Abstract
A crystal chemical investigation of a natural specimen of whitlockite, ideally Ca9Mg(PO4)6[PO3(OH)], from Palermo Mine (USA), was achieved by means of a combination of electron microprobe analysis (EMPA) in WDS mode, single-crystal neutron diffraction probe (NDP) and single-crystal X-ray diffraction (XRD), and Fourier transform infrared (FTIR) spectroscopy. The crystal-chemical characterization resulted in the empirical formula (Ca8.682Na0.274Sr0.045)Σ9.000(Ca0.034□0.996)Σ1.000(Mg0.533Fe2+0.342Mn2+0.062Al0.046)Σ0.983(P1.006O4)6[PO3(OH0.968F0.032)Σ1.000]. Crystal-structure refinement, in the space group R3c, converged to R1 = 7.12% using 3273 unique reflections from NDP data and to R1 = 2.43% using 2687 unique reflections from XRD data. Unit cell parameters from NDP are a = 10.357(3) Å, c = 37.095(15) Å and V = 3446(2) Å3, and from XRD, the parameters are a = 10.3685(4) Å, c = 37.1444(13) Å and V = 3458.2(3) Å3. NDP results allowed a deeper definition of the hydrogen-bond system and its relation with the structural unit [PO3(OH)]. The FTIR spectrum is very similar to that of synthetic tricalcium phosphate Ca3(PO4)2 and displays minor band shifts due to slightly different P-O bond lengths and to the presence of additional elements in the structure. A comparison between whitlockite, isotypic phases from the largest merrillite group, and its synthetic counterpart Ca3(PO4)2 is provided, based on the XRD/NDP and FTIR results.
1. Introduction
The study of calcium phosphates (CaP) is a topic of paramount interest for the scientific community, with these minerals being the natural counterparts of several biomaterials largely employed in biomedical applications such as cements, scaffolds and coatings of components of bone and teeth used in in orthopedics and dentistry [1]. These applications are mostly based on the similarity of the density of these compounds with that of human hard tissues [2]. The most abundant natural CaP belongs to the apatite series Ca10(PO4)6(F,Cl,OH)2 [3,4,5]; however, there is currently an arising interest for the mineral whitlockite, ideally Ca9Mg(PO4)6[PO3(OH)], a rare species found as a secondary phase in some granite pegmatites [6]. The interest for whitlockite, is largely due to its relation with merrillite Ca9NaMg(PO4)7 and keplerite Ca9(Ca0.5□0.5)Mg(PO4)7, two phosphate minerals found in meteorites, which represent the main reservoir of phosphate phosphorus in the Solar System [7]. Whitlockite and merrillite/keplerite are isotypic in the trigonal space group R3c.
The interest of life and material sciences in whitlockite is due to its presence in the human body, inside hyper-mineralized osteocyte lacunae in human alveolar bone [8], in arthritic cartilage and soft-tissue deposits [2], in dental calculi [9] and because it is closely related to the class of synthetic tricalcium phosphates, Ca3(PO4)2 (TCP) [10,11,12]. TPCs are biomaterial particularly wanted for their biocompatibility in human biosystem [1], and for the possibility of different cation replacements at the sites typically occupied by Ca [11], giving rise to different products, employed as commercial cement and paste for bone replacement [13,14]. In this regard, they are preferred to the more common synthetic Ca10(PO4)6(OH)2 hydroxyapatite because of their brittle nature [15,16]. Recently, additional interest has been also devoted to a series of rare-earth-doped TCP that are largely investigated as possible optical materials for biosciences application [10,11,17]. The TCP compound has two well-known polymorphs: a low temperature β-phase and a high-temperature α-phase [2]. The β-TCP phase, most often employed as a biomaterial, is isotypic with whitlockite [12].
In the present study, a whitlockite sample from the Palermo Mine, North Groton, New Hampshire (USA), has been characterized by X-ray and neutron single-crystal diffraction, electron microprobe analysis and (for the first time) infrared spectroscopy, with the aim of defining its crystal-chemical identity.
2. General Chemical and Structural Formula for the Merrillite Group of Minerals
The merrillite group of minerals includes phosphate belonging to the cerite supergroup [18], whose members are all isotypic in the trigonal space group R3c, and whose general chemical formula may be written as:
where A = Ca, Na, Sr, Ce, La and REE; X = □ (vacancy), Ca and Na; M = Mg, Fe2+, Fe3+, Al and Mn2+; T = Si and P; Ø = O and OH; W = □, OH and F. Note that the non-italicized letters A, X, M and T represent groups of cations accommodated at the [8]A(1,3) (coordination site (c.s.): 8), [6]X (c.s: 6), [6]M (c.s: 6), [4]T(1,3) (c.s: 4) crystallographic sites (identified with italicized letters). For T = Si, we have silicates phases belonging to the cerite group, outside the interest of this paper; for T = P, we have phosphate phases belonging to the merrillite group, in turn split into the merrillite subgroup (A = Ca; X = Ca, Na; M = Mg, Fe2+; Ø = O; W = □) and in the whitlockite subgroup (A = Ca, Sr; X = □, Ca; M = Mg, Mn; Ø = O, OH; W = □) [18], respectively. As the Ø-anions are part of a peculiar structural unit and W = □ for the merrillite-group minerals, the general chemical formula for these minerals may be better written as:
with the latter notation defining the general structural formula [19]:
A9 X M [TO3(Φ)]7 W3
A9 X M (TO4)6 [TO3(Φ)]
[A(1)3 A(2)3 A(3)3] X M [T(1) T(2)3 T(3)3] O24
Concerning the whitlockite group, and according to the official list of Mineral Names of the Commission on New Minerals, Nomenclature and Classification (CNMNC) of the International Mineralogical Association (IMA) [http://cnmnc.main.jp (accessed on 1 February 2021)], there are four accredited species for the merrillite subgroup, i.e., merrillite, keplerite, ferromerrillite and mathyite, and four accredited species for whitlockite subgroup, i.e., whitlockite, strontiowhitlockite, wopmayte and hedergaardite: ideal formulas for these minerals are listed in Table 1, whereas significant relations between whitlockite and the other species will be discussed further in the manuscript.

Table 1.
Whitlockite-group minerals approved by the IMA-CNMNC: ideal formula and cation site occupancy according to the structural formula [A(1)3 A(2)3 A(3)3] X M [T(1) T(2)3 T(3)3] O24.
3. Experimental
The whitlockite specimen from the Palermo Mine (USA) here examined was kindly made available by the “Museo Universitario di Scienze della Terra” (MUST), Sapienza University of Rome (Italy), catalogue number 27074/1.
Electron microprobe analysis was obtained by using a wavelength dispersive spectrometer (WDS mode) with a Cameca SX50 instrument, operating at 15 kV, sample current of 15 nA, and with a beam diameter of 1 μm. Natural and synthetic standards (element, emission line) were: fluorapatite (P Ka), baryte (S Ka, Ba La), jadeite (Na Ka), orthoclase (K Ka), vanadinite (V Ka), sylvite (Cl Ka), GaAs (As La), galena (Pb Ma), wollastonite (Si Ka, Ca Ka), celestine (Sr La), rhodonite (Mn Ka), magnetite (Fe Ka) and fluorphlogopite (F Ka).
Single-crystal X-ray diffraction (XRD) data were collected with a Bruker KAPPA APEX-II single-crystal diffractometer (Sapienza University of Rome, Earth Sciences Department) equipped with a CCD area detector (6.2 × 6.2 cm2 active detection area, 512 × 512 pixels) and a graphite crystal monochromator, using MoKα radiation from a fine focus sealed X-ray tube. The sample-to-detector distance was 4 cm. A set of 9473 reflections were integrated and corrected for Lorentz, polarization, background and absorption using the package of software Apex2 [21]. The systematic-absence analysis strongly suggested the choice of the trigonal space group R3c. The crystal structure was refined using SHELXL-2014 [22]. Final anisotropic refinement (by using 143 parameters) converged to R1 = 0.0243 for 2687 unique reflections with I > 2σ(I), and wR2 = 0.0528 for all data, confirming the original structural model of Gopal and Calvo (1972) [23]. Magnesium and Fe were unambiguously located at the M site. Sodium was preliminary assigned over the A(1), A(2), A(3) and X sites: among them, only the A(2) site scattering value suggested the presence of Na. Unit cell parameters are: a = 10.3685(4) Å, c = 37.1444(13) Å and V = 3458.2(3) Å3.
