.; Besenhard, M.O.; Roblegg, E.; Gruber-Woelfler, H.; Khinast, J.G. Continuous sonocrystallization of acetylsalicylic acid (ASA): Control of crystal size. Cryst. Growth Des. 2012, 12, 4733–4738. [Google Scholar] [CrossRef]
- Jordens, J.; Canini, E.; Gielen, B.; Van Gerven, T.; Braeken, L. Ultrasound Assisted Particle Size Control by Continuous Seed Generation and Batch Growth. Crystals 2017, 7, 195. [Google Scholar] [CrossRef]
- Rossi, D. Adipic Acid Sonocrystallization in Continuous Flow Microchannels; Universtiy Colledge London: London, UK, 2017. [Google Scholar]
- Valitov, G.; Jamshidi, R.; Rossi, D.; Gavriilidis, A.; Mazzei, L. Effect of acoustic streaming on continuous flow sonocrystallization in millifluidic channels. Chem. Eng. J. 2020, 379, 122221. [Google Scholar] [CrossRef]
- Jordens, J.; Gielen, B.; Braeken, L.; Van Gerven, T. Determination of the effect of the ultrasonic frequency on the cooling crystallization of paracetamol. Chem. Eng. Process.-Process Intensif. 2014, 84, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Ashokkumar, M.; Kentish, S.E. Influence of mixing and ultrasound frequency on antisolvent crystallisation of sodium chloride. Ultrason. Sonochem. 2014, 21, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Kaur Bhangu, S.; Ashokkumar, M.; Lee, J. Ultrasound Assisted Crystallization of Paracetamol: Crystal Size Distribution and Polymorph Control. Cryst. Growth Des. 2016, 16, 1934–1941. [Google Scholar] [CrossRef]
- Pishchalnikov, Y.A.; Gutierrez, J.; Dunbar, W.W.; Philpott, R.W. Intense cavitation at extreme static pressure. Ultrasonics 2016, 65, 380–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, Y.T.; Pandit, A.B.; Moholkar, V.S. Factors Affecting Cavitation Behavior. In Cavitation Reaction Engineering; Springer: Boston, MA, USA, 1999; pp. 55–83. [Google Scholar] [CrossRef]
- Apfel, R.E. The role of impurities in cavitation-threshold determination. J. Acoust. Soc. Am. 1970, 48, 1179–1186. [Google Scholar] [CrossRef]
- Gaivoronskii, A.N.; Granzhan, V.A. Physicochemical Studies of Systems and Processes Solubility of Adipic Acid in Organic Solvents and Water. Russ. J. Appl. Chem. 2005, 78, 404–408. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Vancleef, A.; Maes, D.; Van Gerven, T.; Thomassena, L.C.J.; Braeken, L. Flow-through microscopy and image analysis for crystallization processes. Chem. Eng. Sci. 2020. accepted. [Google Scholar]
- Randolph, A.D.; Larson, M.A. Theory of Particulate Processes: Analysis and Techniques of Continuous Crystallisation; Elsevier: Amsterdam, The Netherlands, 1971. [Google Scholar]
- Gielen, B.; Jordens, J.; Thomassen, L.; Braeken, L.; Van Gerven, T. Agglomeration Control during Ultrasonic Crystallization of an Active Pharmaceutical Ingredient. Crystals 2017, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Ter Horst, J.H. Crystal nucleation rates from probability distributions of induction times. Cryst. Growth Des. 2011, 11, 256–261. [Google Scholar] [CrossRef]
- Rossi, D.; Jamshidi, R.; Saffari, N.; Kuhn, S.; Gavriilidis, A.; Mazzei, L. Continuous-Flow Sonocrystallization in Droplet-Based Microfluidics. Cryst. Growth Des. 2015, 15, 5519–5529. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Input | Classification Output | ||||||
|---|---|---|---|---|---|---|---|
| Categories | Number | Single Crystals | 2 Nuclei | 3–5 Nuclei | 6–11 Nuclei | 11–20 Nuclei | 21–30 Nuclei |
| Single crystals | 111 | 95 | 13 | 2 | 1 | 0 | 0 |
| 2 nuclei | 43 | 3 | 18 | 8 | 6 | 7 | 1 |
| 3–5 nuclei | 19 | 0 | 1 | 5 | 7 | 3 | 3 |
| 6–10 nuclei | 20 | 0 | 1 | 4 | 7 | 8 | 0 |
| 11–20 nuclei | 20 | 0 | 1 | 4 | 7 | 8 | 0 |
| 21–30 nuclei | 21 | 0 | 1 | 0 | 0 | 7 | 13 |
| Supersaturation Experiments | Residence Time Experiments | Ultrasonic Power Experiments | |
|---|---|---|---|
| Supersaturation (-) | 1.25–1.43–1.6–1.8–2.0 | 1.43 | 1.43 |
| Flow rate (mL/min) | 21 | 11 → 28 (steps of 1)– 30–35–40 | 21 |
| Ultrasound amplitude (%) | 100 | 100 | 20 → 100 (steps of 10) |
| Cooling method | In nucleator | Before nucleator | Before nucleator |
| Thermostatic bath temperature (°C) | 40 | 22 | 22 |
| Nucleator cooling system temperature (°C) | 2 | 21.6 | 21.6 |
| Nucleator outlet temperature (°C) | 22 | 22 | 22 |
| Static pressure (bara) | 1 | 1 | 1 |
| Dilution solution A ratio | 0–0.72–1.4–2.2–3.0 | 1 | 1 |
| Dilution solution B ratio | 1 | 1 | 1 |
| n (sample size) | 3–3–2–2–3 | 1 for each experiment | 1 for each experiment |
| Scale-Up Factor | 1 | 5 |
|---|---|---|
| Flow rate (mL/min) | 21 | 104 |
| Residence time (s) | 2.6 | 2.6 |
| Diameter glass tube (mm) | 2.15 | 4.8 |
| Ultrasonic Amplitude (%) | 100 | 61 |
| Calorimetric ultrasonic power (W) | 2.2 | 10.4 |
| Calorimetric power per unit of volume (W/mL) | 0.4 | 0.4 |
| Electrical power (W) | 33.4 | 35.5 |
| Scale-Up Factor | 1 | 5 |
|---|---|---|
| Flow rate (mL/min) | 21 | 104 |
| Nucleation rate (mL−1s−1) | 1.3 × 105 | 3.8 × 105 |
| Nuclei (mL−1) | 3.3 × 105 | 9.8 × 105 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vancleef, A.; Van Gerven, T.; Thomassen, L.C.J.; Braeken, L. Ultrasound in Continuous Tubular Crystallizers: Parameters Affecting the Nucleation Rate. Crystals 2021, 11, 1054. https://doi.org/10.3390/cryst11091054
Vancleef A, Van Gerven T, Thomassen LCJ, Braeken L. Ultrasound in Continuous Tubular Crystallizers: Parameters Affecting the Nucleation Rate. Crystals. 2021; 11(9):1054. https://doi.org/10.3390/cryst11091054
Chicago/Turabian StyleVancleef, Arne, Tom Van Gerven, Leen C. J. Thomassen, and Leen Braeken. 2021. "Ultrasound in Continuous Tubular Crystallizers: Parameters Affecting the Nucleation Rate" Crystals 11, no. 9: 1054. https://doi.org/10.3390/cryst11091054
APA StyleVancleef, A., Van Gerven, T., Thomassen, L. C. J., & Braeken, L. (2021). Ultrasound in Continuous Tubular Crystallizers: Parameters Affecting the Nucleation Rate. Crystals, 11(9), 1054. https://doi.org/10.3390/cryst11091054