Single-crystal neutron diffraction data were collected at room temperature on a 2 × 2 × 2 mm size crystal of whitlockite on the SXD instrument [24] at the ISIS Neutron and Muon Facility in Harwell (UK), using the time-of-flight (TOF) Laue diffraction method with polychromatic radiation in the range 0.3 to 8 Å. The sample was placed in a chamber evacuated to 10−5 bar in order to avoid contamination of the diffraction pattern from the scattering of air. Data were collected in a series of five different angular orientations of the crystal with exposure time of about 4 h for each orientation. Unit cell parameters and Bragg intensities were extracted by using the line integral fitting procedure implemented in the SXD2001 software [25]. Data were corrected for Lorentz effects, but no absorption correction was applied. The starting model for the structure refinement was based on the atomic coordinates of the non-hydrogen atoms from the corresponding XRD analysis, while H was located from neutron Fourier difference maps. The structure was refined by full matrix least squares on F2 using the SHELXL software [22]. A total of 6650 wavelength-dependent reflections were integrated to give 2024 unique reflections after merging for Fourier (R(σ) = 0.1225, 2θmax = 170.1°). Final statistical factors were: R1merge= 0.0644, R1 = 0.0711, wR2 = 0.1693 with I > 2σ(I) and 156 refined parameters, goodness of fit GooF = 1.02. Hydrogen at the H(10) site was placed at the general position 18b with an occupancy value fixed to that of the O(10) site (= 0.857) because it is bonded to oxygen at O(10) as an (OH) group. Measured unit cell parameters are: a = 10.357(3) Å, c = 37.095(15) Å, V = 3446(2) Å3.
Details of the selected whitlockite crystal, data collection and refinement are given in Table 2. Fractional atom coordinates and equivalent isotropic parameters are reported in Table 3; the crystallographic information files (CIF) can be obtained free of charge from the joint service Cambridge Structure Database (CSD) [26]—Inorganic Crystal Structure Database (ICSD) [27], quoting the depository number CSD2046688 (XRD refinement), and CSD2046635 (NDP refinement), via www.ccdc.cam.ac.uk/getstructures (accessed on 1 February 2021).
Table 2.
Crystal data and structure refinement for whitlockite from Palermo Mine, USA.
Table 3.
Crystallographic site, Wyckoff position (W.p), fractional atom coordinates, equivalent atomic isotropic displacement parameters and site occupancy factors (s.o.f.) for the studied whitlockite.
The powder Fourier Transform Infrared (FTIR) spectrum of whitlockite sample was collected on a Nicolet iS50 FTIR spectrometer equipped with a DTGS detector and a KBr beam splitter; nominal resolution was 4 cm−1 and 64 scans were averaged for both sample and background. The sample was prepared as a KBr disk, mixing 1 mg of sample with 150 mg of KBr. A single crystal was oriented under a polarizing optical microscope by using a spindle stage, embedded in resin and doubly polished to the 100 μm thickness for polarized light FTIR experiments. The spectra were collected by using a Bruker Hyperion 3000 microscope attached to a Vertex V70 bench at INFN (Frascati, Rome, Italy). The system was equipped with a Globar IR source, a KBr beam splitter and an MCT detector. The polarizer was a gold wire grid; the beam diameter was set at 200 μm.
4. Results and Discussion
4.1. Microchemical Composition
Electron microprobe analysis of the present whitlockite detected the following elements (expressed as oxide wt%) in the WDS spectrum: P2O5 = 47.17(33), Al2O3 = 0.22(7), MgO = 2.03(7), CaO = 46.16(23), FeO = 2.32(10), Na2O = 0.80(23), SrO = 0.44(6), F = 0.06(7), H2Ocalc = 0.82; O = F = −0.024; Total = 100.41. Note that the H2O content and the atomic fractions were calculated by charge balance under the assumption of (OH + F) = 1.000 atoms per formula unit (apfu) and 28 anions. The resulting empirical chemical formula is as follows:
A(Ca8.682Na0.274Sr0.045)Σ9.000X(Ca0.034□0.996)Σ1.000M(Mg0.533Fe2+0.342Mn2+0.062Al0.046)Σ0.983(TP1.006O4)6[TPO3(OH0.968F0.032)Σ1.000].
4.2. X-ray and Neutron Structural Studies
The unit cell parameters from neutron and X-ray diffraction measured for the present sample are in the range of those observed in other natural samples from whitlockite and merrillite subgroups from the literature (Table 4).
Table 4.
Unit cell parameters and chemical formulas for merrillite and whitlockite subgroups [18].
For the description of the complex cationic sites within the whitlockite-type structure, we follow the above-reported notation introduced for the general structural formula as there is a self-consistence between the letters in the chemical and structural formula of the merrillite mineral group. Consequently, the following equivalences occur with the notation reported for the isotypic β-TCP phase (a = 10.4352(2) Å; c = 37.4029(5) Å; V = 3527.26 Å3) by [36]: A(1) ≡ M1, A(2) ≡ M2, A(3) ≡ M3, X ≡ M4, M ≡ M5, T(1) ≡ P1, T(2) ≡ P2, T(3) ≡ P3.
Thus, eight non-equivalent cationic sites are present in the whitlockite structure: A(1), A(2) and A(3) are located in the general Wyckoff positions 18b, whereas X and M are placed in the 6a special positions (Table 3). Two P atoms, occupying the T(2) and T(3) sites, and nine O atoms are located at general positions, whereas a third P, occupying the T(1) site, and one O atom, at the O(10) site, are in 6a special positions (Table 3). The X site is only partially occupied; in the refinement of the β-TCP structure its occupancy is usually fixed at 50% for charge balance [12,36]; this is also the case of natural whitlockite and merrillite crystals, where the X site occupancy can be variable to compensate for constituents with different formal ionic charge from Ca2+ such as Na+ [32]. In some β-TCP structures, the X site may be vacant due to the occurrence of trivalent cations, e.g., for the Ca9RE(PO4)7 compound, trivalent rare earth cations substitute for Ca2+ [10,37]. In the description of bond lengths and angles, we will discuss the NDP data only for clarity, with the XRD results being similar, as can be observed in Table 5 and Table 6, where bond distances (Å) and bond valence parameters/sum are reported (BVP/BVS, expressed in valence units = v.u. [38]), respectively, from XRD (Table 5) and NDP refinement (Table 6).
Table 5.
Bond distances (Å) and bond valence/sum (BV/BVS, in valence units = v.u.) for the studied whitlockite (from XRD data). BV/BVS values for the A(2) and M sites calculated according to the single cationic occupancies (Table 3).
Table 6.
Bond distances (Å) and bond valence/sum (BV/BVS, in valence units = v.u.) within the studied whitlockite (from NDP data). BV/BVS values for the A(2) and M sites calculated according to the single cationic occupancies (Table 3).
4.2.1. Polyhedral Coordination
The whitlockite structure consists of three independent T(PO4)3− tetrahedra; two of these, T(2)O4 and T(3)O4, have average <T(1,3)P-O> bond-distances of, respectively, 1.537(4) Å and 1.538(4) Å, in line with the data from natural and synthetic phosphate samples [4,39,40,41]. The last tetrahedron, [T(1)PO3(OH)], has P5+ bonded to the O(10)(OH) group. The [T(1)PO3(OH)]2- unit may flip 180° along the c-axis keeping fixed the position of the O(9) oxygen atoms, but splitting the P-cation position in T(1) and T(1′) about 0.76 Å apart with 85% and 15% occupancy, respectively (Table 3), as displayed in Figure 1. In detail, when T(1) is occupied, the tetrahedron points downward (-c) and the X site is vacant; it should be noted that the actually geometry of the [T(1)PO3(OH)] polyhedron is that of a trigonal pyramid (site symmetry 3) having three O(9)O atoms as the triangular base and O(10)(OH) as the apex. Hydrogen ions at the three H(10) sites complete the bond valence to O(1) via hydrogen bridges. When the T(1′) site is occupied, the tetrahedron points upward (+c) the apex now being the O(10′) site occupied by O2-; the Ca2+ at the X site coordinates three O2- ions at O(1) and three O2- ions at the O(9) site forming a distorted octahedron with very long XCa-O(9) distances (about 2.96 Å) and a vacant H(10) site. The neutron diffraction data allowed the accurate determination of the H(10)H bonded to O at the O(10) site. As O(10) is on the three-fold axis, three H(10) sites are generated by symmetry (Figure 1), with their total occupancy being constrained to that of O(10). It is worth noting that in the final Fourier difference map, no H atom attached to the O at the O(10′) could be found, indicating that the disorder of the phosphate group is not only positional along the symmetry axis, but also compositional, giving rise to the [PO3(OH)] and (PO4) stoichiometries, with the latter occurring in a minor amount (about 15%). In summary, the H(10)H atom is not only bonded to the O(10)O atom, but it is also involved in a strong hydrogen bond with O(1)O (Figure 1) according to the following scheme: O(10)O–H(10)H = 1.03(3) Å; H(10)H···O(1)O = 1.71(3) Å; O(10)O–O(1)O = 2.739(4) Å; (O(10)O–O(10)H···O(1)O) angle = 175(2)°.
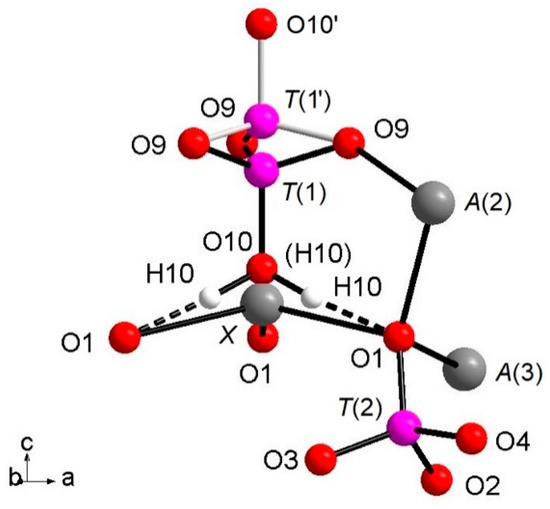
Figure 1.
View of the (T(1)O3OH) group split along c axis; details of O10-H10…O. 1 hydrogen bond and coordination of O1 are also added.
The Ca atoms occupy the A(1,2,3) and X sites, describing, with the first neighboring oxygen atoms, different polyhedra within a bonding threshold of 2.81 Å. The A(1) site displays a complex eight-fold coordination environment (Figure 2), with the shortest and largest bond-distances being A(1)-O(6) = 2.317(4) Å and A(1)-O(3) = 2.766(5) Å, respectively, and with an average distance <A(1)-O> = 2.485(5) Å. In some previous structural studies on whitlockite or synthetic TCP [36], the A(1)O8 polyhedron is described as seven-fold coordinated by excluding the longest A(1)-O(3) distance (see, e.g., [12]). However, the A(1)Ca-O(3)O bond actually exists from a bond valence perspective. Table 6 shows that the bond-valence value for A(1)Ca2+-O(3)O2− = 0.12 valence units (v.u.), is larger than 0.04× the cation valence [38]. As a result, it is reasonable to consider the A(1)Ca-O(3)O bond-distance in the coordination sphere of A(1).
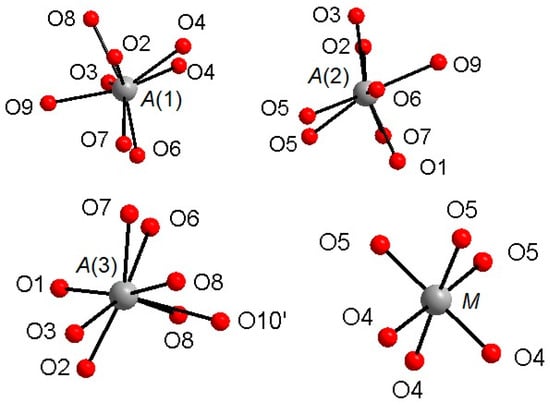
Figure 2.
Coordination environments around the A(1)–A(3) and M cationic sites.
The A(2) and A(3) sites show irregular bonding environments with coordination number = 8 (Figure 2). Regarding the A(2)O8 polyhedron, its shortest and longest bond-distances are, respectively, A(2)-O(2) = 2.373(4) Å and A(2)-O(7) = 2.651(5) Å), with <A(2)-O> = 2.472(5) Å. Considering A(3)O8, its shortest distance is A(3)-O(7) = 2.376(4) Å, whereas its largest distance is A(3)-O(6) = 2.657(5) Å and A(3)-O(10)′ = 2.717(9) Å; <A(3)-O> = 2.512(5) Å (Table 4). Worth of note is the presence of Na at A(2), to date refined for the first time within whitlockite sub-group phases (Table 4). Sodium is a characteristic constituent of merrillite sub-group species, and in the closely related wopmayte Ca6Na3□Mn2+(PO4)3(PO3OH)4 [31], it is up to 10 times more abundant than in the studied whitlockite. In wopmayite, Na is strongly ordered at A(3) [31], with as a result longer mean <A(3)-O> bond lengths. Our whitlockite also shows a longer <A(3)-O> and BVS values slightly lower than 2.00 consistent with the possible presence of a monovalent cation. However, refinement of Na at both A(3), A(1) and X gave negative site occupancy values, thus indicating the occurrence of Na at A(2) only.
Of particular interest for most of the whitlockite structures is the coordination environment of the cation at the X site (Figure 3). This cation is at 6a special position, being involved in a three-fold coordinated regular polyhedron with three equivalent X-O(1) bond distances at about 2.58(4) Å. Due to the disorder of the [T(1)PO3(OH)] unit described above, it is very difficult to characterize the occupancy of the X site, which is located only ~0.6 Å from O(10). Considering that the occupancy of the X site is constrained to compensate the charge balance, X-ray/neutron structure refinements were done at first with only the A(1), A(2) and A(3) sites occupied by Ca. The Fourier difference maps in both X-ray and neutron refinements showed a clear peak at the X-site position. In particular, in the X-rays map, this peak is at 0.65 Å from O(10) with electron density 1.53 e/Å3, while, in the neutrons map, it is at 0.57 Å from O(10) with a density of 1.64 fm/Å3. Adding this peak as X site occupied by Ca to the refinement, a strong correlation between the s.o.f. and the Uiso of X occurred. This correlation was resolved by fixing Uiso of X to 0.01 Å2 (i.e., to the average value refined for the other Ca atoms in the structure) in X-ray/neutron refinements. The resulting s.o.f. is virtually the same in both refinements, 0.058(4) from X-rays and 0.06(2) from neutron, whereas the new refined X position resulted very close to O(10), X-O(10) = 0.51 Å and 0.57 Å in neutron and X-ray refinement, respectively. Its coordination polyhedron XO6 (Figure 3) comprises the three equivalents O(1) at 2.57–2.58 Å (NDP and XRD, respectively), and three equivalents O(9), belonging to the [T(1)PO3(OH)] moiety, at 2.92–2.96 Å. In terms of bond valence, the contribution of XCa2+ can be considered as weak: about 0.20 v.u. and 0.08 v.u. for X-O(1) and X-O(9), respectively (Table 6). The six-fold coordination of the X site is confirmed by the keplerite mineral [7], though this site is usually described as triangular in isotypic β-TCP compounds in that the three X-O(9) interactions tend to disappear being found over 2.96 Å [12].
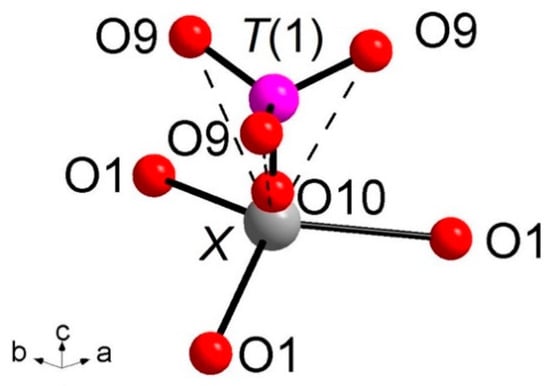
Figure 3.
Coordination environments around the X site. X-O(10) = 0.51 Å (NDP), 0.56 Å (XRD). Dashed bond: X-O(9) weak interactions over 2.90 Å. The H(10) atoms and the T(1′)O3(OH) split moiety are omitted for clarity.
The M site is octahedrally coordinated (Figure 2), with two groups of three equivalent bond distances, M-O(4) = 2.075(4) Å and M-O(5) = 2.096(3) Å, with the average <M-O> distance = 2.086 Å. This value is fully consistent with the occurrence of Mg and Fe as also observed in other whitlockite refinements [28,29,30]. Note that the M site is also characterized by low values of isotropic thermal parameters (Table 3). The M site population provided by EMP, (Mg0.533Fe2+0.342Mn2+0.062Al0.046), is fully consistent with X-ray crystal-structure refinement: occupancy 56% of Mg (+ Al, which has X-ray scattering very similar to Mg) and 46% of Fe (+Mn, which has X-ray scattering very similar to Fe). This excellent agreement reflects the fact that same crystal used for XRD was analyzed with EMP. As for the neutron refinement, the M site occupancy, 80% of Mg and 20% of Fe, shows a slight deviation from what is provided by XRD. This deviation might be ascribed to a possible chemical inhomogeneity in the much bigger volume (about 8 mm3) of the specimen used for the neutron diffraction.
Bond valence sum (BVS) calculations are in close agreement with the expected values from the empirical formula: 2.07 (v.u.) at A(1), 2.06 at A(2), 1.94 at A(3), 4.98 at T(2) and 4.97 at T(3). Of particular interest are the T(1), T(1′), X and M sites. With regard to T(1), we have to consider the previously mentioned split configuration of the [T(1)PO3(OH)]2- structural unit, which provides a separate contribution to the BVS = 5.07 x 85% + 5.43 × 15% = 5.12 v.u. The BVS incidents at the X site = 0.81 v.u., smaller than what was expected for Ca2+, indicates the occurrence of bond strain and reflects the presence of the split configuration for O(10). Finally, the value of BVS at the M site (2.16 v.u.) is consistent with occurrence of dominant divalent cations (Mg and Fe) and minor trivalent cations (Al3+) at this site, as reported in the empirical chemical formula of the studied whitlockite.
4.2.2. Three-dimensional Framework
The three-dimensional arrangement of whitlockite can be described by the repeat of two columnar arrays, indicated as ‘I’ and ‘II’ in Figure 4, running along the c axis. The I array consists of T(1)O4–MO6 sequence of polyhedra, while the II array is formed by T(3)O4–A(1)O8–A(3)O8–A(2)O8–T(2)O4–T(3)O4 sequence. In the I array, the MO6 polyhedron and the P5+ at T(1) and T(1′) do not share any atom; the MO6 octahedron is engaged, via corner sharing with T(2)O4 and T(3)O4 tetrahedra, in the [M(TO4)16] pinwheel unit [32] (see inset (a) in Figure 4), which can be considered the building block of the whitlockite framework, as later illustrated. All cations in the I array, i.e., those at the X, M and T(1)/T(1′), are in special position (x = y = 0, Table 3), and hence, they are constrained along the c axis. This constraint prevents any positional disorder except that along the z direction, as previously discussed for T(1) and O(10).
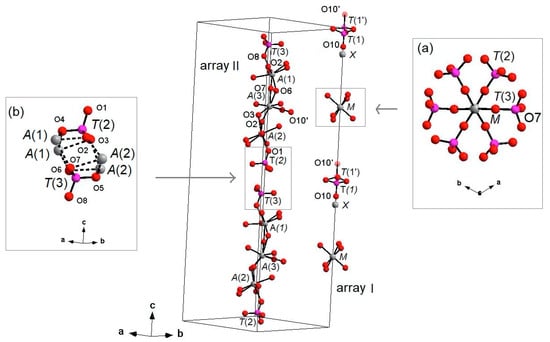
Figure 4.
‘I’ and ‘II’ arrays in whitlockite. Inset (a): [M(PO4)6] pinwheel units [32]. Inset (b): detail of II-II arrays joining (M-O bonds dashed). H atoms omitted for clarity. X-O(1) bonds omitted for clarity.
On the other hand, in the II array, all the cations at A(1), A(2), A(3), T(2) and T(3) are located in general positions (Table 3) and may, thus, cause disorder to give a denser arrangement. Two II arrays are joined together, thus making up a column, via A(1)-O-A(2) bonds, with A(1) and A(2) belonging to adjacent arrays (see the inset (b) in Figure 4).
Each I array is surrounded by six II arrays, whereas each II array is surrounded by two I and four II arrays (Figure 5). The [M(TO4)6] pinwheel units assure the link between one I column and the six adjacent II columns, being the pinwheels flat in the c axis and held together by intralayer A(1)Ca cations, forming layers perpendicular to c axis (Figure 6). These anionic units form a perfectly closest-packed planar arrangement [32]: these layers are linked by an interlayer of cations at A(2) and A(3) from II columns, making up the whitlockite three dimensional framework, further compacted by the interactions formed by the split T(1)O4 tetrahedron, which share O(9) with A(2)O8 and A(1)O8 from different II arrays, and the O(10′) vertex with the A(3)O8 polyhedron. This second joining mechanism among I and II arrays, formed by split T(1)O4 tetrahedron, is conditioned by the site symmetry of T(1) (Table 3). In fact, as previously described, the T(1)O4 tetrahedron forms a triangular pyramid, whose three O(9)O base-vertices are shared at the same time both with one A(1)O8 polyhedron from an adjacent II array and with a A(2)O8 polyhedron belonging to another II array. Owing to the site symmetry, the joining mechanism is repeated three times, while the split along c of the tetrahedral unit causes the presence of O(10′) position, joined to three A(3)O8 polyhedra from adjacent II arrays. The framework is completed by the X-O bonds, considered as weak interactions owing to the low cation occupancy of the site, X(Ca0.034□0.996)Σ1.000.
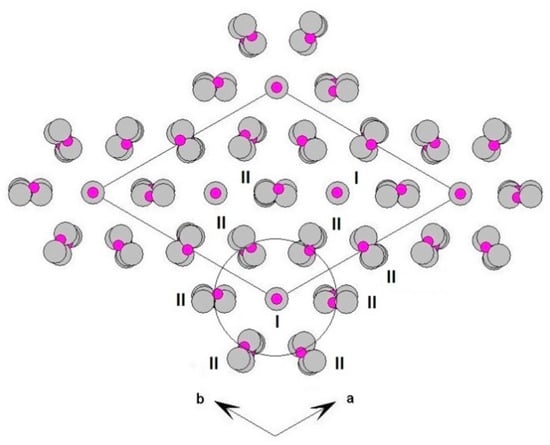
Figure 5.
Three-dimensional framework of whitlockite. Detail of the I and II columnar arrays, viewed down c.
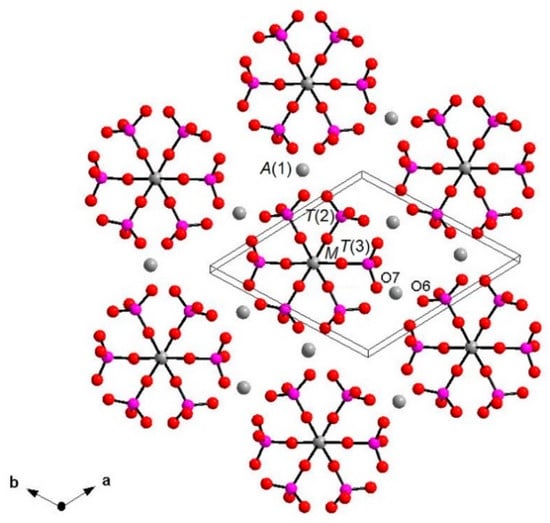
Figure 6.
Layer perpendicular to c showing [M(PO4)6] pinwheel units [32]. A(2), A(3) and X sites omitted for clarity. A(1)-O, A(2)-O, A(3)-O and X-O bonds omitted for clarity.
The whitlockite arrangement, as previously stated, is strictly related to that of isotypic β-TCP [32]. The occurrence of a trivalent RE-cation such as Cr3+ [42], Fe3+ [43] or more frequently REE3+ [10] in the β-TCP structure leads to the formula Ca9RE3+(PO4)7, characterized, compared to whitlockite, by a fully vacant X-site and by the presence of the [T(1)PO3(O)] unit according to the deprotonation-substitution mechanism RE3+ + O2− → R2+ + (OH)− [11]. In the case of Cr3+ and Fe3+, the mechanism of dopant distribution occurs exclusively at the M site [42,43]. In the case of Ca9RE(PO4)7 β-TCP, the rare earth distribution was discussed in [11] on the basis of the RE ionic radii. In detail, for ‘low’ RE (La up to Gd), the dopant element is disordered over the A(1,2,3) sites, whereas the M site is fully occupied by Ca. As for the ‘heavy’ RE (from Tb to Lu) the dopant is disordered over A(1), A(2) and M site, with A(3) being fully occupied by Ca [11].
4.3. Relations within Whitlockite Group Minerals
Phosphate phases from whitlockite subgroup are generally rare terrestrial minerals, usually found as secondary phases in paragenesis with other minerals. Wopmayite, ideally Ca6Na3□Mn(PO4)3(PO3OH)4, is a secondary mineral from the Tanco mine, Bernic Lake, Manitoba (Canada); it crystallizes together with rhodochrosite, quartz, whitlockite, apatite, and other phases after dissolution of primary lithiophosphate by hydrothermal solutions. Wopmayite has a structural unit consisting of an [M2+(PO4)6] arrangement that is topologically the same as the structural units in the whitlockite and merrillite structures; it is related to whitlockite by the substitution Na + H → Ca + □, whereby Na is incorporated primarily at the A(3) site and H attaches to the T(1)O4 tetrahedron to produce an acid-phosphate group. Thus, merrillite contains no acid-phosphate group, whitlockite contains a single acid-phosphate group at T(3), and wopmayite contains acid-phosphate groups at both T(1) and T(3) [31].
Minor details are available for strontiowhitlockite, ideally Sr9Mg(PO3OH)(PO4)6, the Sr-dominant analog of whitlockite, found in Kovdor deposit, Kola Pensinsula, Russia. The mineral, together to other phosphates such as carbonate fluoroapatite, occurs in a vein of dolomite carbonatite cutting host pyroxenites. At the time of its discovery, it was studied only with powder XRD technique, allowing no structure solution; its powder pattern was really close to that of whitlockite and of synthetic β-TCP [30]. Few details are available for hedegaardite, ideally (Ca,Na)9(Ca,Na)Mg(PO3OH)(PO4)6, in the CMMNC Newsletter [20]. Lastly, the fluorine dominant whitlockite, ideally Ca9Mg(PO4)6(PO3F), was approved in 2008 as a new mineral, with the name “bobdownsite” [44], but was later discredited being F-free and then indistinguishable from whitlockite [45].
The de-hydrogenated extra-terrestrial phases belong to the merrillite subgroup and are merrillite, ideally Ca9NaMg(PO4)7, and keplerite, ideally Ca9(Ca0.5□0.5)Mg(PO4)7 (Table 1). Merrillite is typical of genetic environments of high temperature and pressure, having been found in Moon rocks and meteorites [29]: in this phase, Na+ is refined at the X site, whereas Mg and Fe2+ are refined at M, analogously to whitlockite [33]. Keplerite is a recent new mineral, typical of high-temperature environments characterized by extreme depletion of Na, such as pallasite and angrite meteorites and, on Earth, as pyrometamorphic rocks. It was recently interpreted as the Ca-dominant counterpart of merrillite, and both minerals are related by the heterovalent substitution at the X-site of the crystal structure: 2Na+ (merrillite) ↔ Ca2+ + □ (keplerite) [7].
Ferromerrillite, ideally Ca9NaFe2+(PO4)7, occurs as a common accessory phase in several Martian meteorites, classified as basaltic and olivine-phyric shergottites. Besides the Fe2+ presence at the M site, the samples were described as heavily deformed for the shock events during the entry in the atmosphere [34]. Minor details are available for mathyite, ideally Ca9(Ca0.5□0.5)Fe2+(PO4)7, characterized by the absence of Na at the A(1,2,3) sites, and reported as the product of metasomatic process in the D’Orbigny angrite meteorite [35].
4.4. FTIR Spectroscopy
The powder FTIR spectrum of whitlockite in the 4000–400 cm−1 wavenumber region is compared in Figure 7 with that of pure β-TCP [11]. Both show a complex pattern in the P-O antisymmetric (1150–1000 cm−1) and symmetric (1000–900 cm−1) stretching regions, where bands assigned to the vibrational modes of distorted (PO4) tetrahedra are observed [10,11].
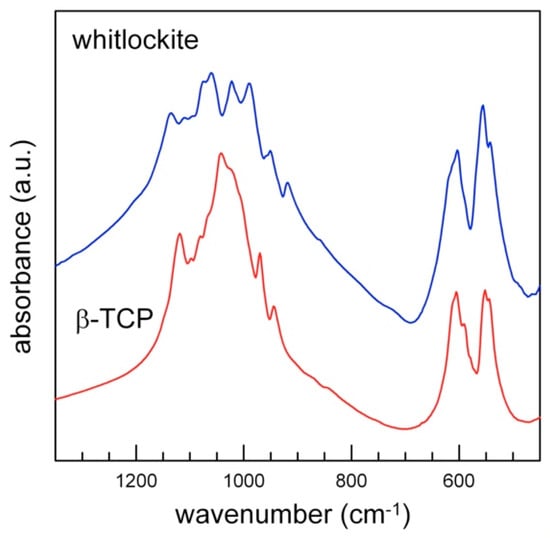
Figure 7.
Powder FTIR spectra of whitlockite (blue) and TCP (red, sample from [11]): detail of the tetrahedral modes region (1600–400 cm−1).
Despite the significant interest for synthetic whitlockite that has emerged during the last years in biomaterial sciences [46], a complete FTIR study of natural whitlockite is missing in the literature. We can, thus, use for comparison the data known for the spectrum of the isotypical β-TCP, which has been widely investigated in terms of site-symmetry and factor group analysis [47,48], see also [39,40,49]. According to [47,48,50], the free phosphate ion (PO4)3− with ideal Td point symmetry displays four vibrational modes: (1) the antisymmetric stretching ν3 at 1082 cm−1; (2) the symmetric stretching ν1 at 980 cm−1; (3) the antisymmetric bending ν4 at 567 cm−1; (4) the symmetric bending ν2 at 420 cm−1. In accordance with the selection rules, the triply degenerate antisymmetric stretching and bending modes (F2) are both Raman and infrared active, whereas the non-degenerate symmetric stretching (A1) and the doubly degenerate symmetric bending (E) are Raman active only. When the symmetry of the (PO4)3− ion is lowered from Td, band splitting, shifts of the absorption/scattering bands and appearance of non-active IR/Raman modes are observed [50].
The FTIR spectrum of whitlockite in the phosphate modes region (<1200 cm−1, Figure 7) is similar to that of synthetic β-TCP, and shows the same band multiplicity, in agreement with their very similar phosphate framework; the only differences are observed in the wavenumber shift of some peaks, due to the slightly different P-O bond lengths (Figure 7, Table 7), and to the presence of additional minor constituents. The prominent features in the ν4 antisymmetric bending region (500–700 cm−1) are also almost identical, in terms of both band multiplicity and position.
Table 7.
Measured absorption in the FTIR spectrum and relative assignments for present whitlockite in comparison with data of β-TCP [11].
The polarized spectra collected on the single-crystal (hk0) section are displayed in Figure 8. The section was prepared by orienting the crystal under the polarizing microscope using a spindle stage [51], which was then transferred onto the grain on a glass slide for cutting and the section was polished twice. Although the thickness was reduced (100 μm), the absorbance was below 1300 cm−1 due to the tetrahedral P-O modes, and is evidently out of scale; in the OH-stretching region, two broad bands are observed, centered at 2905 and 2415 cm−1, respectively (Figure 8). Cooper et al. (2013) [31] presented a very similar pattern for the closely related wopmayite and assigned the former component to the (P)-OH stretching mode, while the latter, following the work of Moraes et al. (2006) [52] was assigned to an overtone of the P-O modes. This assignment is supported, although not conclusively, by the polarized spectra displayed in Figure 8 that show a significant reduction of the intensity of the 2905 cm−1 component with respect the 2415 cm−1 band when the electric vector (E) is ⊥ c. The intensity of the 2905 cm−1 band for both polarization directions is in agreement with the alignment of the O-H vector along a direction intermediate between the crystallographic axes [53].
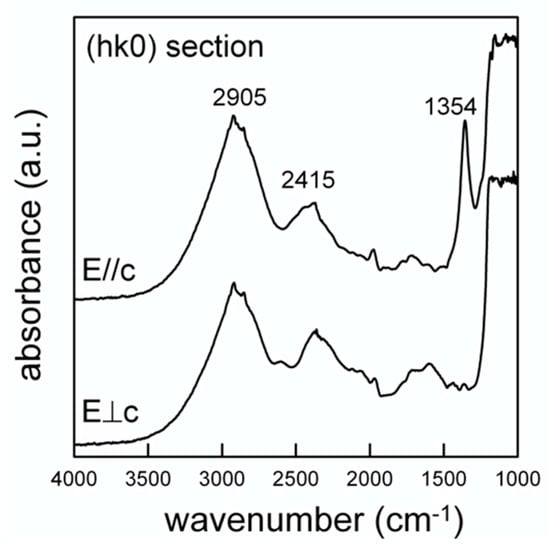
Figure 8.
Single-crystal polarized light FTIR spectra of whitlockite. Doubly polished 100 μm thick (hk0) section.
The structural data show that the hydroxyl oxygen at O(10) is involved in a strong hydrogen bridge with O(1) (Figure 1); neutron diffraction provide the geometry of the hydrogen bond system to be D-H = 1.03(3) Å, H...A = 1.71(3) Å, D...A = 2.739(4) Å and (DHA) = 175(2)°, where D-H = O(10)O–H(10)H, H...A = H(10)H···O(1)O, D...A = O(10)O–O(1)O and (DHA) = (O(10)O–O(10)H···O(1)O) angle. By using the D…A distance, from the relation given by Libowitzki (1999) [54], a band position of 3290 cm−1 is calculated for the stretching mode. Although somehow higher than the observed band (2905 cm−1), this value is in good agreement with the correlation between the O-H stretching frequency and the O-H…O bond distance presented in [54] once we consider the strong incertitude in the curve for strong hydrogen-bonded systems.
The data of Figure 8 also show the appearance of an intense and relatively sharp peak at 1354 cm−1 for E//c. According to [31] this peak can be assigned to the (P)-O-H bending. An additional element supporting this inference comes from the NIR (near infrared) region from 6000–4000 cm−1 where absorptions due to the combination between the OH stretching and bending modes are observed [55]. Regarding the hypothesis of a stretching band at 2905 cm−1 and the (P)-O-H bending at 1354 cm−1, a combination band around 4300 cm−1 is expected in the NIR spectrum; this component is in fact resolvable as a weak and broad band in Figure 9 (arrowed).
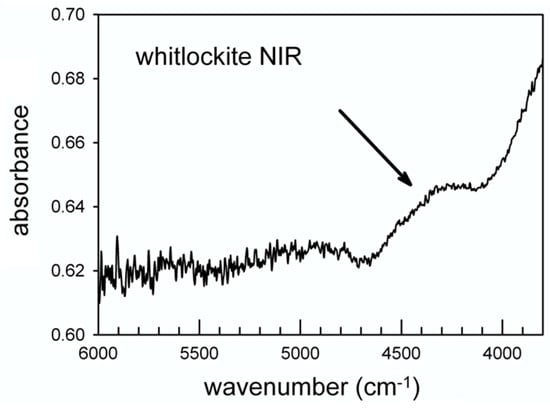
Figure 9.
Single-crystal NIR spectrum of whitlockite.
5. Conclusions
In this work, we provide an in-depth crystal-chemical investigation of the rare natural calcium phosphate whitlockite, ideal formula Ca9Mg(PO4)6[PO3(OH)]. EMP analysis highlighted the non-negligible presence of Na and Fe replacing Ca and Mg, respectively, in the formula. X-ray and neutron refinements showed the presence of a disordered [T(1)PO3(OH)]2− structural unit, split along the c-crystallographic axis. FTIR analysis showed that the powder MIR spectrum in the P-O framework mode region of whitlockite is very similar to that of synthetic β-TCP in terms of band multiplicity, in agreement with the essentially similar structural arrangement between the two compounds. The single-crystal FTIR data are consistent with the band assignment proposed for isotypic wopmayite [31] and show a single and very broad absorption due to an O-H group involved in a strong hydrogen bridge with the surrounding oxygen.
Author Contributions
All the authors conceived and designed the main ideas together, and wrote and supervised the whole work. F.B. provided the sample, the XRD data and the WDS analysis; F.C. contributed to XRD structural refinement; S.C.C. contributed to NDP structural refinement; G.D.V. and F.R. contributed to FTIR spectroscopy. All authors have read and agreed to the published version of the manuscript.
Funding
G.D.V. was supported by MIUR-Italy Dipartimenti di Eccellenza, ARTICOLO 1, COMMI 314–337 LEGGE 232/2016.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data contained in the crystallographic information files (CIF) are openly available at the joint service CSD-ICSD (www.ccdc.cam.ac.uk/getstructures (accessed on 1 February 2021)), quoting the depository number CSD2046688 (XRD refinement), and CSD2046635 (NDP refinement).
Acknowledgments
Neutron data were collected at Beamline SXD, ISIS Neutron and Muon Suorce, Didcot (UK), owing to SXD Xpress Proposal 1990072 ‘Structure investigation of natural whitlockite’ (2019). F.C. is grateful to Francesco Baldassarre (Institute of Crystallography—CNR, Bari, Italy) for helpful discussion on the calcium phosphate topic.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Habraken, W.; Habibovic, P.; Epple, M.; Bohner, M. Calcium phosphates in biomedical applications: Materials for the future? Mater. Today 2016, 19, 69–87. [Google Scholar] [CrossRef]
- Dorozkhin, S.V. Calcium orthophosphates bioceramics. Ceram. Int. 2015, 41, 13913–13966. [Google Scholar] [CrossRef]
- Pasero, M.; Kampf, A.R.; Ferraris, C.; Pekov, I.V.; Rakovan, J.; White, T.J. Nomenclature of the apatite supergroup minerals. Eur. J. Mineral. 2010, 22, 163–179. [Google Scholar] [CrossRef]
- Rossi, M.; Ghiara, M.R.; Chita, G.; Capitelli, F. Crystal-chemical and structural characterization of fluorapatites in ejecta from Somma-Vesuvius volcanic complex. Am. Miner. 2011, 96, 1828–1837. [Google Scholar] [CrossRef]
- Hughes, J.M.; Rakovan, J.F. Structurally robust, chemically diverse: Apatite and apatite supergroup minerals. Elements 2015, 11, 165–170. [Google Scholar] [CrossRef]
- Adcock, C.; Tschauner, O.; Hausrath, E.; Udry, A.; Luo, S.N.; Cai, Y.; Ren, M.; Lanzirotti, A.; Newville, M.; Kunz, M.; et al. Shock-transformation of whitlockite to merrillite and the implications for meteoritic phosphate. Nat. Commun. 2017, 8, 14667. [Google Scholar] [CrossRef]
- Britvin, S.N.; Galuskina, I.O.; Vlasenko, N.S.; Vereshchagin, O.S.; Bocharov, V.N.; Krzhizhanovskaya, M.G.; Shilovskikh, V.V.; Galuskin, E.V.; Vapnik, Y.; Obolonskaya, E.V. Keplerite, Ca9(Ca0.5□0.5)Mg(PO4)7, a new meteoritic and terrestrial phosphate isomorphous with merrillite, Ca9NaMg(PO4)7. Am. Mineral. 2021, 106. [Google Scholar] [CrossRef]
- Shah, F.A.; Lee, B.E.J.; Tedesco, J.; Larsson Wexell, C.; Persson, C.; Thomsen, P.; Grandfield, K.; Palmquist, A. Micrometer-Sized Magnesium Whitlockite Crystals in Micropetrosis of Bisphosphonate-Exposed Human Alveolar Bone. Nano Lett. 2017, 17, 6210–6216. [Google Scholar] [CrossRef]
- Sakae, T.; Yamamoto, H.; Mishima, H.; Matsumoto, T.; Kozawa, Y. Morphology and chemical composition of dental calculi mainly composed of whitlockite. Scanning Microsc. 1989, 3, 855–860. [Google Scholar] [PubMed]
- El Khouri, A.; Elaatmani, M.; Della Ventura, G.; Sodo, A.; Rizzi, R.; Rossi, M.; Capitelli, F. Synthesis, structure refinement and vibrational spectroscopy of new rare-earth tricalcium phosphates Ca9RE(PO4)7 (RE = La,-Pr, Nd, Eu, Gd, Dy, Tm, Yb). Ceram. Int. 2017, 43, 15645–15653. [Google Scholar] [CrossRef]
- Capitelli, F.; Rossi, M.; El Khouri, A.; Elaatmani, M.; Corriero, N.; Sodo, A.; Della Ventura, G. Synthesis, structural model and vibrational spectroscopy of lutetium tricalcium phosphate Ca9Lu(PO4)7. J. Rare Earths 2018, 36, 1162–1168. [Google Scholar] [CrossRef]
- Altomare, A.; Rizzi, R.; Rossi, M.; El Khouri, A.; Elaatmani, M.; Paterlini, V.; Della Ventura, G.; Capitelli, F. New Ca2.90(Me2+)0.10(PO4)2 β-tricalcium Phosphates with Me2+ = Mn, Ni, Cu: Synthesis, Crystal-Chemistry, and Luminescence Properties. Crystals 2019, 9, 288. [Google Scholar] [CrossRef]
- Horowitz, R.A.; Mazor, Z.; Foitzik, C.; Prasad, H.; Rohrer, M.; Palti, A. β-tricalcium phosphate as bone substitute material: Properties and clinical applications. J. Osseointegration 2010, 1, 61–68. [Google Scholar]
- Guo, X.; Liu, X.; Gao, H.; Shi, X.; Zhao, N.; Wang, Y. Hydrothermal growth of whitlockite coating on β-tricalcium phosphate surfaces for enhancing bone repair potential. J. Mater. Sci. Technol. 2018, 34, 1054–1059. [Google Scholar] [CrossRef]
- Baldassarre, F.; Altomare, A.; Corriero, N.; Mesto, E.; Lacalamita, M.; Bruno, G.; Sacchetti, A.; Dida, B.; Karaj, D.; Della Ventura, G.D.; et al. Crystal Chemistry and Luminescence Properties of Eu-Doped Polycrystalline Hydroxyapatite Synthesized by Chemical Precipitation at Room Temperature. Crystals 2020, 10, 250. [Google Scholar] [CrossRef]
- Paterlini, V.; Bettinelli, M.; Rizzi, R.; El Khouri, A.; Rossi, M.; Della Ventura, G.; Capitelli, F. Characterization and Luminescence of Eu3+- and Gd3+-Doped Hydroxyapatite Ca10(PO4)6(OH)2. Crystals 2020, 10, 806. [Google Scholar] [CrossRef]
- Bessière, A.; Ait Benhamou, R.; Wallez, G.; Lecointre, A.; Viana, B. Site occupancy and mechanism of thermally stimulated luminescence in Ca9Ln(PO4)7 (Ln = Lanthanide). Acta Mater. 2012, 60, 6641–6649. [Google Scholar] [CrossRef]
- Atencio, D.; de Almeida Azzi, A. Cerite: A new supergroup of minerals and cerite-(La) renamed ferricerite-(La). Mineral. Mag. 2017, 84, 928–931. [Google Scholar] [CrossRef]
- Bosi, F.; Biagioni, C.; Oberti, R. On the chemical identification and classification of minerals. Minerals 2019, 9, 591. [Google Scholar] [CrossRef]
- Witzke, T.; Phillips, B.L.; Woerner, W.; Countinho, J.M.V.; Farber, G.; Contreira Filho, R.R. Hedegaardite, IMA 2014- 069. CNMNC Newsletter No. 23, February 2015, page 54. Mineral. Mag. 2015, 79, 51–58. [Google Scholar]
- Bruker AXS Inc. APEX 2. Bruker Advanced X-ray Solutions; Bruker AXS Inc.: Madison, WI, USA, 2004. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Gopal, R.; Calvo, C. Structural relationship of whitlockite and β-Ca3(PO4)2. Nat. Phys. Sci. 1972, 237, 30–32. [Google Scholar] [CrossRef]
- Keen, D.A.; Gutmann, M.J.; Wilson, C.C. SXD-the single-crystal diffractometer at the ISIS spallation neutron source. J. Appl. Cryst. 2006, 39, 714–722. [Google Scholar] [CrossRef]
- Gutmann, M. SXD2001-a program for treating data from TOF neutron single-crystal diffraction. Acta Cryst. A 2005, 61, c164. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Inorganic Crystal Structure Database (ICSD); Version 2018–2; Fachinformationszentrum: Karlsruhe, Germany, 2018.
- Calvo, C.; Gopal, R. The crystal structure of whitlockite from the Palermo quarry. Am. Mineral. 1975, 60, 120–133. [Google Scholar]
- Hughes, J.M.; Jolliff, B.L.; Rakovan, J.F. The crystal chemistry of whitlockite and merrillite and the dehydrogenation of whitlockite to merrillite. Am. Mineral. 2008, 93, 1300–1305. [Google Scholar] [CrossRef]
- Britvin, S.N.; Pakhomovskii, Y.A.; Bogdanova, A.N.; Skiba, V.I. Strontiowhitlockite, Sr9Mg(PO3OH)(PO4)6. A new mineral species from the Kovdor Deposit, Kola-Peninsula, SSR. Can. Mineral. 1991, 29, 87–93. [Google Scholar]
- Cooper, M.A.; Hawthorne, F.C.; Abdu, Y.A.; Ball, N.A.; Ramik, R.A.; Tait, K.T. Wopmayite, Ideally Ca6Na3□Mn(PO4)3(PO3OH)4, A New Phosphate Mineral From The Tanco Mine, Bernic Lake, Manitoba: Description And Crystal Structure. Can. Mineral. 2013, 51, 93–106. [Google Scholar] [CrossRef]
- Hughes, J.M.; Jolliff, B.L.; Gunter, M.E. The atomic arrangement of merrillite from the Fra Mauro Formation, Apollo 14 lunar mission: The first structure of merrillite from the Moon. Am. Mineral. 2006, 91, 1547–1552. [Google Scholar] [CrossRef]
- Xie, X.; Yang, H.; Gu, X.; Downs, R.T. Chemical composition and crystal structure of merrillite from the Suizhou meteorite. Am. Mineral. 2015, 100, 2753–2756. [Google Scholar] [CrossRef]
- Britvin, S.N.; Krivovichev, S.V.; Armbruster, T. Ferromerrillite, Ca9NaFe2+(PO4)7, a new mineral from the Martian meteorites, and some insights into merrillite-tuite transformation in shergottites. Eur. J. Mineral. 2016, 28, 125–136. [Google Scholar] [CrossRef]
- Hwang, S.L.; Shen, P.Y.; Chu, H.T.; Yui, T.F.; Varela, M.E.; Iizuka, Y. New minerals tsangpoite Ca5(PO4)2(SiO4) and matyhite Ca9(Ca0.5□0.5)Fe(PO4)7 from the D’Orbigny angrite. Mineral. Mag. 2019, 83, 293–313. [Google Scholar] [CrossRef]
- Yashima, M.; Sakai, A.; Kamiyama, T.; Hoshikawa, A. Crystal structure analysis of β-tricalcium phosphate Ca3(PO4)2 by neutron powder diffraction. J. Solid State Chem. 2003, 175, 272–277. [Google Scholar] [CrossRef]
- Piccinelli, F.; Trevisani, M.; Plaisier, J.R.; Bettinelli, M. Structural study of Yb3+, Eu3+ and-Pr3+ doped Ca9Lu(PO4)7. J. Rare Earths 2015, 33, 977–982. [Google Scholar] [CrossRef]
- Brown, I.D.; Altermatt, D. Bond-valence parameters obtained from a systematic analysis of the Inorganic Crystal Structure Database. Acta Cryst. B 1985, 41, 244–247. [Google Scholar] [CrossRef]
- Capitelli, F.; Khaoulaf, R.; Harcharras, M.; Ennaciri, A.; Habyby, S.H.; Valentini, V.; Mattei, G.; Bertolasi, V. Crystal structure and vibrational spectroscopy of the new acidic diphosphate (NH4)2Zn(H2P2O7)2·2H2O. Z. Kristallogr. 2005, 220, 25–30. [Google Scholar]
- Capitelli, F.; Chita, G.; Ghiara, M.R.; Rossi, M. Crystal chemical investigation of Fe3(PO4)2∙8H2O vivianite minerals. Z. Kristallogr. 2012, 227, 92–101. [Google Scholar] [CrossRef]
- Capitelli, F.; Harcharras, M.; Assaaoudi, H.; Ennaciri, A.; Moliterni, A.G.G.; Bertolasi, V. Crystal structure of new hexahydrate dicobalt pyrophosphate Co2P2O7·6H2O: Comparison with Co2P2O7·2H2O, α-, β- and γ- Co2P2O7. Z. Kristallogr. 2013, 218, 345–350. [Google Scholar] [CrossRef]
- Zatovsky, I.V.; Strutynska, N.Y.; Baumer, V.N.; Shishkin, O.V.; Slobodyanik, N.S. The whitlockite-related-phosphate Ca9Cr(PO4)7. Acta Crystallogr. E 2007, 63, i180–i181. [Google Scholar] [CrossRef]
- Lazoryak, B.I.; Morozov, V.A.; Belik, A.A.; Khasanov, S.S.; Shekhtman, V.S. Crystal structures and characterization of Ca9Fe(PO4)7 and Ca9FeH0.9(PO4)7. J. Solid State Chem. 1996, 122, 15–21. [Google Scholar] [CrossRef]
- Tait, K.T.; Barkley, M.C.; Thomson, R.C.; Origlieri, M.J.; Evans, C.T.; Yang, H. Bobdownsite, a new mineral species from Big Fish River, Yukon, Canada, and its structural relationship with whitlockite-type compounds. Can. Mineral. 2011, 49, 1065–1078. [Google Scholar] [CrossRef]
- McCubbin, F.M.; Phillips, B.L.; Adcock, C.T.; Tait, K.T.; Steele, A.; Vaughn, J.S.; Fries, M.D.; Atudorei, V.; Kaaden, K.E.V.; Hausrath, E.M. Discreditation of bobdownsite and the establishment of criteria for the identification of minerals with essential monofluorophosphate (PO3F)2-. Am. Mineral. 2018, 103, 1319–1328. [Google Scholar] [CrossRef]
- Batool, S.; Liaqat, U.; Hussain, Z.; Sohail, M. Synthesis, characterization and process optimization of bone whitlockite. Nanomaterials 2020, 10, 1856. [Google Scholar] [CrossRef] [PubMed]
- De Aza, P.N.; Guitián, F.; Santos, C.; De Aza, S.; Cuscó, R.; Artús, L. Vibrational Properties of Calcium Phosphate Compounds. 2. Comparison between Hydroxyapatite and β-Tricalcium-Phosphate. Chem. Mater. 1997, 9, 916–922. [Google Scholar] [CrossRef]
- Jillavenkatesa, A.; Condrate, R.A., Sr. The Infrared and Raman Spectra of α- and β-Tricalcium-Phosphate (Ca3(PO4)2). Spectrosc. Lett. 1998, 31, 1619–1634. [Google Scholar] [CrossRef]
- Harcharras, M.; Ennaciri, A.; Capitelli, F.; Mattei, G. Vibrational spectra and thermal dehydration of Co2P2O7·6H2O diphosphate. Vib. Spectrosc. 2003, 33, 189–196. [Google Scholar] [CrossRef]
- Ross, S.D. Phosphates and other oxy-anions of group V. In Mineralogical Society Monograph 4: The Infrared Spectra of Minerals; Farmer, V.C., Ed.; Mineralogical Society of Great Britain and Ireland: London, UK, 1974. [Google Scholar]
- Radica, F.; Della Ventura, G.; Bellatreccia, F.; Cinque, G.; Cestelli Guidi, M. The diffusion kinetics of CO2 in cordierite: An HT-FTIR micro-spectroscopy study. Contrib. Mineral. Petrol. 2016, 171, 12. [Google Scholar] [CrossRef]
- Moraes, A.P.A.; Romano, R.; Souza Filho, A.G.; Freire, P.T.C.; Mendes Filho, J.; Alves, O.L. Vibrational spectra of α-Ge(HPO4)2·H2O compound. Vib. Spectrosc. 2006, 40, 209–212. [Google Scholar] [CrossRef]
- Della Ventura, G.; Bellatreccia, F.; Radica, F.; Chopin, C.; Oberti, R. The arrojadite enigma III. The incorporation of volatiles: A polarised FTIR spectroscopy study. Eur. J. Mineral. 2014, 26, 679–688. [Google Scholar] [CrossRef]
- Libowitzky, E. Correlation of O-H stretching frequencies and O-H...O hydrogen bond lengths in minerals. Monat. Chem. 1999, 130, 1047–1059. [Google Scholar]
- Della Ventura, G.; Gatta, D.; Redhammer, G.; Bellatreccia, F.; Loose, A.; Parodi, G.C. Single-crystal polarized FTIR spectroscopy and neutron diffraction refinement of cancrinite. Phys. Chem. Miner. 2009, 36, 193–206. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).